
Promoter Motifs in NCLDVs: An Evolutionary Perspective



 , and
, and 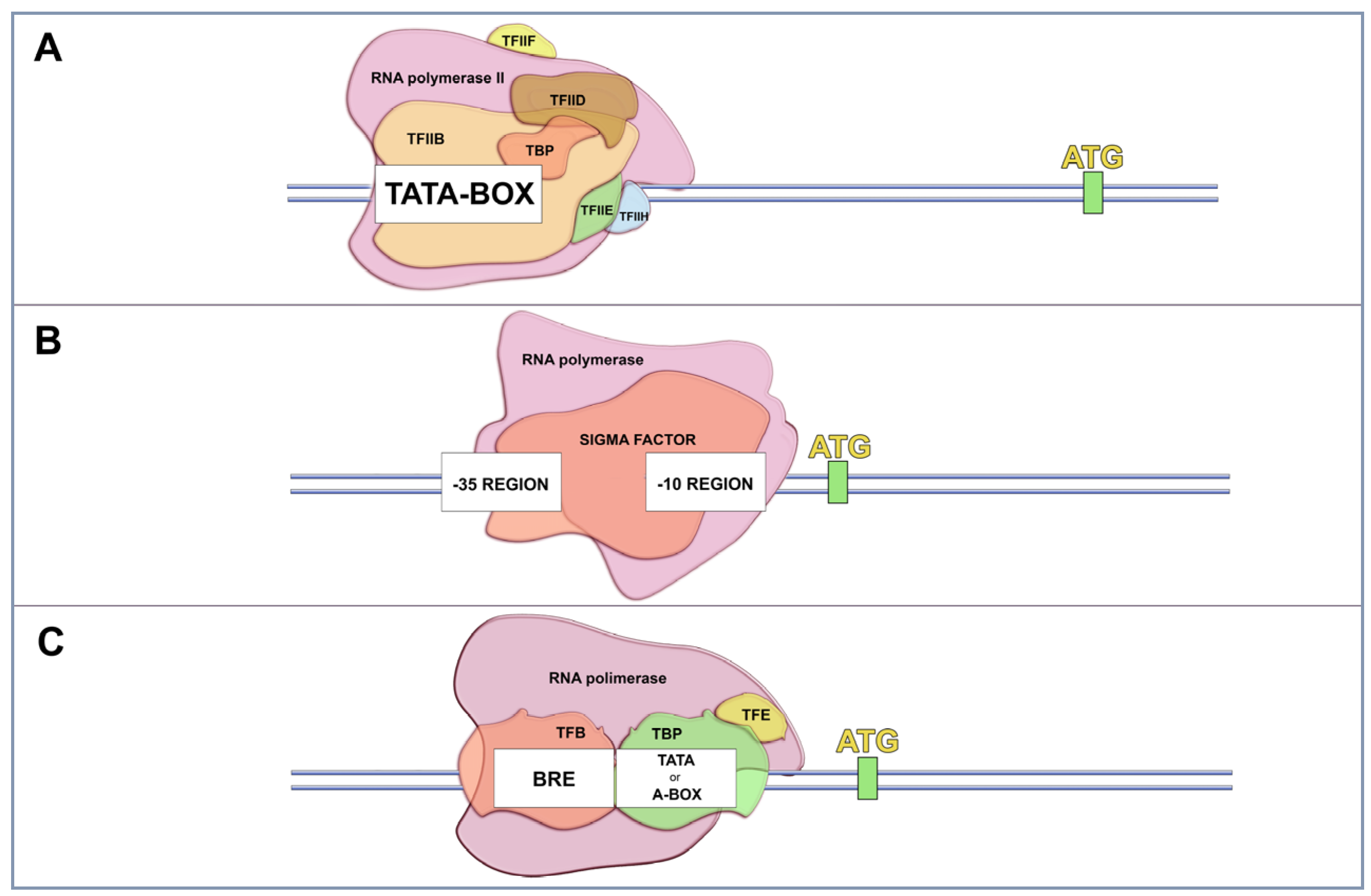{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
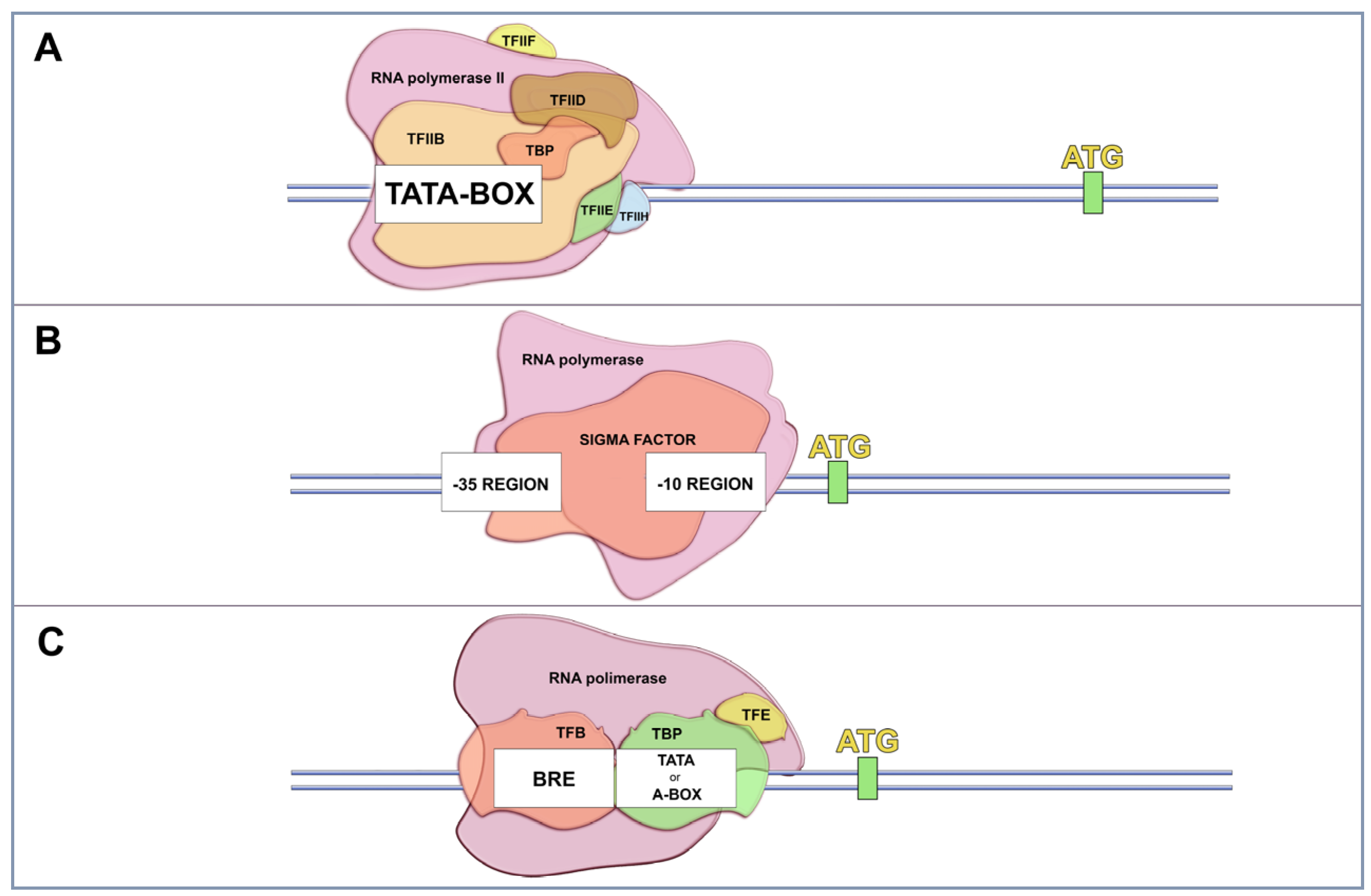
2. Gene Expression in Cells
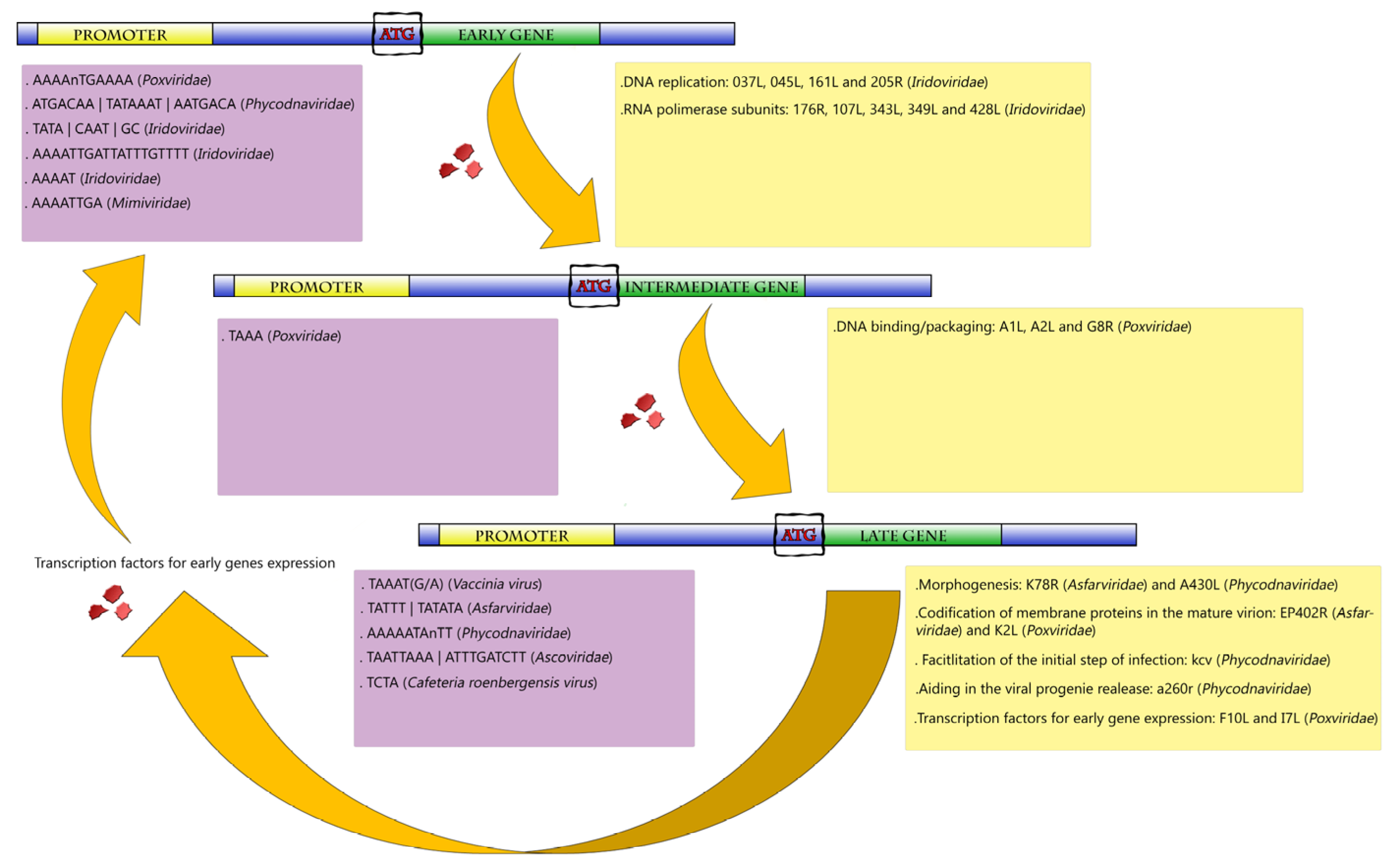
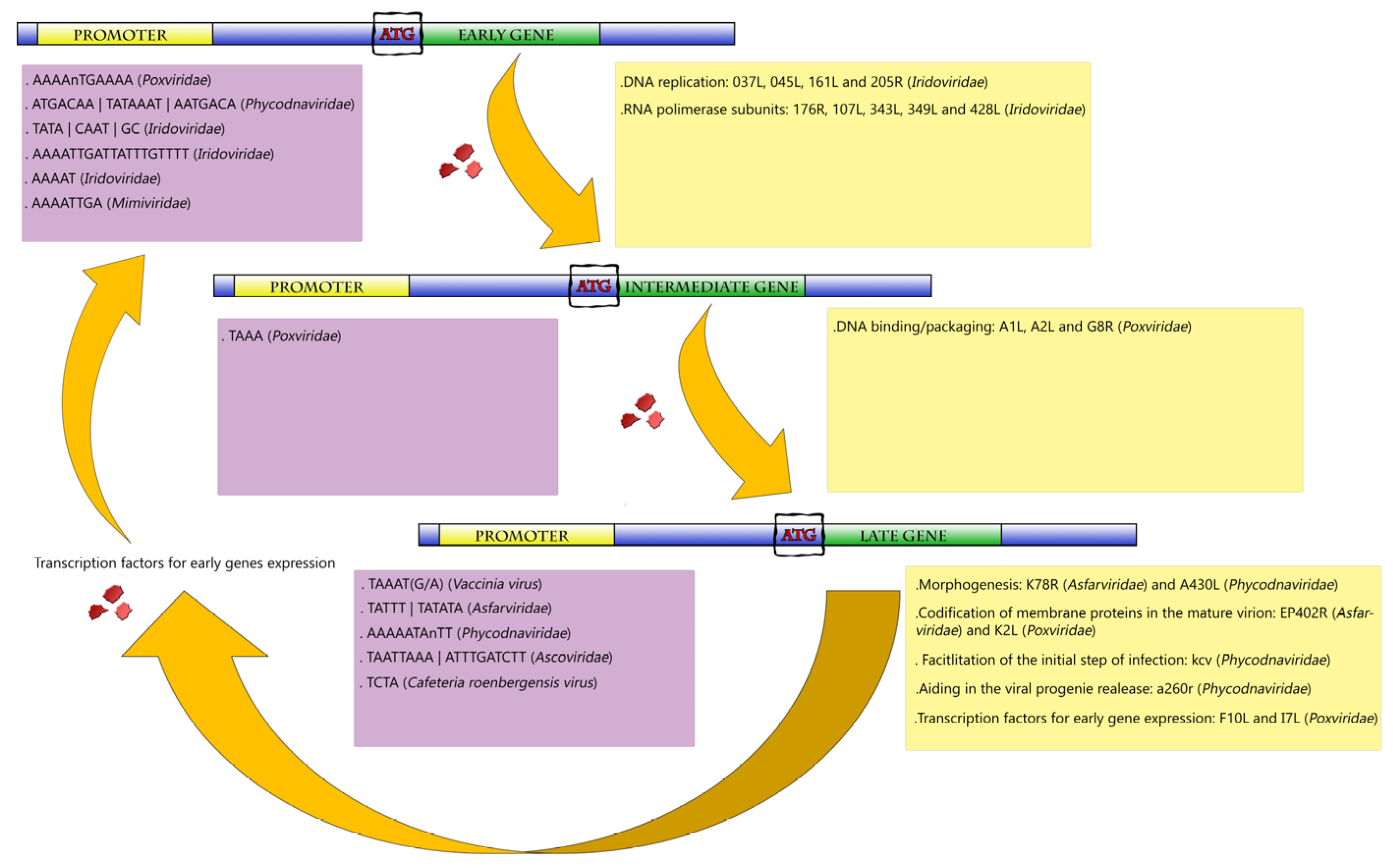
Gene Expression in NCLDVs
3. Poxviridae Family
4. Asfarviridae
5. Phycodnaviridae
6. Iridoviridae
7. Ascoviridae
8. Mimiviridae and Other Amoebal Giant Viruses
9. MEGA-Box: A Putative Promoter Region in the Common Ancestor of Megavirales
10. What Comes Next?
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lwoff, A. The concept of virus. J. Gen. Microbiol. 1957, 17, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Durzyńska, J.; Goździcka-Józefiak, A. Viruses and cells intertwined since the dawn of evolution. Virol. J. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001, 75, 11720. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.M.; Raoult, D. A giant virus in amoebae. Science 2003, 299. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; de Lamballerie, X.; Fournous, G.; Raoult, D. Reclassification of giant viruses composing a fourth domain of life in the new order Megavirales. Intervirology 2012, 55, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Colson, P.; Chabrol, O.; Pontarotti, P.; Raoult, D. Pithovirus sibericum, a new bona fide member of the “Fourth TRUC” club. Front. Microbiol. 2015, 4, 722. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Colson, P.; Chabrol, O.; Scheid, P.; Pontarotti, P.; Raoult, D. Welcome to pandoraviruses at the “Fourth TRUC” club. Front. Microbiol. 2015, 18, 423. [Google Scholar]
- Scheid, P. A strange endocytobiont revealed as largest virus. Curr. Opin. Microbiol. 2016, 31, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Benamar, S.; Reteno, D.G.; Bandaly, V.; Labas, N.; Raoult, D.; la Scola, B. Faustoviruses: Comparative genomics of new Megavirales family members. Front. Microbiol. 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Filée, J. Genomic comparison of closely related Giant Viruses supports an accordion-like model of evolution. Front. Microbiol. 2015, 16, 593. [Google Scholar] [CrossRef] [PubMed]
- Suzan-Monti, M.; la Scola, B.; Raoult, D. Genomic and evolutionary aspects of Mimivirus. Virus Res. 2006, 117, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M. Viruses take center stage in cellular evolution. Genome Biol. 2006, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; López-García, P. Comment on “The 1.2-megabase genome sequence of Mimivirus”. Science 2005, 20, 1114. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Wolf, Y.I.; Koonin, E.V. Origin of giant viruses from smaller DNA viruses not from a fourth domain of cellular life. Virology 2014, 38–52, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; la Scola, B.; Suzan, M.; Claverie, J.M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Boyer, M.; Madoui, M.A.; Gimenez, G.; La Scola, B.; Raoult, D. Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4 domain of life including giant viruses. PLoS ONE 2010, 2, e15530. [Google Scholar] [CrossRef] [PubMed]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.-Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Fuda, N.J.; Ardehali, M.B.; Lis, J.T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 2009, 10, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Lacadie, S.A.; Ibrahim, M.M.; Gokhale, S.A.; Ohler, U. Divergent transcription and epigenetic directionality of human promoters. FEBS J. 2016, 283, 4214–4222. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Lenhard, B. Promoter architectures and developmental gene regulation. Semin. Cell Dev. Biol. 2016, 57, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Gladstone, L. A mammalian system for the incorporation of cytidine triphosphate into ribonucleic acid. J. Am. Chem. Soc. 1959, 81, 4118–4119. [Google Scholar] [CrossRef]
- Thomas, M.C.; Chiang, C.M. The general transcription machinery and generalcofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178. [Google Scholar] [CrossRef] [PubMed]
- Sabin, L.R.; Delás, M.J.; Hannon, G.J. Dogma derailed: The many influences of RNA on the genome. Mol. Cell 2013, 49, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Vannini, A.; Ringel, R.; Kusser, A.G.; Berninghausen, O.; Kassavetis, G.A.; Cramer, P. Molecular basis of RNA polymerase III transcription repression by Maf1. Cell 2010, 143, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.A.; Taylor, M.S.; Engström, P.G.; Frith, M.C. Genome-wide analysis of mammalian promoter architecture andevolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.J.; Trinklein, N.D.; Anton, E.D.; Nguyen, L.; Myers, R.M. Comprehensive analysis of transcriptional promoter structure and functionin 1% of the human genome. Genome Res. 2006, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T.; Kadonaga, J.T. The RNA polymerase II core promoter. Annu. Rev. Biochem. 2003, 72, 449–479. [Google Scholar] [CrossRef] [PubMed]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genom. Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.K.; Tjian, R. TAFs and TFIIA mediate differential utilization of thetandem Adh promoters. Cell 1995, 82, 565–575. [Google Scholar] [CrossRef]
- Ren, B.; Maniatis, T. Regulation of Drosophila Adh promoter switching by aninitiator-targeted repression mechanism. EMBO J. 1998, 17, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.F.; Busby, S.J. Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. 2016, 14, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.R.; Travers, A.A.; Dunn, J.J.; Bautz, E.K. Factor stimulating transcription by RNA polymerase. Nature 1969, 221, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Minchin, S.D.; Busby, S.J. Activating transcription in bacteria. Annu. Rev. Microbiol. 2012, 66, 125–152. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.S.; Masuda, S.; Campbell, E.A.; Muzzin, O.; Darst, S.A. Structural basis of transcription initiation: An RNA polymerase holoenzyme–DNA complex. Science 2002, 296, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.F.; Busby, S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004, 2, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Hausner, W.; Wettach, J.; Hethke, C.; Thomm, M. Two transcription factors related with the eucaryal transcription factors TATA binding protein and transcription factor IIB direct promoter recognition by an archaeal RNA polymerase. J. Biol. Chem. 1996, 271, 30144–30148. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.D.; Jaxel, C.; Nadal, M.; Kosa, P.F.; Jackson, S.P. Temperature template topology, and factor requirements of archaeal transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 15218–15222. [Google Scholar] [CrossRef] [PubMed]
- Darcy, T.J.; Hausner, W.; Awery, D.E.; Edwards, A.M.; Thomm, M.; Reeve, J.N. Methanobacterium thermoautotrophicum RNA polymerase and transcription in vitro. J. Bacteriol. 1999, 181, 4424–4429. [Google Scholar] [PubMed]
- Reiter, W.D.; Hudepohl, U.; Zillig, W. Mutational analysis of an archae bacterial promoter—Essential role of a TATA Box for transcription efficiency and start-site selection in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 9509–9513. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.R.; Daniels, C.J. In vivo definition of an archaeal promoter. J. Bact. 1995, 177, 1844–1849. [Google Scholar] [CrossRef] [PubMed]
- Kyrpides, N.C.; Ouzounis, C.A. Transcription in Archaea. Proc. Natl. Acad. Sci. USA 1999, 96, 8545–8550. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. DNA-binding proteins and evolution of transcription regulation in the Archaea. Nucleic Acids Res. 1999, 27, 4658–4670. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.D.; Jackson, S.P. Mechanism and regulation of transcription in Archaea. Curr. Opin. Microbiol. 2001, 4, 208–213. [Google Scholar] [CrossRef]
- Werner, F.; Grohmann, D. Evolution of multisubunit RNA polymerases in the three domains of life. Nat. Rev. Microbiol. 2011, 9, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S. Viral Replication Strategies. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2014; Volume 2, p. 2160. [Google Scholar]
- Abergel, C.; Rudinger-Thirion, J.; Giege, R.; Claverie, J.M. Virus-encoded aminoacyl-tRNA synthetases: Structural and functional characterization of mimivirus TyrRS and MetRS. J. Virol. 2007, 81, 12406–12417. [Google Scholar] [CrossRef] [PubMed]
- Broyles, S.S.; Knutson, B.A. Poxvirus transcription. Future Virol. 2010, 5, 639–650. [Google Scholar] [CrossRef]
- Moss, B. Poxviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2014; Volume 2, p. 2129. [Google Scholar]
- Legendre, M.; Audic, S.; Poirot, O.; Hingamp, P.; Seltzer, V.; Byrne, D.; Lartigue, A.; Lescot, M.; Bernadac, A.; Poulain, J.; et al. mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Res. 2010, 20, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Audic, S.; Claverie, J.M. Mimivirus gene promoters exhibit an unprecedented conservation among all eukaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 14689–14693. [Google Scholar] [CrossRef] [PubMed]
- Damon, I.K. Poxviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2014; Volume 2, p. 2160. [Google Scholar]
- Baroudy, B.M.; Moss, B. Purification and characterization of a DNA dependent RNA polymerase from vaccinia virions. J. Biol. Chem. 1980, 255, 4372–4380. [Google Scholar] [PubMed]
- Knutson, B.A.; Broyles, S.S. Expansion of poxvirus RNA polymerase subunits sharing homology with corresponding subunits of RNA polymerase II. Virus Genes 2008, 36, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Moss, B. The structure of vaccinia virus early promoters. J. Mol. Biol. 1989, 210, 749–769. [Google Scholar] [CrossRef]
- Baldick, C.J.; Keck, J.G.; Moss, B. Mutational analysis of the core, spacer and initiator regions of vaccinia virus intermediate class promoters. J. Virol. 1992, 66, 4710–4719. [Google Scholar] [PubMed]
- Knutson, B.A.; Liu, X.; Oh, J. Vaccinia virus intermediate and late promoter elements are targeted by the TATA-binding protein. J. Virol. 2006, 80, 6784–6793. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bruno, D.P.; Martens, C.A.; Porcella, S.F.; Moss, B. Genome-wide analysis of the 5’and 3’ends of vaccinia virus early mRNAs delineates regulatory sequences of annotated and anomalous transcripts. J. Virol. 2011, 85, 5897–5909. [Google Scholar] [CrossRef] [PubMed]
- Keck, J.G.; Baldick, C.J.; Moss, B. Role of DNA replication in vaccinia virus gene expression: A naked template is required for transcription of three late transactivator genes. Cell 1990, 61, 801–809. [Google Scholar] [CrossRef]
- Yang, Z.; Reynolds, S.E.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 2011, 85, 9899–9908. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Pervasive initiation and 3’-end formation of poxvirus postreplicative RNAs. J. Biol. Chem. 2012, 287, 31050–31060. [Google Scholar] [CrossRef] [PubMed]
- Broyles, S.S.; Liu, X.; Zhu, M.; Kremer, M. Transcription factor YY1 is a vaccinia virus late promoter activator. J. Biol. Chem. 1999, 274, 35662–35667. [Google Scholar] [CrossRef] [PubMed]
- Hänggi, M.; Bannwarth, W.; Stunnenberg, H.G. Conserved TAAAT motif in vaccinia virus late promoters: Overlapping TATA box and site of transcription initiation. EMBO J. 1986, 5, 1071–1076. [Google Scholar] [PubMed]
- International Committee on Taxonomy of Viruses. Available online: http://www.ictvonline.org/taxonomyHistory.asp?taxnode_id=20151927&taxa_name=Asfarviridae (accessed on 22 September 2016).
- Sogo, J.M.; Almendral, J.M.; Talavera, A.; Vinuela, E. Terminal and internal inverted repetitions in African swine fever virus DNA. Virology 1984, 133, 271–275. [Google Scholar] [CrossRef]
- Tulman, E.R.; Delhon, G.A.; Ku, B.K.; Rock, D.L. African swine fever virus. Curr. Top. Microbiol. Immunol. 2009, 328, 43–87. [Google Scholar] [PubMed]
- Kuznar, J.; Salas, M.L.; Vinuela, E. DNA-dependent RNA polymerase in African swine fever virus. Virology 1980, 101, 169–175. [Google Scholar] [CrossRef]
- Salas, J.; Salas, M.L.; Vinuela, E. Effect of inhibitors of the host cell RNA polymerase II on African swine fever virus multiplication. Virology 1988, 164, 280–283. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Salas, M.L. African swine fever virus transcription. Virus Res. 2013, 173, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc. Natl. Acad. Sci. USA 1996, 15, 11341–11348. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Salas, M.L.; Vinuela, E. Intermediate class of mRNAs in African swine fever virus. J. Virol. 1996, 70, 8584–8589. [Google Scholar] [PubMed]
- Garcia-Escudero, R.; Vinuela, E. Structure of African swine fever virus late promoters: Requirement of a TATA sequence at the initiation region. J. Virol. 2000, 74, 8176–8182. [Google Scholar] [CrossRef] [PubMed]
- Yates, P.R.; Dixon, L.K.; Turner, P.C. Promoter analysis of an African swine fever virus gene encoding a putative elongation factor. Biochem. Soc. Trans. 1995, 23, 139S. [Google Scholar] [CrossRef]
- Van Etten, J.L.; Graves, M.V.; Müller, D.G.; Boland, W.; Delaroque, N. Phycodnaviridae—Large DNA algal viruses. Arch. Virol. 2002, 147, 1479–1516. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Meints, R.H. Giant viruses infecting algae. Annu. Rev. Microbiol. 1999, 53, 447–494. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; van Etten, J.L.; Allen, M.J. The Phycodnaviridae: The story of how tiny giants rule the world. Curr. Top. Microbiol. Immunol. 2009, 328, 1–42. [Google Scholar] [PubMed]
- International Committee on Taxonomy of Viruses. Available online: http://www.ictvonline.org/taxonomyHistory.asp?taxnode_id=20153552&taxa_name=Phycodnaviridae (accessed on 22 September 2016).
- Kang, M.; Graves, M.; Mehmel, M.; Moroni, A.; Gazzarrini, S.; Thiel, G.; Gurnon, J.R.; van Etten, J.L. Genetic diversity in chlorella viruses flanking kcv, a gene that encodes a potassium ion channel protein. Virology 2004, 326, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Tanaka, M.; Fujie, M.; Usami, S.; Yamada, T. Immediate early genes expressed in chlorovirus infections. Virology 2004, 318, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Chuchird, N.; Nishida, K.; Kawasaki, T.; Fujie, M.; Usami, S.; Yamada, T. A variable region on the chlorovirus CVK2 genome contains five copies of the gene for Vp260, a viral-surface glycoprotein. Virology 2002, 10, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Hiramatsu, S.; Songsri, P.; Fujie, M. Alternative expression of a chitosanase gene produces two different proteins in cells infected with Chlorella virus CVK2. Virology 1997, 14, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Graves, M.V.; Meints, R.H. Characterization of the major capsid protein and cloning of its gene from algal virus PBCV-1. Virology 1992, 188, 198–207. [Google Scholar] [CrossRef]
- Sugimoto, I.; Hiramatsu, S.; Murakami, D.; Fujie, M.; Usami, S.; Yamada, T. Algal-lytic activities encoded by Chlorella virus CVK2. Virology 2000, 10, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, L.A.; Graves, M.V.; Li, X.; Feldblyum, T.; Hartigan, J.; van Etten, J.L. Sequence and annotation of the 314-kb MT325 and the 321-kb FR483 viruses that infect Chlorella Pbi. Virology 2007, 20, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, L.A.; Graves, M.V.; Li, X.; Feldblyum, T.; Nierman, W.C.; van Etten, J.L. Sequence and annotation of the 369-kb NY-2A and the 345-kb AR158 viruses that infect Chlorella NC64A. Virology 2007, 20, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, L.A.; Boucher, P.T.; Yanai-Balser, G.M.; Suhre, K.; Graves, M.V.; van Etten, J.L. Putative gene promoter sequences in the chlorella viruses. Virology 2008, 25, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L. Unusual life style of giant chlorella viruses. Ann. Rev. Genet. 2003, 37, 153–195. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Schroeder, D.C.; Allen, M.J.; Holden, M.T.; Parkhill, J.; Barrell, B.G.; Churcher, C.; Hamlin, N.; Mungall, K.; Norbertczak, H.; et al. Complete genome sequence and lytic phase transcription profile of a Coccolithovirus. Science 2005, 12, 1090–1092. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses. Available online: http://www.ictvonline.org/taxonomyHistory.asp.taxnode_id=20153003&taxa_name=Iridoviridae (accessed on 22 September 2016).
- Darai, G.; Delius, H.; Clarke, J.; Apfel, H.; Schnitzler, P.; Flugel, R.M. Molecular cloning and physical mapping of the genome of fish lymphocystis disease virus. Virology 1985, 146, 292–301. [Google Scholar] [CrossRef]
- Williams, T.; Barbosa-Solomieu, V.; Chinchar, V.G. A decade of advances in Iridovirus research. Adv. Virus Res. 2005, 65, 174–248. [Google Scholar]
- Jakob, N.J.; Muller, K.; Bahr, U.; Darai, G. Analysis of the first complete DNA sequence of an invertebrate iridovirus: Coding strategy of the genome of Chilo iridescent virus. Virology 2001, 286, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Chinchar, V.G.; Yu, K.H.; Jancovich, J.K. The molecular biology of frog virus 3 and other iridoviruses infecting cold-blooded vertebrates. Viruses 2011, 3, 1959–1985. [Google Scholar] [CrossRef] [PubMed]
- Barray, S.; Devauchelle, G. Protein synthesis in cells infected by Chilo iridescent virus: Evidence for temporal control of three classes of induced polypeptides. Virology 1987, 138, 253–261. [Google Scholar]
- D’Costa, S.M.; Yao, H.; Bilimoria, S.L. Transcription and temporal cascade in Chilo iridescent virus infected cells. Arch. Virol. 2001, 146, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, S.M.; Yao, H.J.; Bilimoria, S.L. Transcriptional mapping in Chilo iridescent virus infections. Arch. Virol. 2004, 149, 723–742. [Google Scholar] [CrossRef] [PubMed]
- Willis, D.B. DNA sequences required for trans activation of an immediate- early frog virus 3 gene. Virology 1987, 161, 1–7. [Google Scholar] [CrossRef]
- Beckman, W.; Tham, T.N.; Aubertin, A.M.; Willis, D.B. Structure and regulation of the immediate-early frog virus 3 gene that encodes ICR489. J. Virol. 1988, 62, 1271–1277. [Google Scholar] [PubMed]
- Willis, D.B.; Granoff, A. Transactivation of an immediateearly frog virus 3 promoter by a virion protein. J. Virol. 1985, 56, 495–501. [Google Scholar] [PubMed]
- Pallister, J.; Goldie, S.; Coupar, B.; Hyatt, A. Promoter activity in the 59 flanking regions of the Bohle iridovirus ICP 18, ICP 46 and major capsid protein genes. Arch. Virol. 2005, 150, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Nalcacioglu, R.; Ince, I.A.; Vlak, J.M.; Demirbag, Z.; van Oers, M.M. The Chilo iridescent virus DNA polymerase promoter contains an essential AAAAT motif. J. Gen. Virol. 2007, 88, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Nalcacioglu, R.; Demirbag, Z.; Vlak, J.M.; van Oers, M.M. Promoter analysis of the Chilo iridescent virus DNA polymerase and major capsid protein genes. Virology 2003, 317, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Dizman, Y.A.; Demirbag, Z.; Ince, I.A.; Nalcacioglu, R. Transcriptomic analysis of Chilo iridescent virus immediate early promoter. Virus Res. 2012, 167, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Goorha, R. Frog virus 3 requires RNA polymerase II for its replication. J. Virol. 1981, 37, 496–499. [Google Scholar] [PubMed]
- Goorha, R.; Willis, D.B.; Granoff, A. Macromolecular synthesis in cells infected by frog virus 3. VI. Frog virus 3 replication is dependent on the cell nucleus. J. Virol. 1977, 21, 802–805. [Google Scholar] [PubMed]
- Goorha, R.; Murti, G.; Granoff, A.; Tirey, R. Macromolecular synthesis in cells infected by frog virus 3. VIII. The nucleus is a site of frog virus 3 DNA and RNA synthesis. Virology 1978, 84, 32–50. [Google Scholar] [CrossRef]
- Baroudy, B.M.; Moss, B. Sequence homologies of diverse length tandem repetitions near ends of vaccinia virus genome suggest unequal crossing over. Nucleic Acids Res. 1982, 25, 5673–5679. [Google Scholar] [CrossRef]
- Tidona, C.A.; Darai, G. Molecular anatomy of lymphocystis disease virus. Arch. Virol. Suppl. 1997, 13, 49–56. [Google Scholar] [PubMed]
- Federici, B.A.; Bigot, Y.; Granados, R.R.; Hamm, J.J.; Miller, L.K.; Newton, I.; Stasiak, K.; Vlak, J.M. Family Ascoviridae. In Virus Taxonomy: 8th Report of the International Committee on Taxonomy of Viruses; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2005; pp. 261–265. [Google Scholar]
- International Committee on Taxonomy of Viruses. Available online: http://www.ictvonline.org/taxonomyHistory.asp?taxnode_id=20151919&taxa_name=Ascoviridae (accessed on 22 September 2016).
- Cheng, X.W.; Wang, L.; Carner, G.R.; Arif, B.M. Characterization of three ascovirus isolates from cotton insects. J. Invertebr. Pathol. 2005, 89, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Asgari, S.; Davis, J.; Wood, D.; Wilson, P.; McGrath, A. Sequence and organization of the Heliothis virescens ascovirus genome. J. Gen. Virol. 2007, 88, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Bigot, Y.; Rabouille, A.; Sizaret, P.Y.; Hamelin, M.H.; Periquet, G. Particle and genomic characterization of a new member of the Ascoviridae, Diadromus pulchellus ascovirus. J. Gen. Virol. 1997, 78, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.W.; Carner, G.R.; Arif, B.M. A new ascovirus from Spodoptera exigua and its relatedness to the isolate from Spodoptera frugiperda. J. Gen. Virol. 2000, 81, 3083–3092. [Google Scholar] [CrossRef] [PubMed]
- Salem, T.Z.; Turney, C.M.; Wang, L.; Xue, J.; Wan, X.-F.; Cheng, X.-W. Transcriptional analysis of a major capsid protein gene from Spodoptera exigua ascovirus 5a. Arch. Virol. 2008, 153, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Cui, L.W. Molecular characterization of the major virion protein gene from the Trichoplusia ni ascovirus. Virus Genes 2003, 27, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, K.; Renault, S.; Demattei, M.V.; Bigot, Y.; Federici, B.A. Evidence for the evolution of ascoviruses from iridoviruses. J. Gen. Virol. 2003, 84, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Chinchar, V.G.; Hyatt, A.; Miyazaki, T.; Williams, T. Family Iridoviridae: Poor viral relations no longer. Curr. Top. Microbiol. Immunol. 2009, 328, 123–170. [Google Scholar] [PubMed]
- Piégu, B.; Asgari, S.; Bideshi, D.; Federici, B.A.; Bigot, Y. Evolutionary relationships of iridoviruses and divergence of ascoviruses from invertebrate iridoviruses in the superfamily Megavirales. Mol. Phylogenet. Evol. 2015, 84, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zauberman, N.; Mutsafi, Y.; Halevy, D.B.; Shimoni, E.; Klein, E.; Xiao, C.; Sun, S.; Minsky, A. Distinct DNA exit and packaging portals in the virus Acanthamoeba polyphaga mimivirus. PLoS Biol. 2008, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Kuznetsov, Y.G.; Sun, S.; Hafenstein, S.L.; Kostyuchenko, V.A.; Chipman, P.R.; Suzan-Monti, M.; Raoult, D.; McPherson, A.; Rossmann, M.G. Structural studies of the giant mimivirus. PLoS Biol. 2009, 28, e92. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.A.; dos Santos Silva, L.K.; Dornas, F.P.; de Oliveira, D.B.; Magalhães, T.F.; Santos, D.A.; Costa, A.O.; de Macêdo Farias, L.; Magalhães, P.P.; Bonjardim, C.A.; et al. Mimivirus fibrils are important for viral attachment to the microbial world by a diverse glycoside interaction repertoire. J. Virol. 2015, 89, 11812–11819. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Santini, S.; Rico, A.; Abergel, C.; Claverie, J.M. Breaking the 1000-gene barrier for Mimivirus using ultra-deep genome and transcriptome sequencing. Virol. J. 2011, 4, 8–99. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 9, 19508–19513. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; de Lamballerie, X.; Yutin, N.; Asgari, S.; Bigot, Y.; Bideshi, D.K.; Cheng, X.W.; Federici, B.A.; van Etten, J.L.; Koonin, E.V.; et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 2013, 158, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Reteno, D.G.; Benamar, S.; Khalil, J.B.; Andreani, J.; Armstrong, N.; Klose, T.; Rossmann, M.; Colson, P.; Raoult, D.; La Scola, B. Faustovirus, an asfarvirus-related new lineage of giant viruses infecting amoebae. J. Virol. 2015, 89, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [PubMed]
- Levasseur, A.; Andreani, J.; Delerce, J.; Bou Khalil, J.; Robert, C.; La Scola, B.; Raoult, D. Comparison of a modern and fossil pithovirus reveals its genetic conservation and evolution. Genome Biol. Evol. 2016, 25, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alempic, J.M.; Ramus, C.; Bruley, C.; Labadie, K.; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, 5327–5335. [Google Scholar] [CrossRef] [PubMed]
- McLysaght, A.; Baldi, P.F.; Gaut, B.S. Extensive gene gain associated with adaptive evolution of poxviruses. Proc. Natl. Acad. Sci. USA 2003, 23, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Friedman, R. Poxvirus genome evolution by gene gain and loss. Mol. Phylogenet. Evol. 2005, 35, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Elde, N.C.; Child, S.J.; Eickbush, M.T.; Kitzman, J.O.; Rogers, K.S.; Shendure, J.; Geballe, A.P.; Malik, H.S. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 2012, 17, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Portugal, R.; Coelho, J.; Höper, D.; Little, N.S.; Smithson, C.; Upton, C.; Martins, C.; Leitão, A.; Keil, G.M. Related strains of African swine fever virus with different virulence: Genome comparison and analysis. J. Gen. Virol. 2015, 96, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Huang, X.; Liu, H.; Gong, J.; Ouyang, Z.; Cui, H.; Cao, J.; Zhao, Y.; Wang, X.; Jiang, Y.; et al. Complete sequence determination of a novel reptile iridovirus isolated from soft-shelled turtle and evolutionary analysis of Iridoviridae. BMC Genomics 2009, 14, 224. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Krupovic, M.; Yutin, N. Evolution of double-stranded DNA viruses of eukaryotes: From bacteriophages to transposons to giant viruses. Ann. N. Y. Acad. Sci. 2015, 1341, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Kim, K.M.; Caetano-Anolles, G. Giant viruses coexisted with the cellular ancestors and represent a distinct supergroup along with superkingdoms Archaea, Bacteria and Eukarya. BMC Evol. Biol. 2012, 24. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Reflections on the early development of poxvirus vectors 2013. Vaccine 2013, 6, 4220–4222. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, G.P.; Andrade, A.C.d.S.P.; Rodrigues, R.A.L.; Arantes, T.S.; Boratto, P.V.M.; Silva, L.K.d.S.; Dornas, F.P.; Trindade, G.D.S.; Drumond, B.P.; La Scola, B.; et al. Promoter Motifs in NCLDVs: An Evolutionary Perspective. Viruses 2017, 9, 16. https://doi.org/10.3390/v9010016
Oliveira GP, Andrade ACdSP, Rodrigues RAL, Arantes TS, Boratto PVM, Silva LKdS, Dornas FP, Trindade GDS, Drumond BP, La Scola B, et al. Promoter Motifs in NCLDVs: An Evolutionary Perspective. Viruses. 2017; 9(1):16. https://doi.org/10.3390/v9010016
Chicago/Turabian StyleOliveira, Graziele Pereira, Ana Cláudia dos Santos Pereira Andrade, Rodrigo Araújo Lima Rodrigues, Thalita Souza Arantes, Paulo Victor Miranda Boratto, Ludmila Karen dos Santos Silva, Fábio Pio Dornas, Giliane De Souza Trindade, Betânia Paiva Drumond, Bernard La Scola, and et al. 2017. "Promoter Motifs in NCLDVs: An Evolutionary Perspective" Viruses 9, no. 1: 16. https://doi.org/10.3390/v9010016
APA StyleOliveira, G. P., Andrade, A. C. d. S. P., Rodrigues, R. A. L., Arantes, T. S., Boratto, P. V. M., Silva, L. K. d. S., Dornas, F. P., Trindade, G. D. S., Drumond, B. P., La Scola, B., Kroon, E. G., & Abrahão, J. S. (2017). Promoter Motifs in NCLDVs: An Evolutionary Perspective. Viruses, 9(1), 16. https://doi.org/10.3390/v9010016