
Mx Is Not Responsible for the Antiviral Activity of Interferon-α against Japanese Encephalitis Virus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Interferon
2.2. Virus Infection and Titration
2.3. Antibodies
2.4. Plasmids
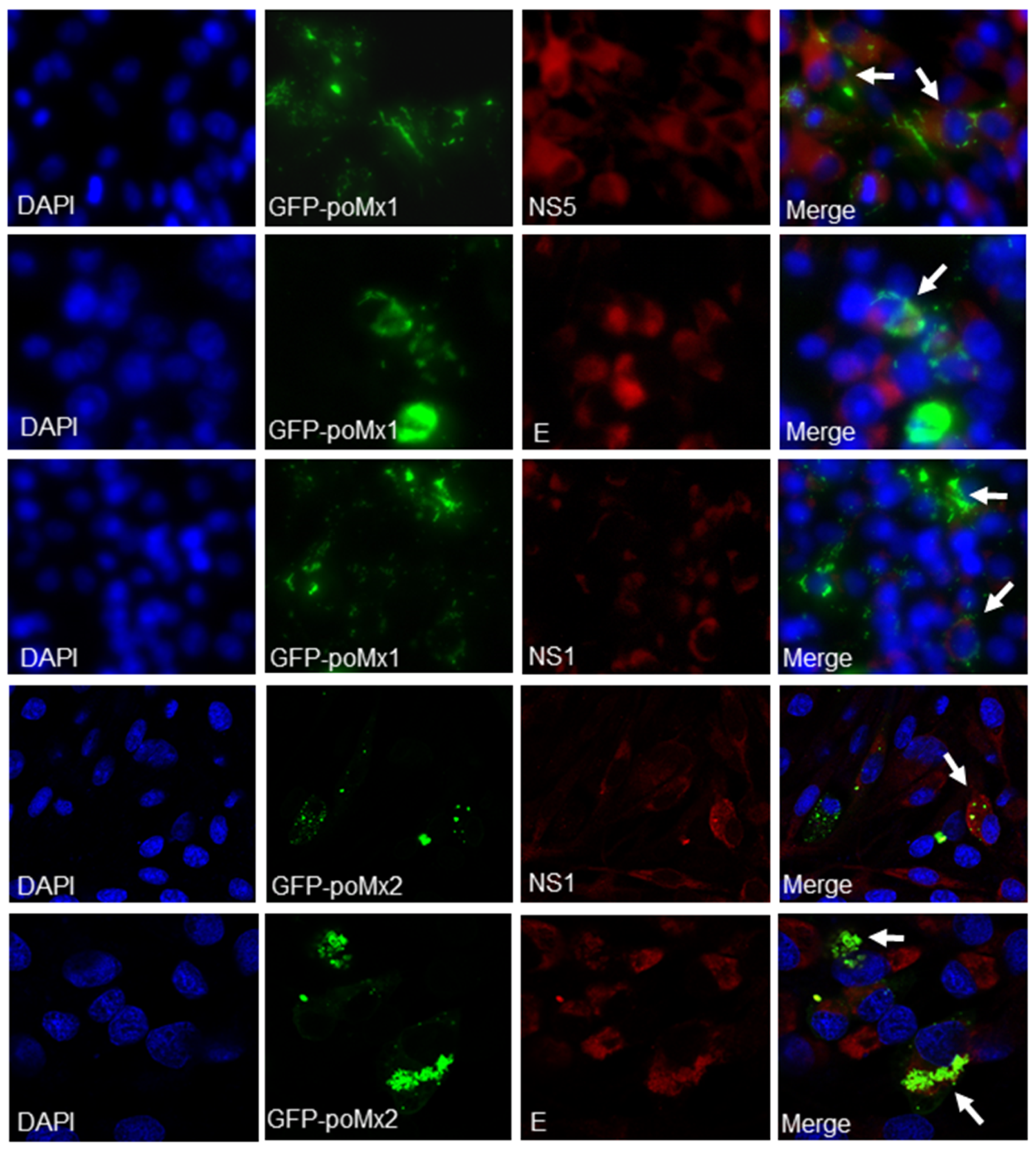
2.5. Immunofluorescence Assay
2.6. Western Blot Analysis
2.7. Brefeldin A (BFA) Treatment
2.8. Knockdown Experiments
2.9. RT-qPCR
2.10. Statistical Analysis
3. Results
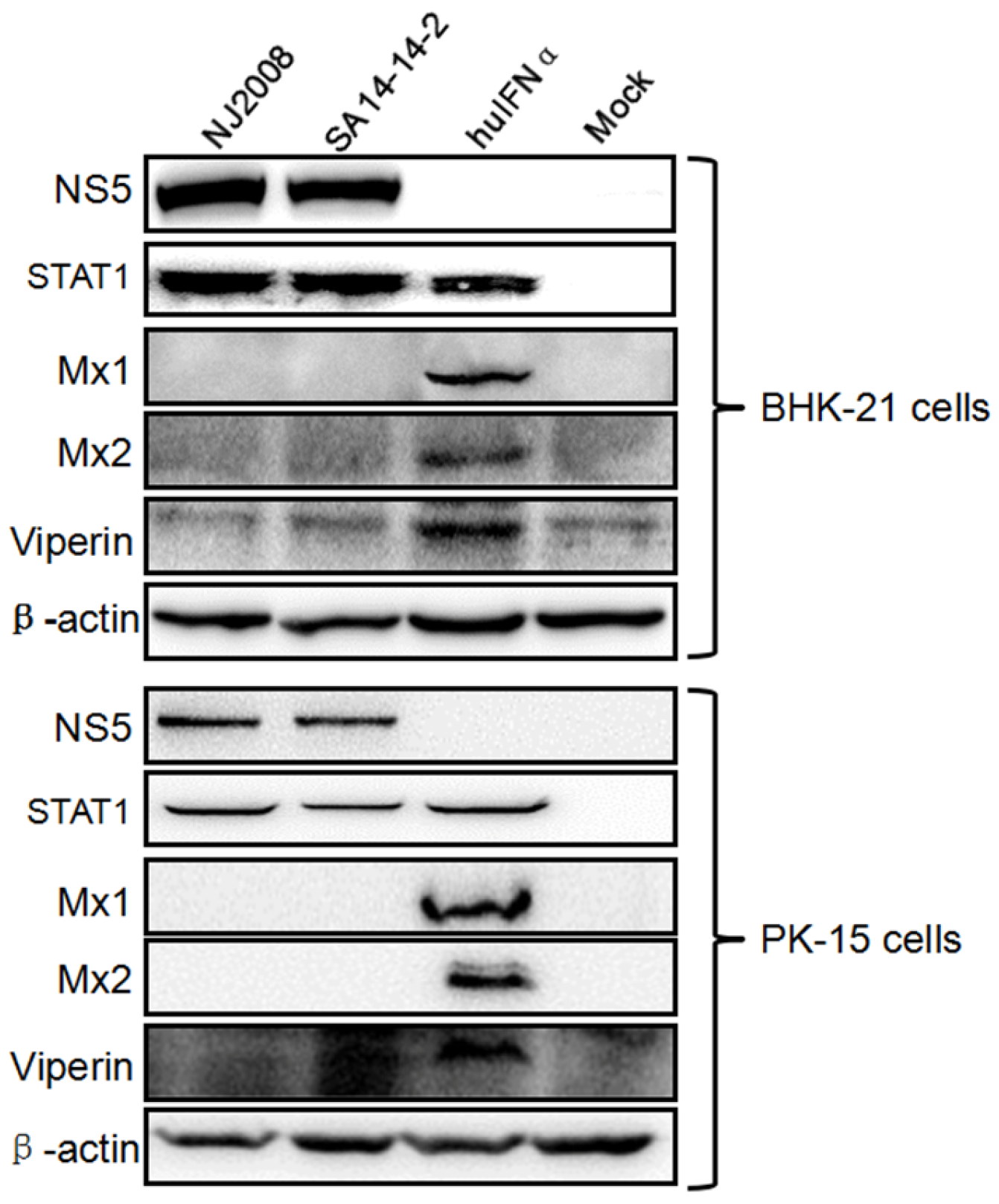
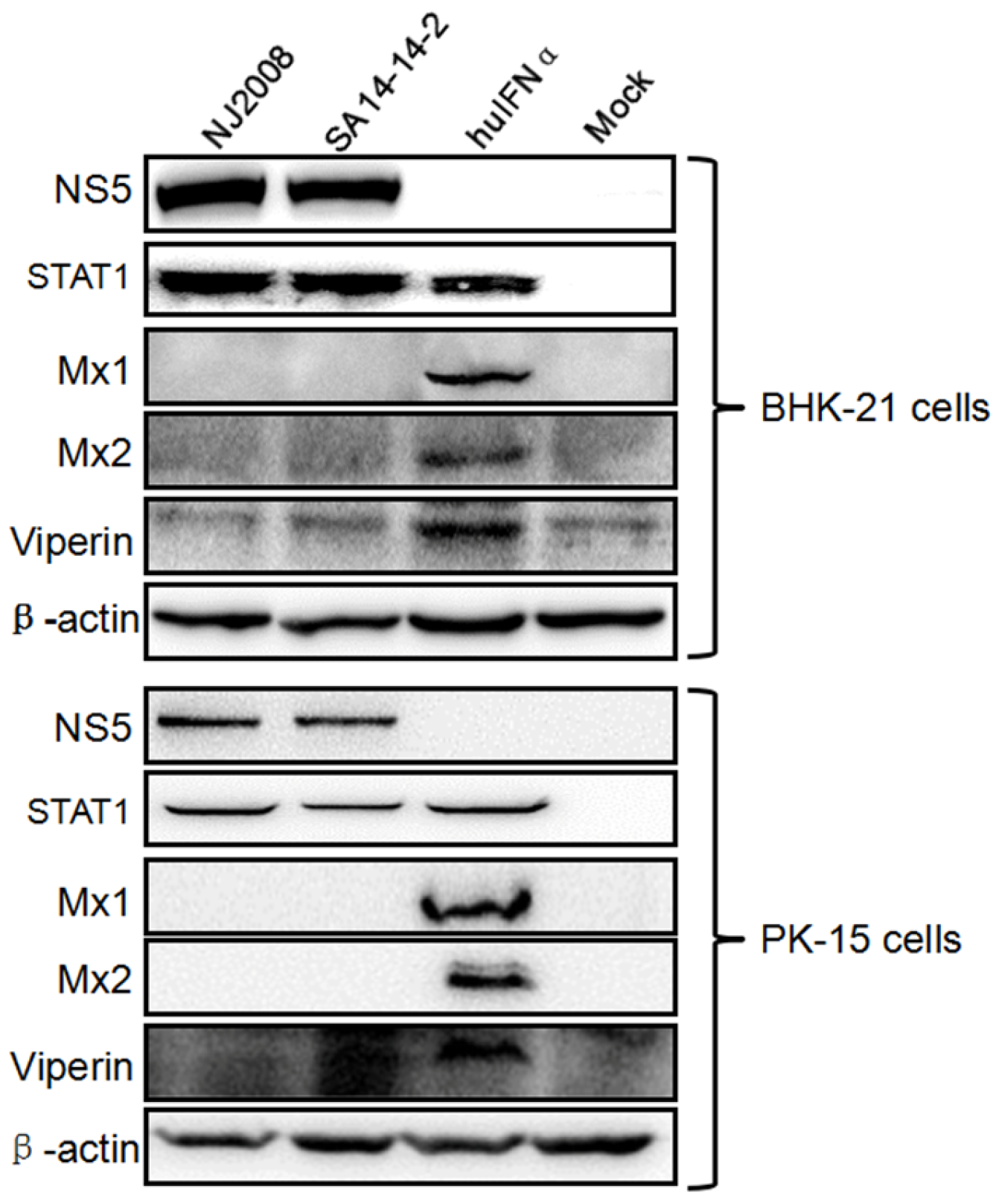
3.1. Mx Proteins Were Not Detectable in JEV-Infected Cells
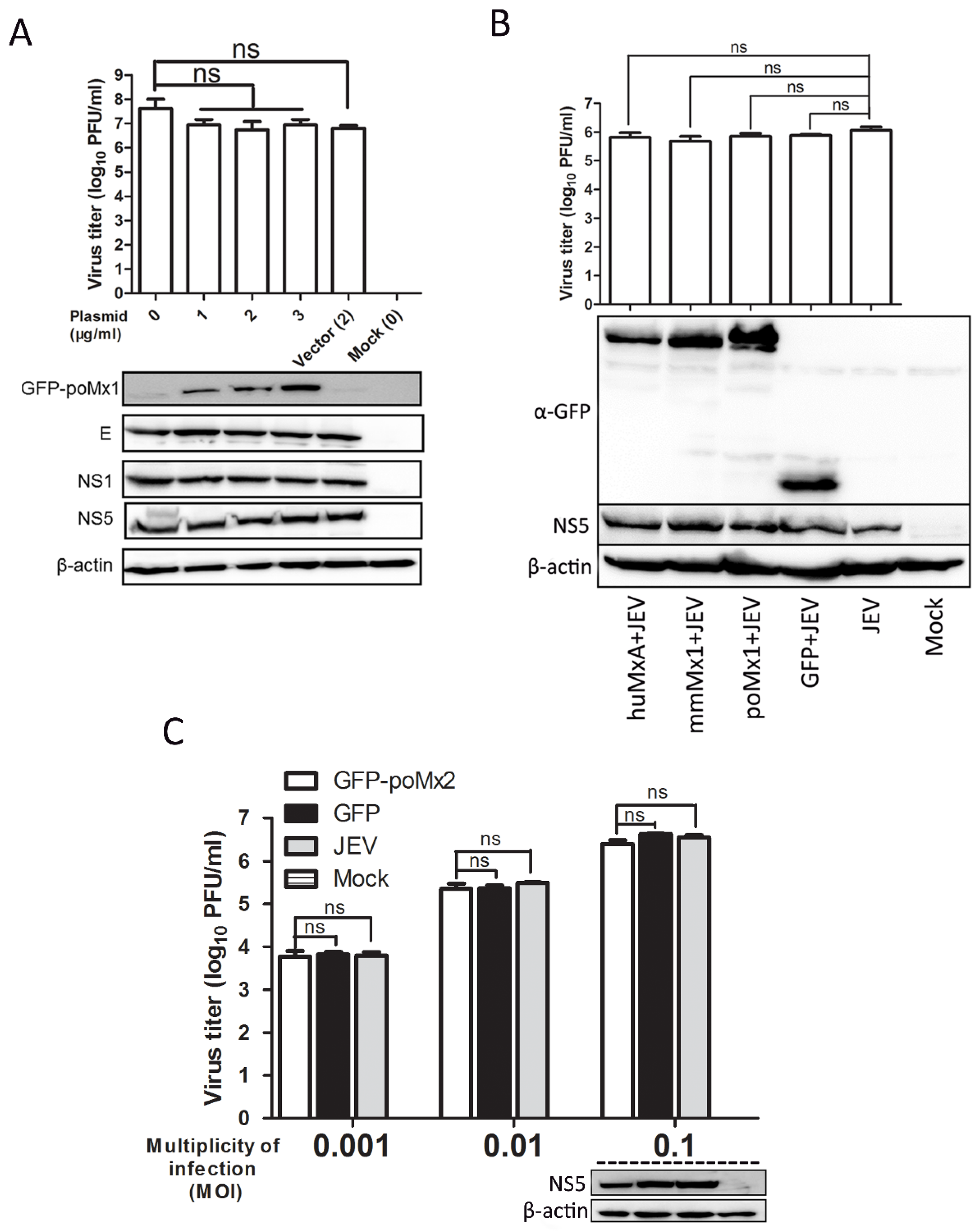
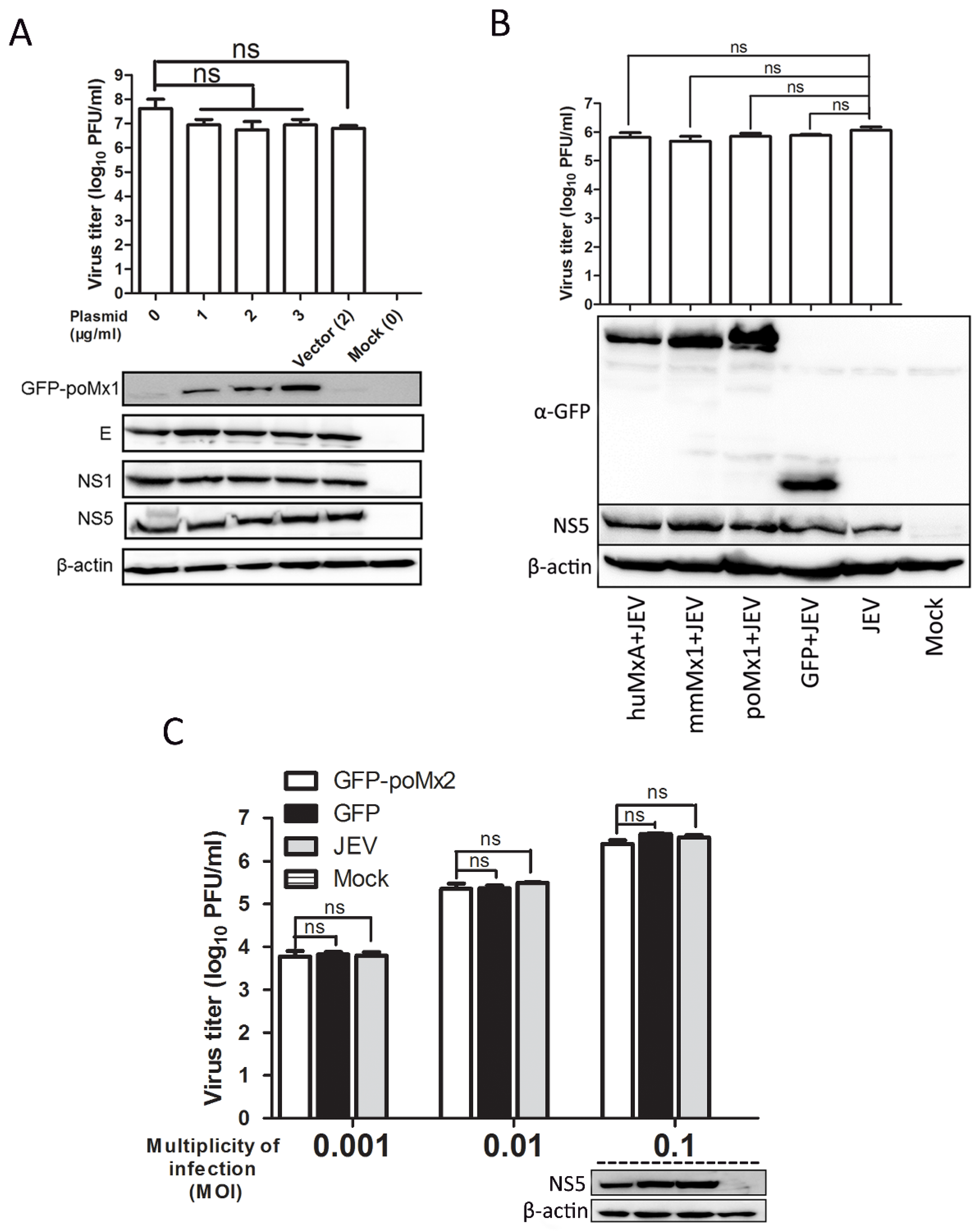
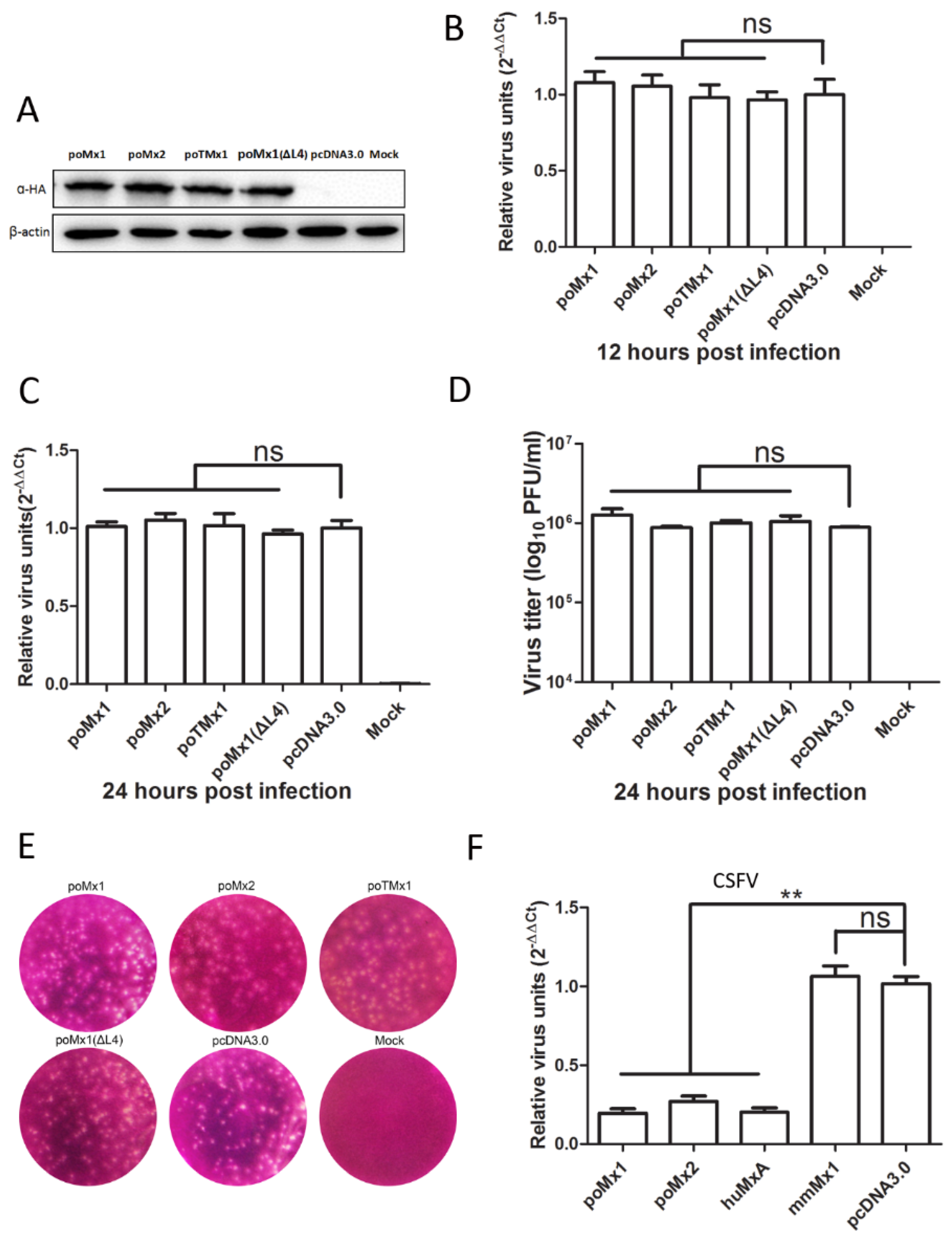
3.2. Exogenous Mx Proteins Have No Anti-JEV Activity in Infected Cells
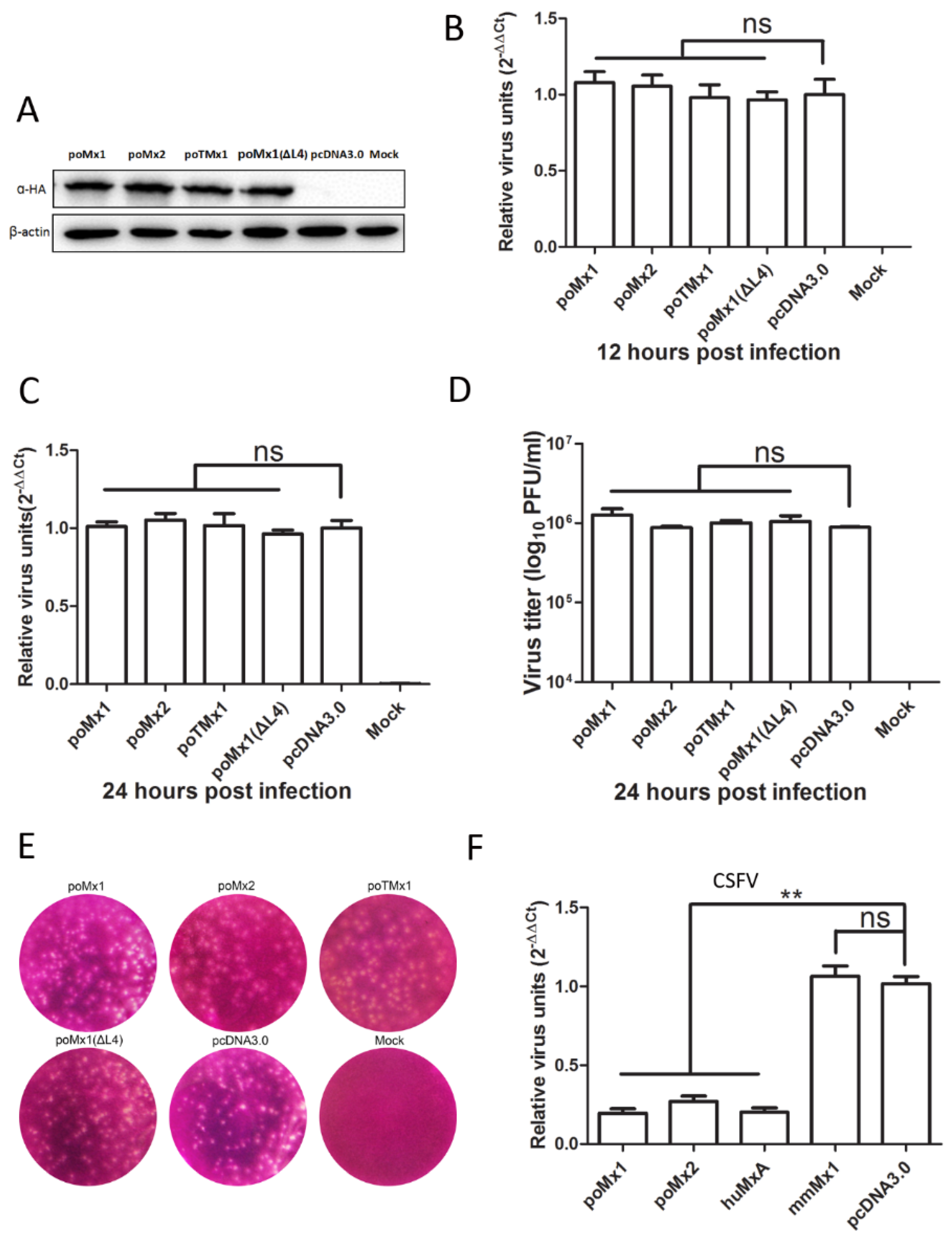
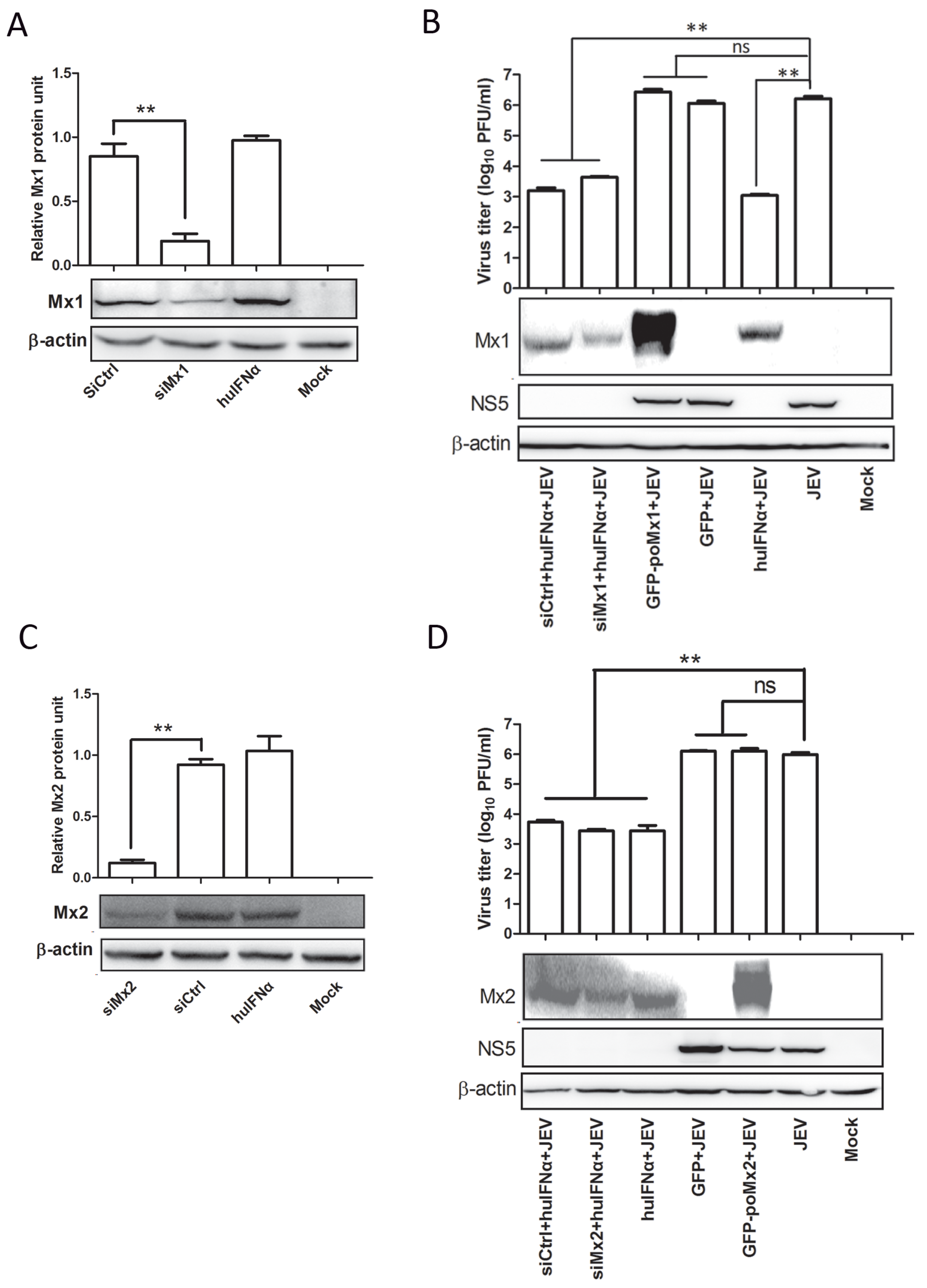
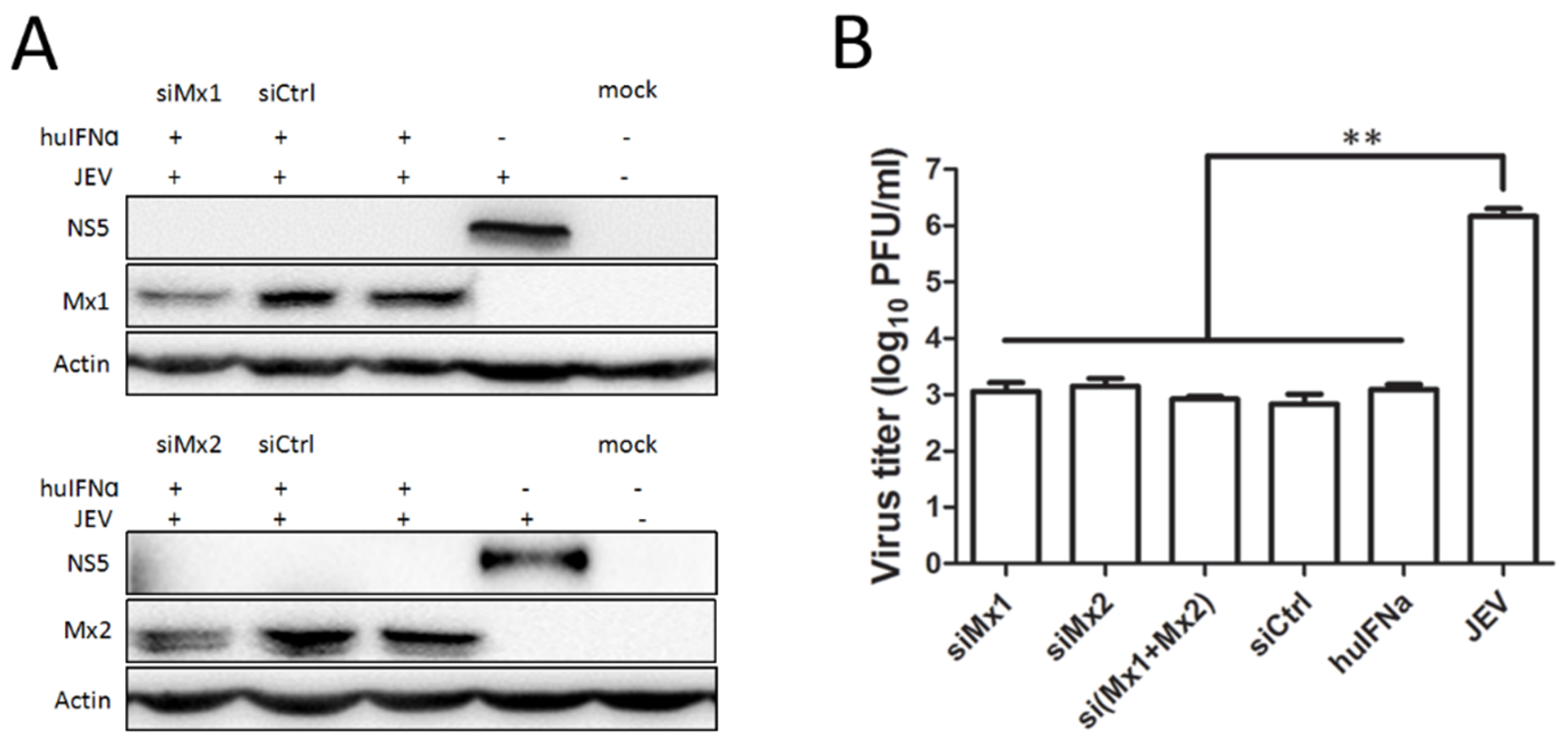
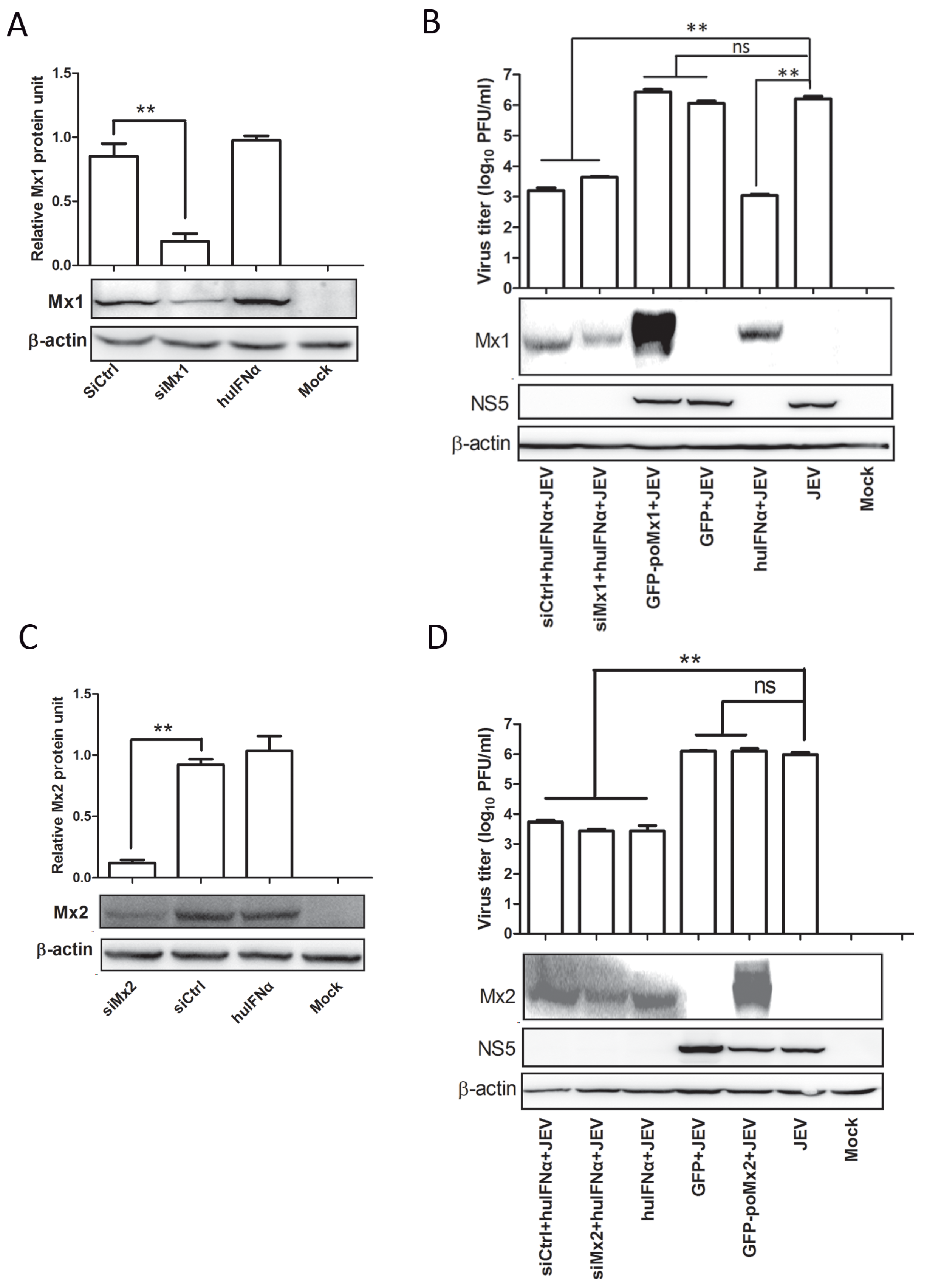
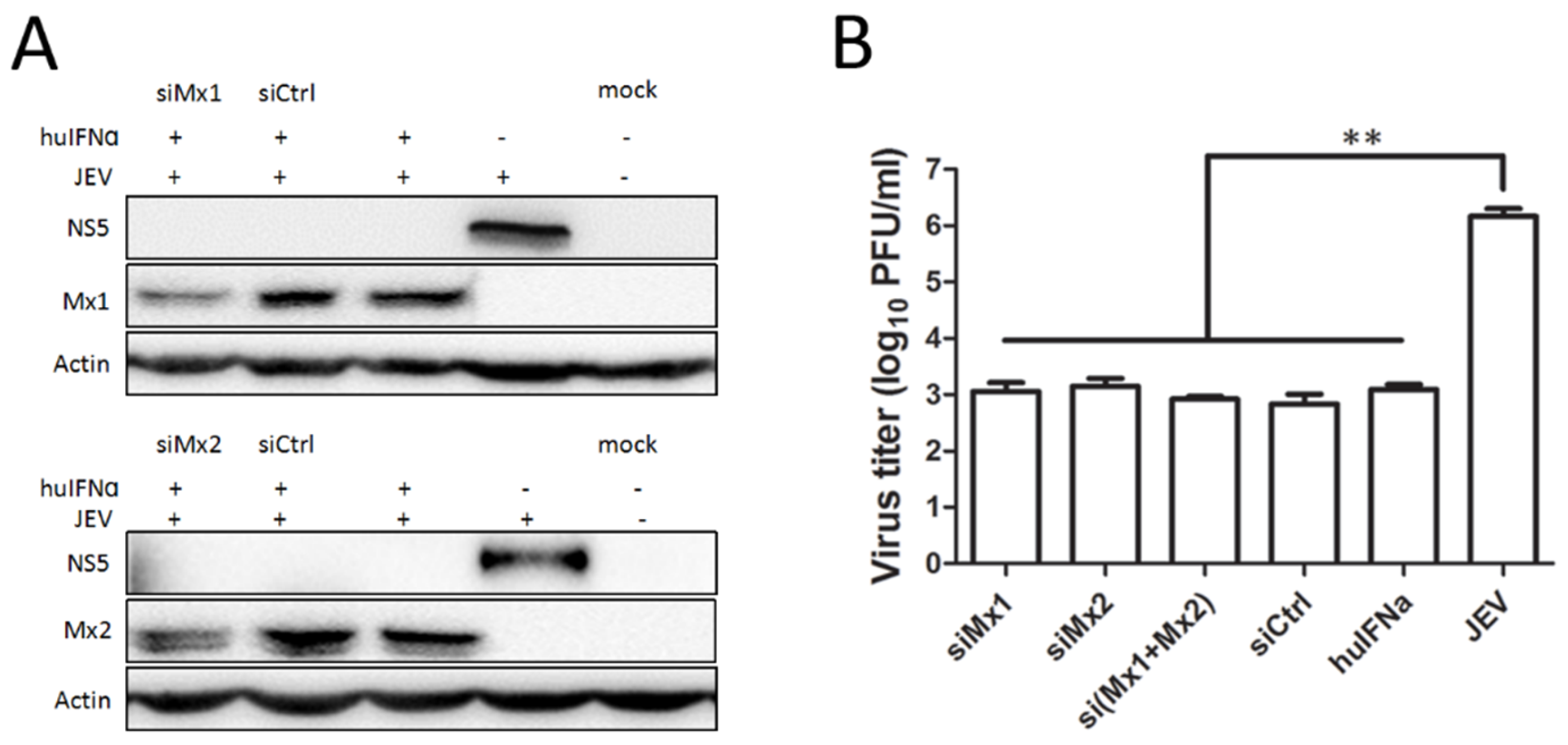
3.3. Mx1 or Mx2 Depletion Did Not Affect the Antiviral Activity of IFN
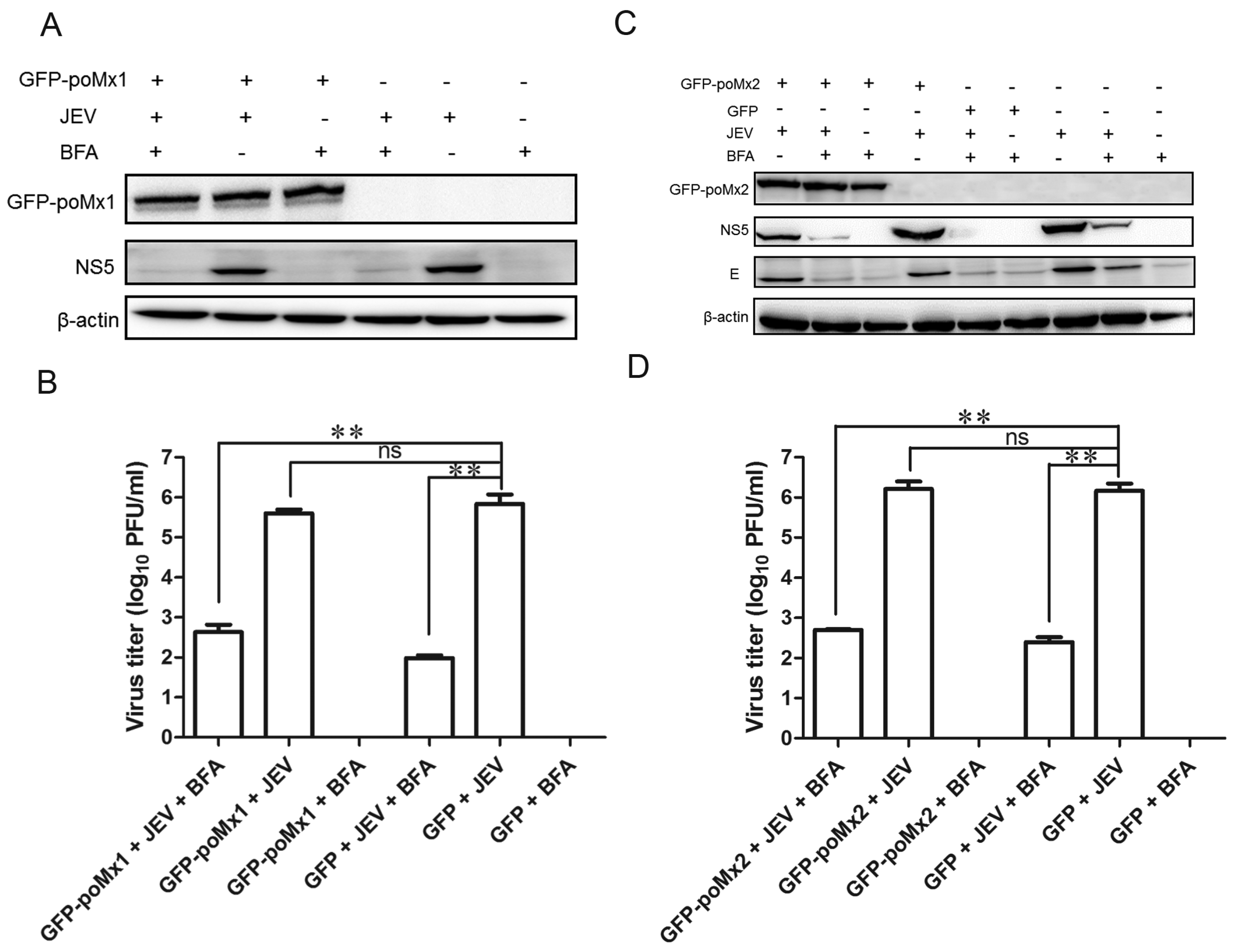
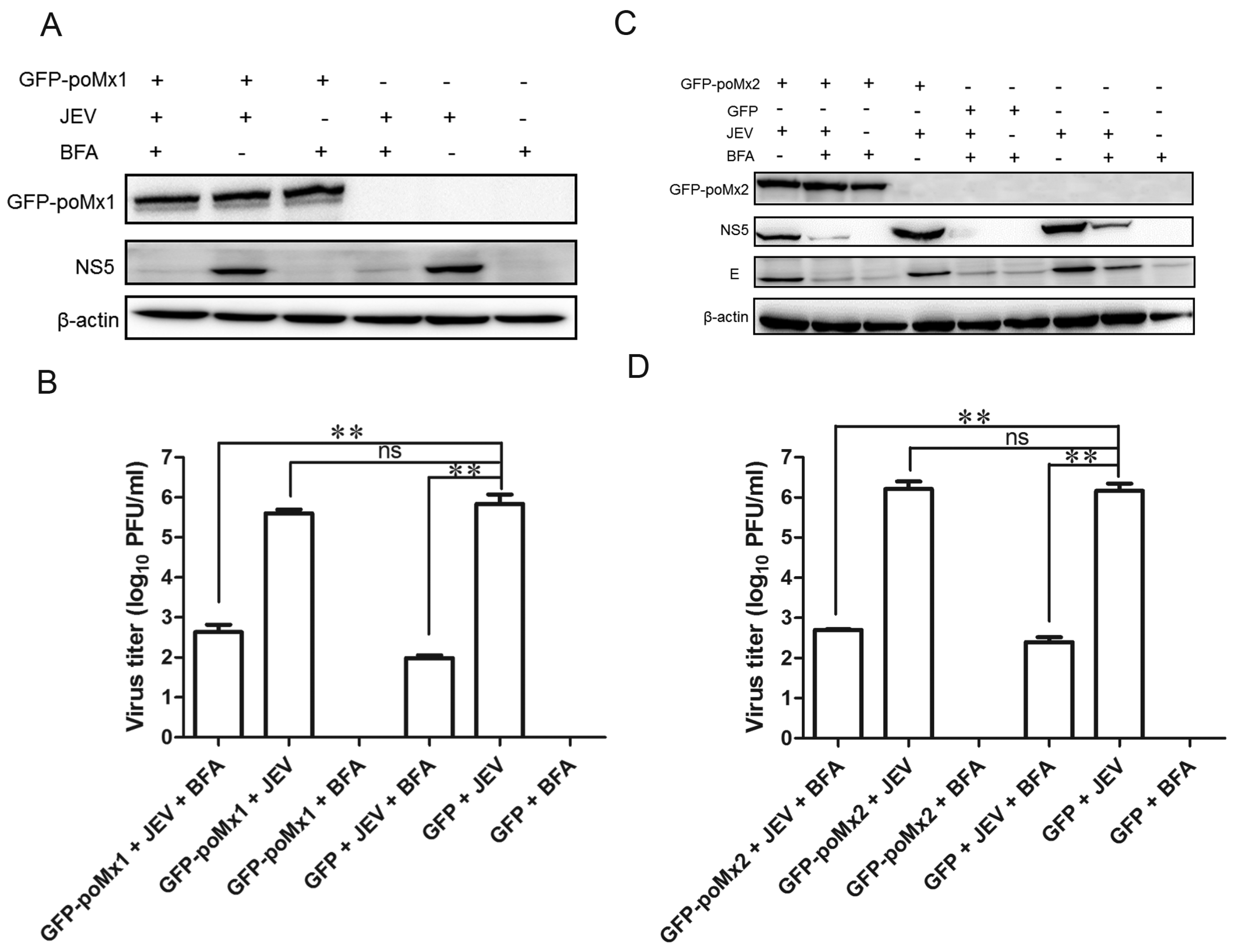
3.4. Mx Does Not Inhibit JEV Replication in Cells Treated with BFA
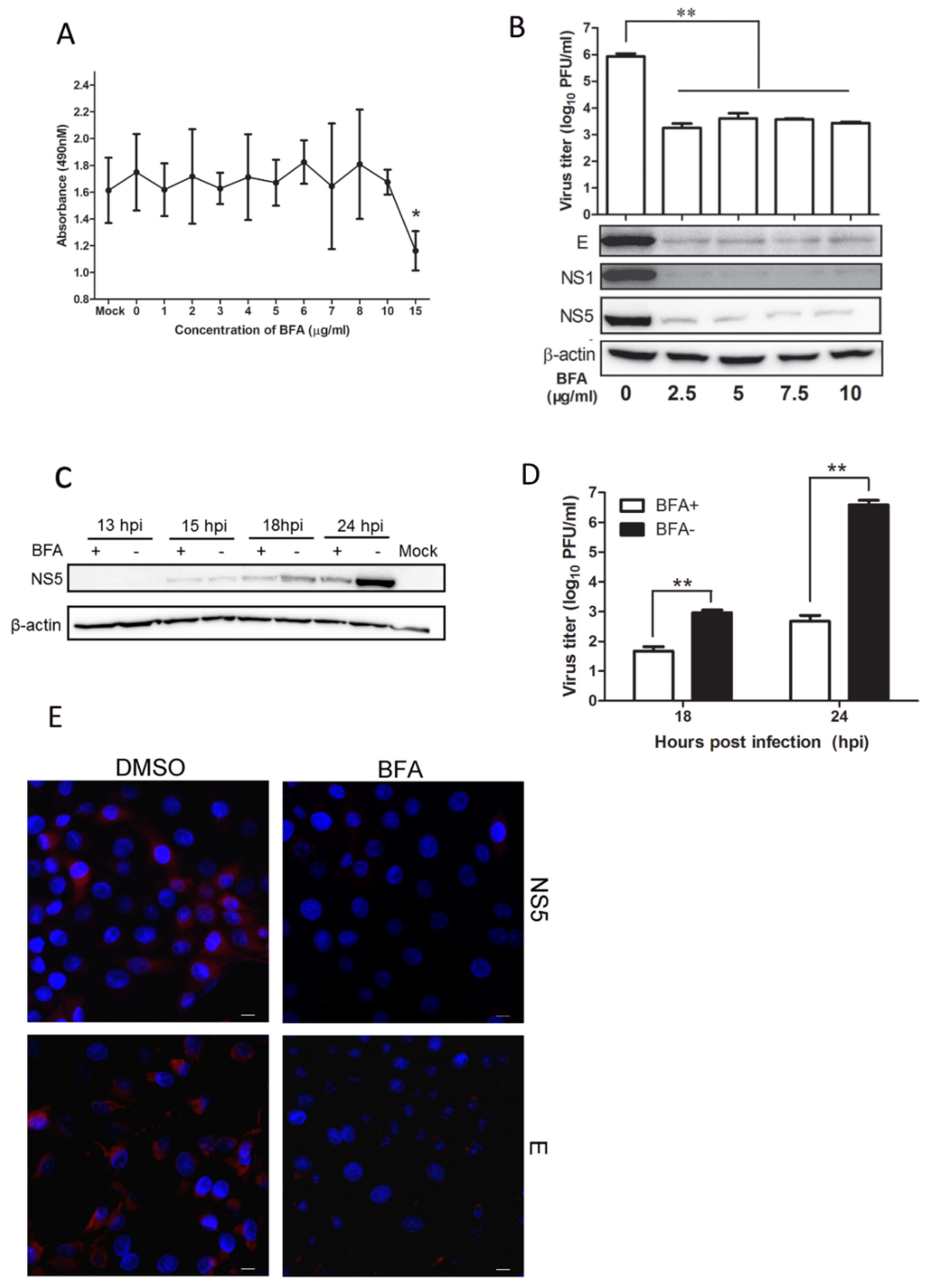
3.5. BFA Effectively Inhibits JEV Replication
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van den Hurk, A.F.; Ritchie, S.A.; Mackenzie, J.S. Ecology and geographical expansion of Japanese encephalitis virus. Annu. Rev. Entomol. 2009, 54, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Takasaki, T.; Fu, S.H.; Sun, X.H.; Zhang, H.L.; Wang, Z.X.; Hao, Z.Y.; Zhang, J.K.; Tang, Q.; Kotaki, A.; et al. Molecular epidemiological analysis of Japanese encephalitis virus in China. J. Gen. Virol. 2007, 88, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.; He, D.N.; Zhou, B.; Liu, K.; Zhao, J.; Zhang, X.M.; Chen, P.Y. In vitro inhibition of Japanese encephalitis virus replication by capsid-targeted virus inactivation. Antivir. Res. 2013, 97, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Verma, R. Japanese encephalitis vaccine: Need of the hour in endemic states of India. Hum. Vaccines Immunother. 2012, 8, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Doughty, L.; Nguyen, K.; Durbin, J.; Biron, C. A role for IFN-alpha beta in virus infection-induced sensitization to endotoxin. J. Immunol. 2001, 166, 2658–2664. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Liao, X.; Zhou, B.; Yao, H.; Fan, S.; Chen, P.; Miao, D. Porcine alpha interferon inhibit Japanese encephalitis virus replication by different ISGs in vitro. Res. Vet. Sci. 2013, 95, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhu, D.; Lian, X.; Liu, W.; Cao, R.; Chen, P. Porcine 2′,5′-oligoadenylate synthetases inhibit Japanese encephalitis virus replication in vitro. J. Med. Virol. 2016, 88, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Charleston, B.; Stewart, H.J. An interferon-induced Mx protein: cDNA sequence and high-level expression in the endometrium of pregnant sheep. Gene 1993, 137, 327–331. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G. Interferon-induced Mx proteins: Dynamin-like GTPases with antiviral activity. Traffic 2002, 3, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Stertz, S.; Kochs, G. The Mx GTPase family of interferon-induced antiviral proteins. Microbes Infect. 2007, 9, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, J.; Hulpiau, P.; Saelens, X. Mx proteins: Antiviral gatekeepers that restrain the uninvited. Microbiol. Mol. Biol. Rev. 2013, 77, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jing, J.; Li, W.; Liu, K.; Shi, B.; Xu, Q.; Ma, Z.; Zhou, B.; Chen, P. Porcine Mx1 fused to HIV Tat protein transduction domain (PTD) inhibits classical swine fever virus infection in vitro and in vivo. BMC Vet. Res. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Asano, A.; Ko, J.H.; Morozumi, T.; Hamashima, N.; Watanabe, T. Polymorphisms and the antiviral property of porcine Mx1 protein. J. Vet. Med. Sci. 2002, 64, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, E.; Morozumi, T.; Tsukamoto, K.; Watanabe, T.; Plastow, G.; Mitsuhashi, T. A naturally occurring variant of porcine Mx1 associated with increased susceptibility to influenza virus in vitro. Biochem. Genet. 2007, 45, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Palm, M.; Leroy, M.; Thomas, A.; Linden, A.; Desmecht, D. Differential anti-influenza activity among allelic variants at the Sus scrofa Mx1 locus. J. Interferon Cytokine Res. 2007, 27, 147–155. [Google Scholar] [CrossRef] [PubMed]
- He, D.N.; Zhang, X.M.; Liu, K.; Pang, R.; Zhao, J.; Zhou, B.; Chen, P.Y. In vitro inhibition of the replication of classical swine fever virus by porcine Mx1 protein. Antivir. Res. 2014, 104, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Fang, H.; Shen, C.; Zheng, C. Expression of porcine Mx1 with FMDV IRES enhances the antiviral activity against foot-and-mouth disease virus in PK-15 cells. Arch. Virol. 2015, 160, 1989–1999. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Fu, Q.; Ren, Y.; Wang, D.; Qiao, J.; Wang, P.; Zhang, H.; Chen, C. Both foot-and-mouth disease virus and bovine viral diarrhea virus replication are inhibited by Mx1 protein originated from porcine. Anim. Biotechnol. 2015, 26, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Patzina, C.; Haller, O.; Kochs, G. Structural requirements for the antiviral activity of the human MxA protein against Thogoto and influenza A virus. J. Biol. Chem. 2014, 289, 6020–6027. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Gao, S.; von der Malsburg, A.; Daumke, O.; Kochs, G. Dynamin-like MxA GTPase: Structural insights into oligomerization and implications for antiviral activity. J. Biol. Chem. 2010, 285, 28419–28424. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human Mx2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. Mx2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, P.; Cao, R.; Gu, J. Mutation of putative N-linked glycosylation sites in Japanese encephalitis virus premembrane and envelope proteins enhances humoral immunity in BALB/C mice after DNA vaccination. Virol. J. 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.F.; Lee, C.J.; Liao, C.L.; Dwek, R.A.; Zitzmann, N.; Lin, Y.L. Antiviral effects of an iminosugar derivative on flavivirus infections. J. Virol. 2002, 76, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Ponten, A.; Sick, C.; Weeber, M.; Haller, O.; Kochs, G. Dominant-negative mutants of human MxA protein: Domains in the carboxy-terminal moiety are important for oligomerization and antiviral activity. J. Virol. 1997, 71, 2591–2599. [Google Scholar] [PubMed]
- Von der Malsburg, A.; Abutbul-Ionita, I.; Haller, O.; Kochs, G.; Danino, D. Stalk domain of the dynamin-like MxA GTPase protein mediates membrane binding and liposome tubulation via the unstructured L4 loop. J. Biol. Chem. 2011, 286, 37858–37865. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, A.; Liu, W.; Kochs, G.; Khromykh, A.A.; Mackenzie, J.M. West Nile virus-induced cytoplasmic membrane structures provide partial protection against the interferon-induced antiviral MxA protein. J. Gen. Virol. 2007, 88, 3013–3017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, K.; Wills, E.; Tang, L.; Baines, J.D. A mutation in the DNA polymerase accessory factor of herpes simplex virus 1 restores viral DNA replication in the presence of raltegravir. J. Virol. 2014, 88, 11121–11129. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bai, J.; Fan, B.; Li, Y.; Zhang, Q.; Jiang, P. The Interferon-Induced Mx2 inhibits porcine reproductive and respiratory syndrome virus replication. J. Interferon Cytokine Res. 2016, 36, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; He, D.N.; Zhou, B.; Pang, R.; Liu, K.; Zhao, J.; Chen, P.Y. In vitro inhibition of vesicular stomatitis virus replication by purified porcine Mx1 protein fused to HIV-1 Tat protein transduction domain (PTD). Antivir. Res. 2013, 99, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Chang, B.L.; Yu, H.P.; Liao, C.L.; Lin, Y.L. Blocking of interferon-induced JAK-STAT signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Pang, D.; Wang, T.; Yang, X.; Wu, R.; Ren, L.; Yuan, T.; Huang, Y.; Ouyang, H. Human MxA protein inhibits the replication of classical swine fever virus. Virus Res. 2011, 156, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Westaway, E.G. Markers for trans-Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flavivirus-infected cells. J. Virol. 1999, 73, 9555–9567. [Google Scholar] [PubMed]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef] [PubMed]
- Reid, E.; Juleff, N.; Windsor, M.; Gubbins, S.; Roberts, L. Type I and III IFNs produced by plasmacytoid dendritic cells in response to a member of the Flaviviridae suppress cellular immune responses. J. Immunol. 2016, 196, 4214–4226. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Huang, Y.L.; Ma, S.H.; Yeh, C.T.; Chiou, S.Y.; Chen, L.K.; Liao, C.L. Inhibition of Japanese encephalitis virus infection by nitric oxide: Antiviral effect of nitric oxide on RNA virus replication. J. Virol. 1997, 71, 5227–5235. [Google Scholar] [PubMed]
- Ma, D.; Jiang, D.; Qing, M.; Weidner, J.M.; Qu, X.; Guo, H.; Chang, J.; Gu, B.; Shi, P.Y.; Block, T.M.; et al. Antiviral effect of interferon lambda against West Nile virus. Antivir. Res. 2009, 83, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Pinto, A.K.; Vogt, M.R.; Gale, M., Jr.; Diamond, M.S. Beta interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 2011, 85, 7186–7194. [Google Scholar] [CrossRef] [PubMed]
- Pulit-Penaloza, J.A.; Scherbik, S.V.; Brinton, M.A. Type 1 IFN-independent activation of a subset of interferon stimulated genes in West Nile virus Eg101-infected mouse cells. Virology 2012, 425, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, N.W.; Chen, J.W.; Yang, T.C.; Orloff, G.M.; Wu, Y.Y.; Lai, C.H.; Lan, Y.C.; Lin, C.W. ISG15 over-expression inhibits replication of the Japanese encephalitis virus in human medulloblastoma cells. Antivir. Res. 2010, 85, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.L.; Chang, T.H.; Liao, C.L.; Lin, Y.L. The cellular antiviral protein viperin is attenuated by proteasome-mediated protein degradation in Japanese encephalitis virus-infected cells. J. Virol. 2008, 82, 10455–10464. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, A.; Gillespie, L.; Morgan, G.; van der Heide, P.; Khromykh, A.; Mackenzie, J. The West Nile virus assembly process evades the conserved antiviral mechanism of the interferon-induced MxA protein. Virology 2014, 448, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Nuchtern, J.G.; Bonifacino, J.S.; Biddison, W.E.; Klausner, R.D. Brefeldin A implicates egress from endoplasmic reticulum in class I restricted antigen presentation. Nature 1989, 339, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Bennink, J.R. Brefeldin A specifically inhibits presentation of protein antigens to cytotoxic T lymphocytes. Science 1989, 244, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Banfield, B.W.; Tufaro, F. Brefeldin A arrests the maturation and egress of herpes simplex virus particles during infection. J. Virol. 1991, 65, 1893–1904. [Google Scholar] [PubMed]
- Macovei, A.; Zitzmann, N.; Lazar, C.; Dwek, R.A.; Branza-Nichita, N. Brefeldin A inhibits pestivirus release from infected cells, without affecting its assembly and infectivity. Biochem. Biophys. Res. Commun. 2006, 346, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Cureton, D.K.; Burdeinick-Kerr, R.; Whelan, S.P. Genetic inactivation of COPI coatomer separately inhibits vesicular stomatitis virus entry and gene expression. J. Virol. 2012, 86, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Cuconati, A.; Molla, A.; Wimmer, E. Brefeldin A inhibits cell-free, de novo synthesis of poliovirus. J. Virol. 1998, 72, 6456–6464. [Google Scholar] [PubMed]
- Maynell, L.A.; Kirkegaard, K.; Klymkowsky, M.W. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 1992, 66, 1985–1994. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′ → 3′) |
|---|---|
| pEGFP-poMx2-F | TGACAAGCTTACCATGCCTAAACCCCGCATGTCG |
| pEGFP-poMx2-R | TGACCTCGAGTTACCCCTGTAATGACTGAGC |
| pcDNA3.0-poMx2-F | TGACAAGCTTACCATGCCTAAACCCCGCATGTCG |
| pcDNA3.0-poMx2-R | TGACCTCGAGTTAAGCGTAGTCTGGGACGTCGTATGGGTAC CCCTGTAATGACTGAGC |
| pEGFP-huMxA-F | TGACCTCGAGCTACCATGGTTGTTTCCGAAGTGGACATC |
| pEGFP-huMxA-R | TGACGGATCCACCGGGGAACTGGGCAAGCCGGCG |
| pEGFP-mmMx1-F | TGACCTCGAGCTACCATGGATTCTGTGAATAATCTGTGC |
| pEGFP-mmMx1-R | TGACGGATCCATCGGAGAATTTGGCAAGCTTCTG |
| pcDNA3.0-poMx1-F | TGACAAGCTTACCATGGTTTATTCCAGCTGTG |
| pcDNA3.0-poMx1-R | TGACCTCGAGTTAAGCGTAGTCTGGGACGTCGTATGGGTAGC CTGGGAACTTGGCGA |
| pcDNA3.0-poTMx1-F | TGACAAGCTTACCATGGACAAGGAGTTCCTGGAGGCTCCTAA GAAGAAGAGAAAGGTTGAGTTCAGAATTGTTTATTCC |
| AACTGTGAAAGTAAAGAACCTGATTCAGTT | |
| pcDNA3.0-poTMx1-R | TGACCTCGAGTTAAGCGTAGTCTGGGACGTCGTATGGGTAGCC TGGGAACTTGGCGA |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Wang, S.-Q.; Wei, J.-C.; Zhang, X.-M.; Gao, Z.-C.; Liu, K.; Ma, Z.-Y.; Chen, P.-Y.; Zhou, B. Mx Is Not Responsible for the Antiviral Activity of Interferon-α against Japanese Encephalitis Virus. Viruses 2017, 9, 5. https://doi.org/10.3390/v9010005
Zhou J, Wang S-Q, Wei J-C, Zhang X-M, Gao Z-C, Liu K, Ma Z-Y, Chen P-Y, Zhou B. Mx Is Not Responsible for the Antiviral Activity of Interferon-α against Japanese Encephalitis Virus. Viruses. 2017; 9(1):5. https://doi.org/10.3390/v9010005
Chicago/Turabian StyleZhou, Jing, Shi-Qi Wang, Jian-Chao Wei, Xiao-Min Zhang, Zhi-Can Gao, Ke Liu, Zhi-Yong Ma, Pu-Yan Chen, and Bin Zhou. 2017. "Mx Is Not Responsible for the Antiviral Activity of Interferon-α against Japanese Encephalitis Virus" Viruses 9, no. 1: 5. https://doi.org/10.3390/v9010005
APA StyleZhou, J., Wang, S.-Q., Wei, J.-C., Zhang, X.-M., Gao, Z.-C., Liu, K., Ma, Z.-Y., Chen, P.-Y., & Zhou, B. (2017). Mx Is Not Responsible for the Antiviral Activity of Interferon-α against Japanese Encephalitis Virus. Viruses, 9(1), 5. https://doi.org/10.3390/v9010005