
Measles Virus Host Invasion and Pathogenesis


 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
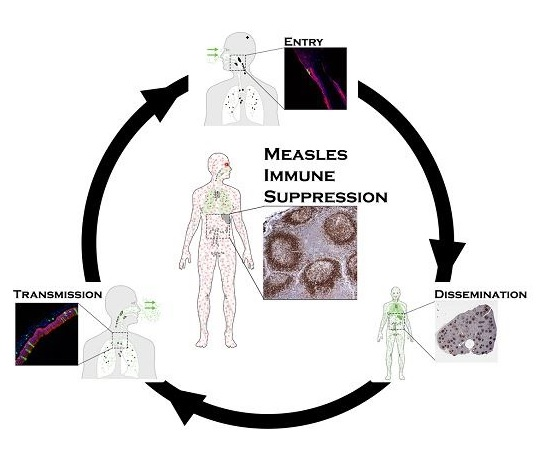
1. Introduction
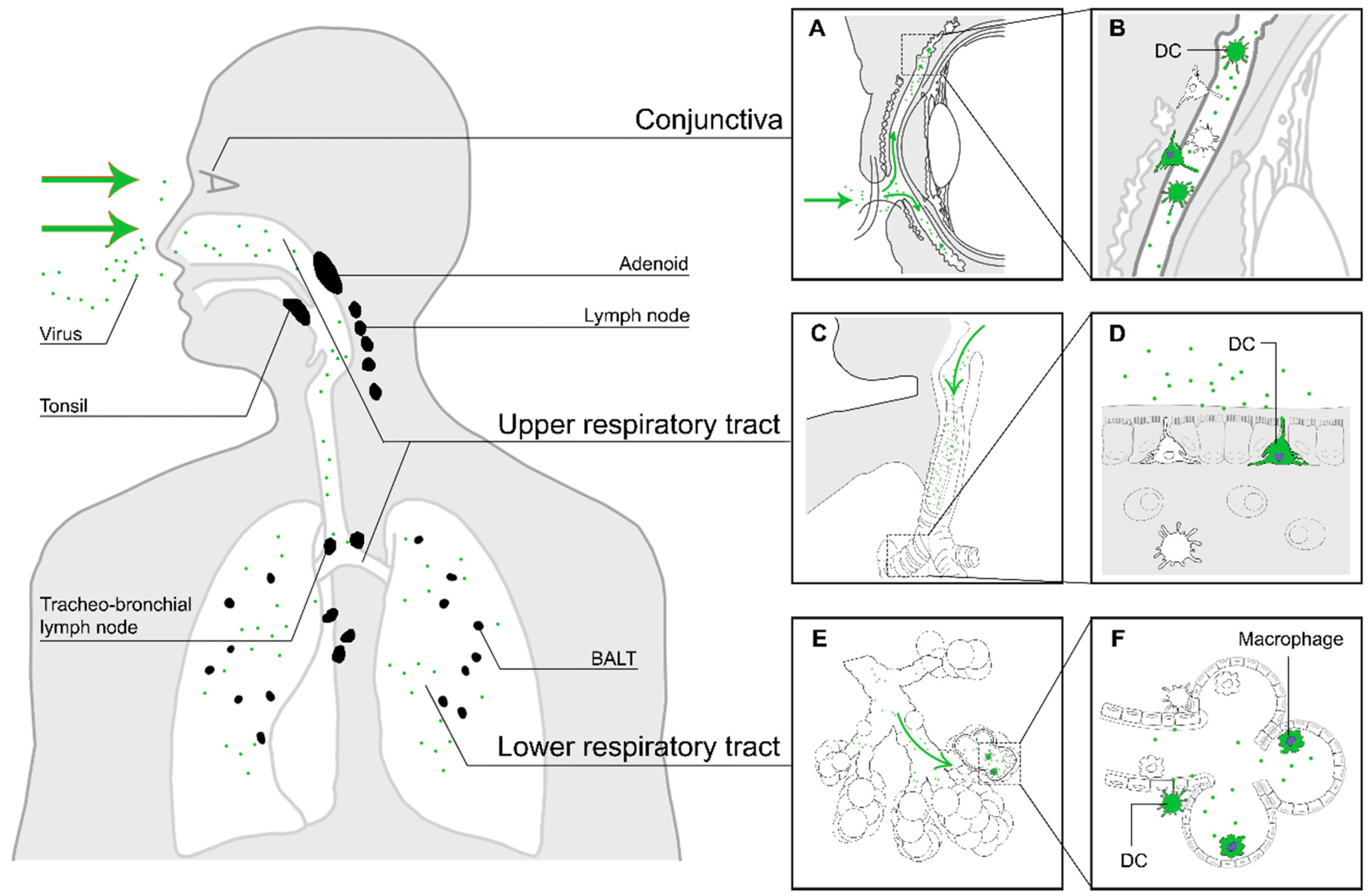
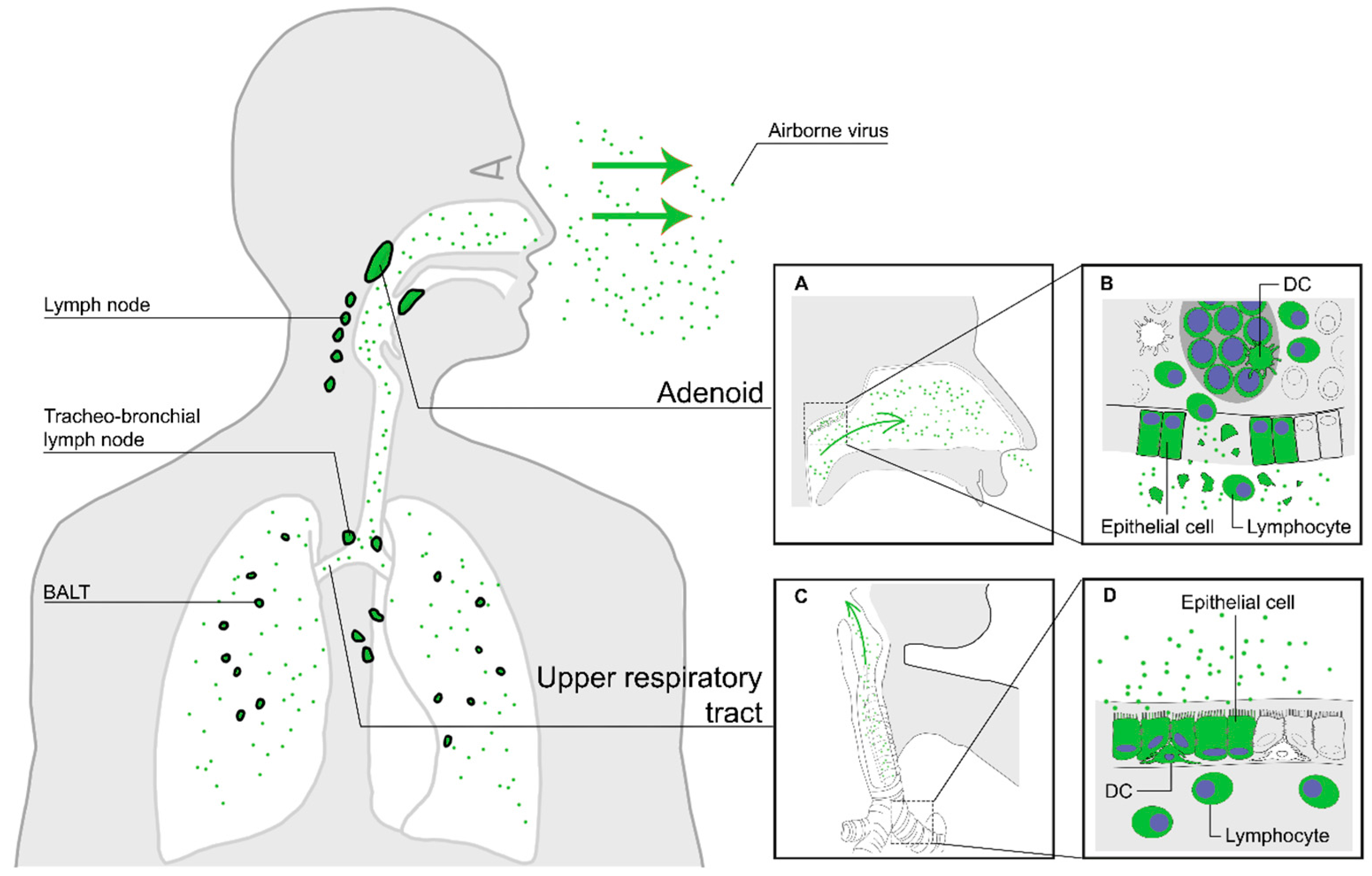
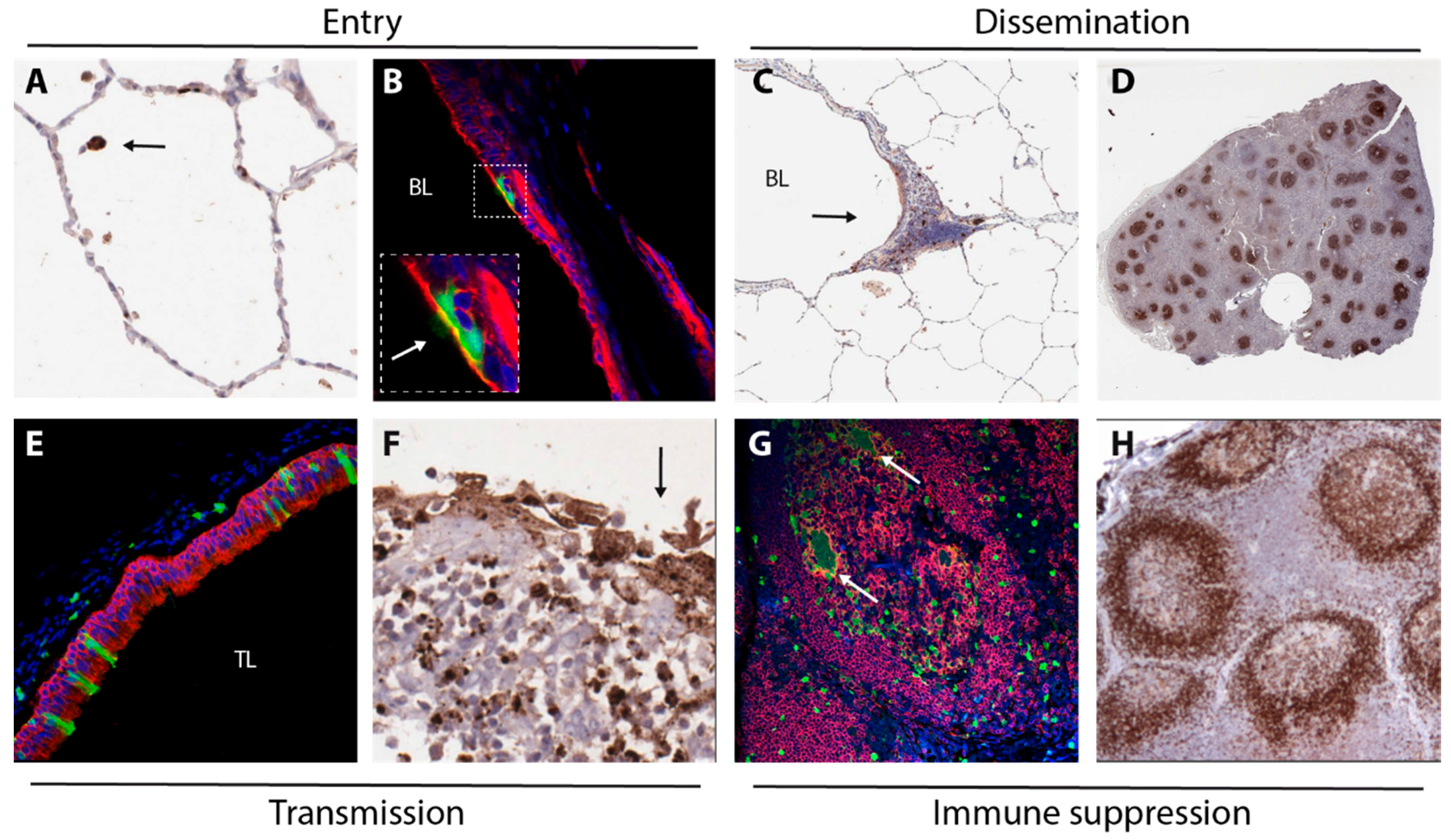
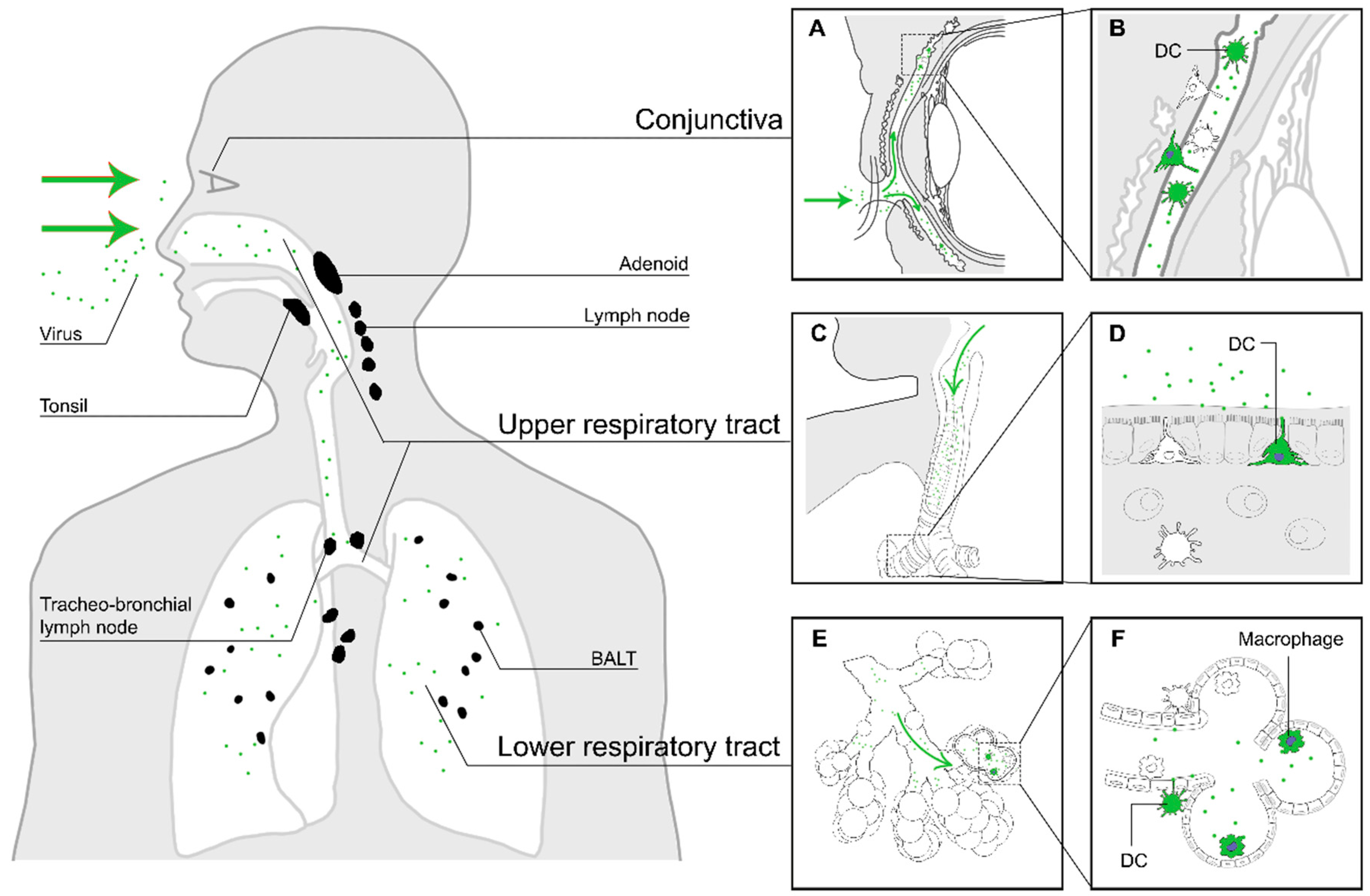
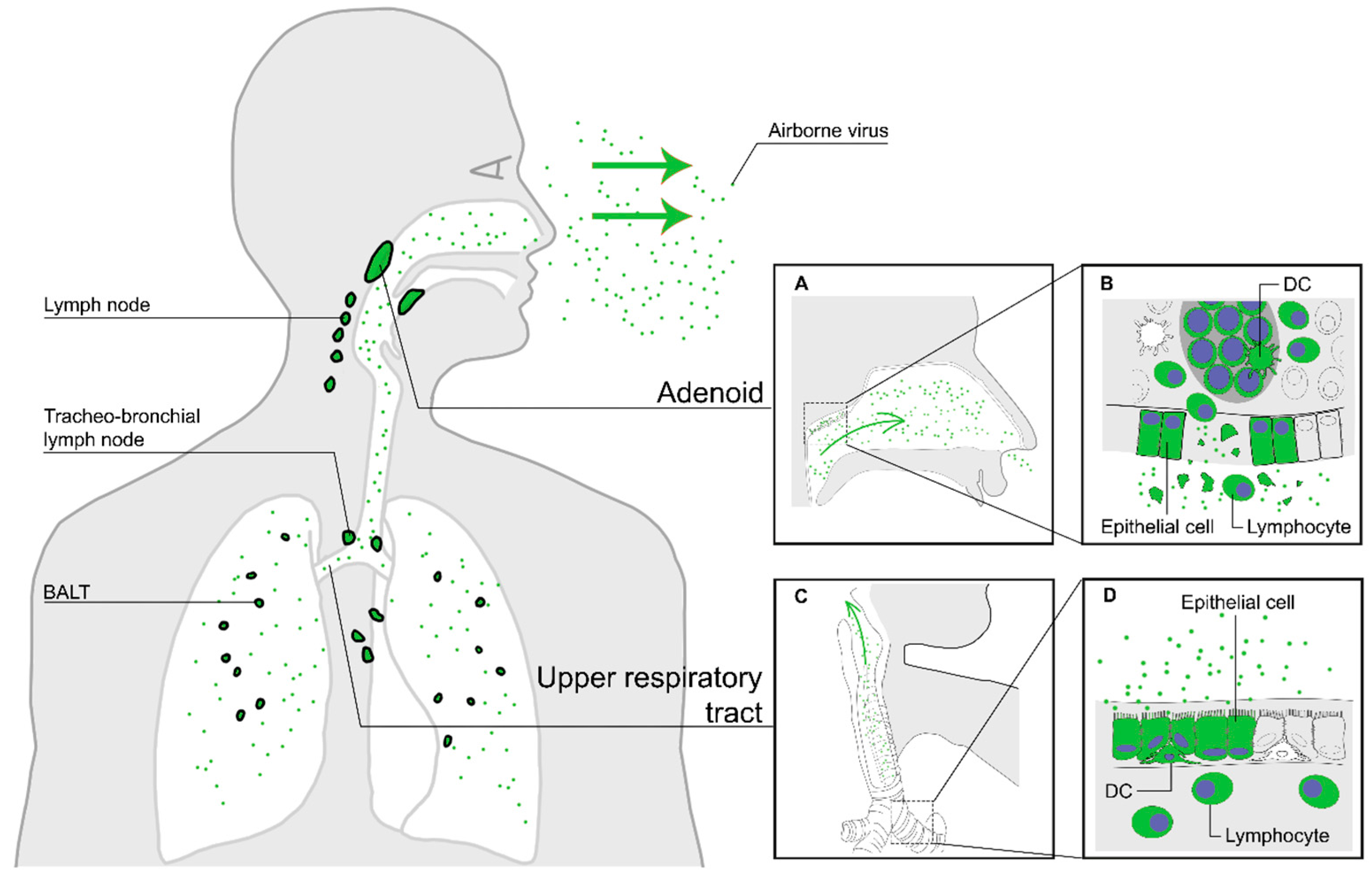
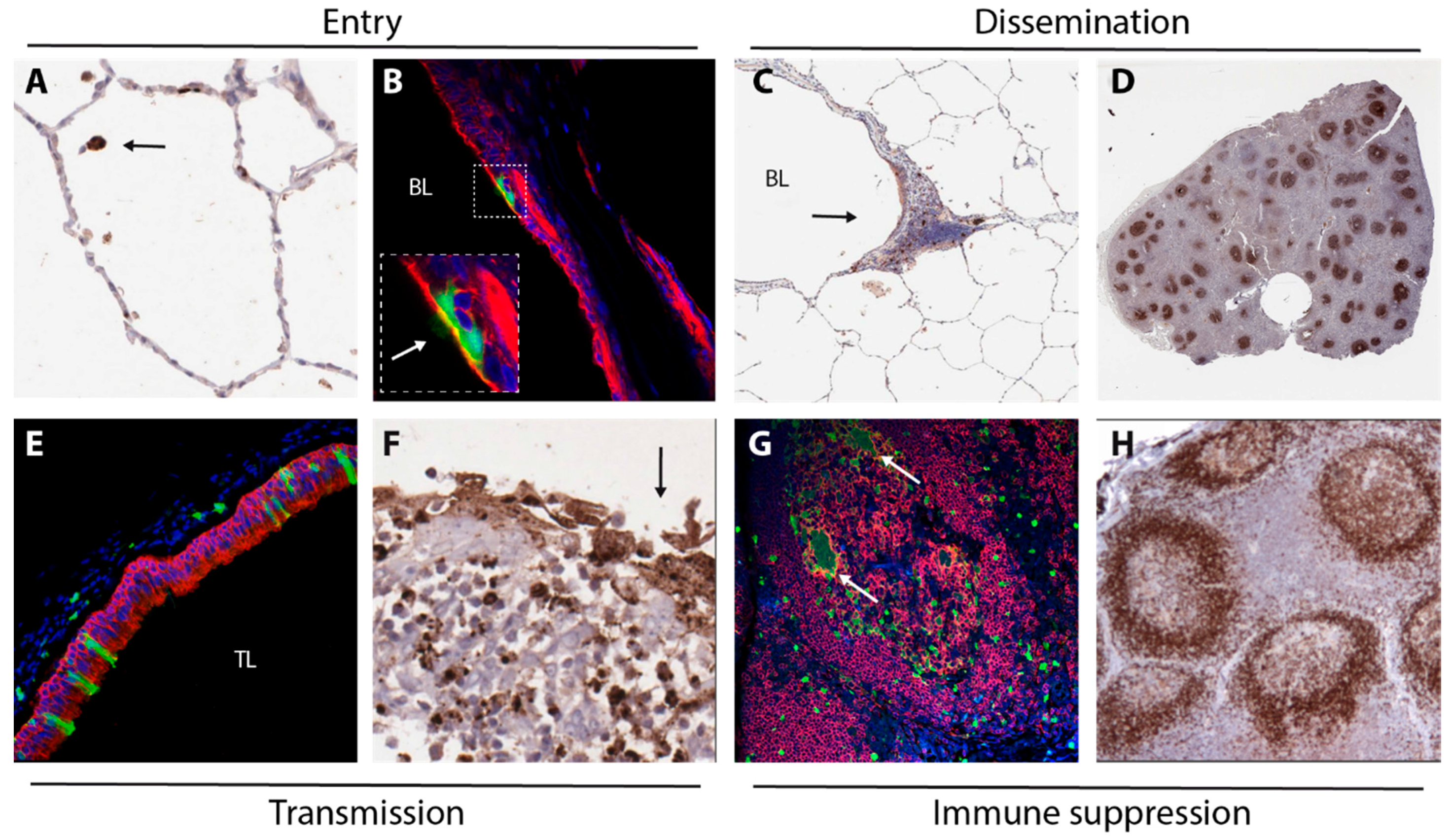
2. Entry
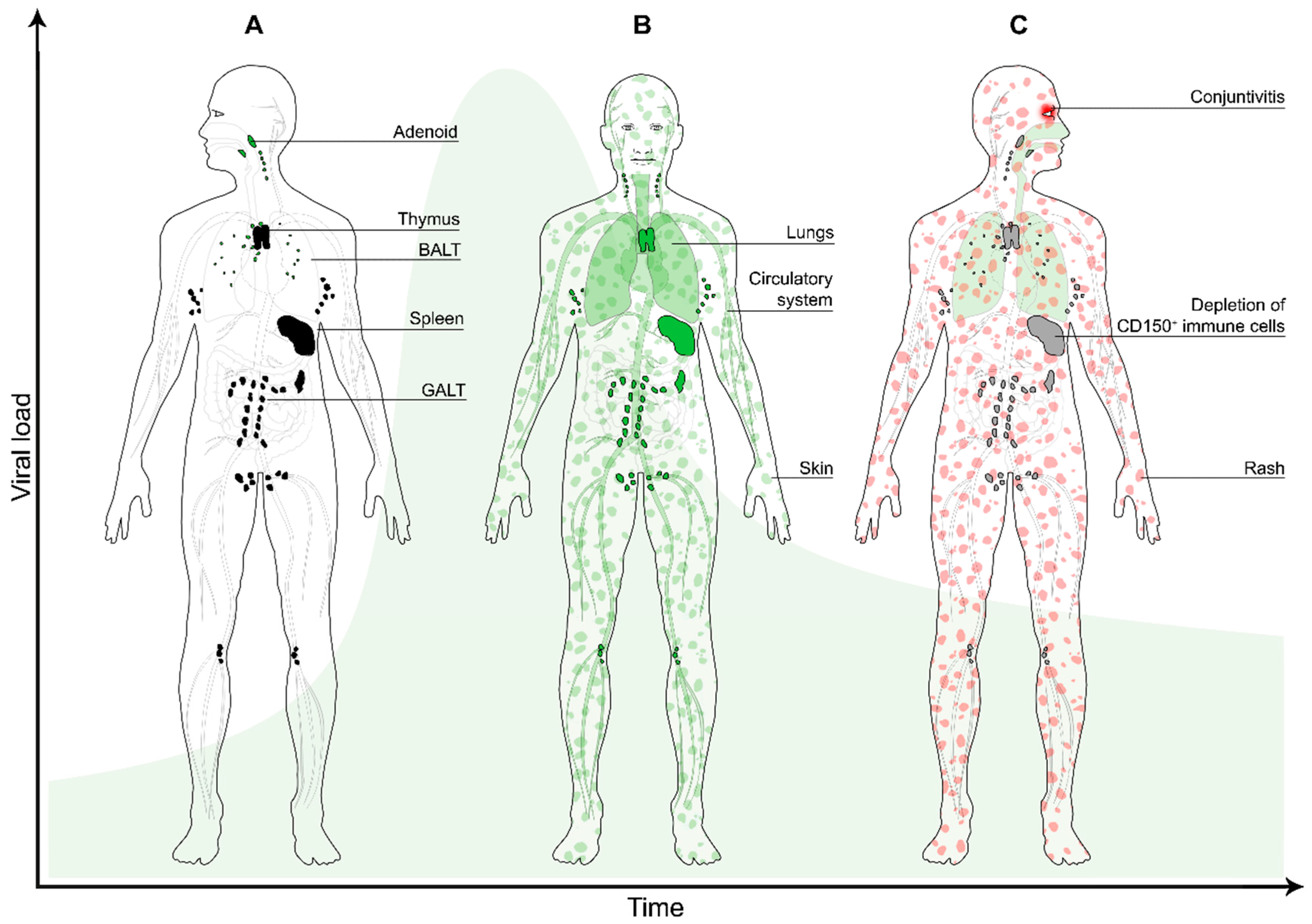
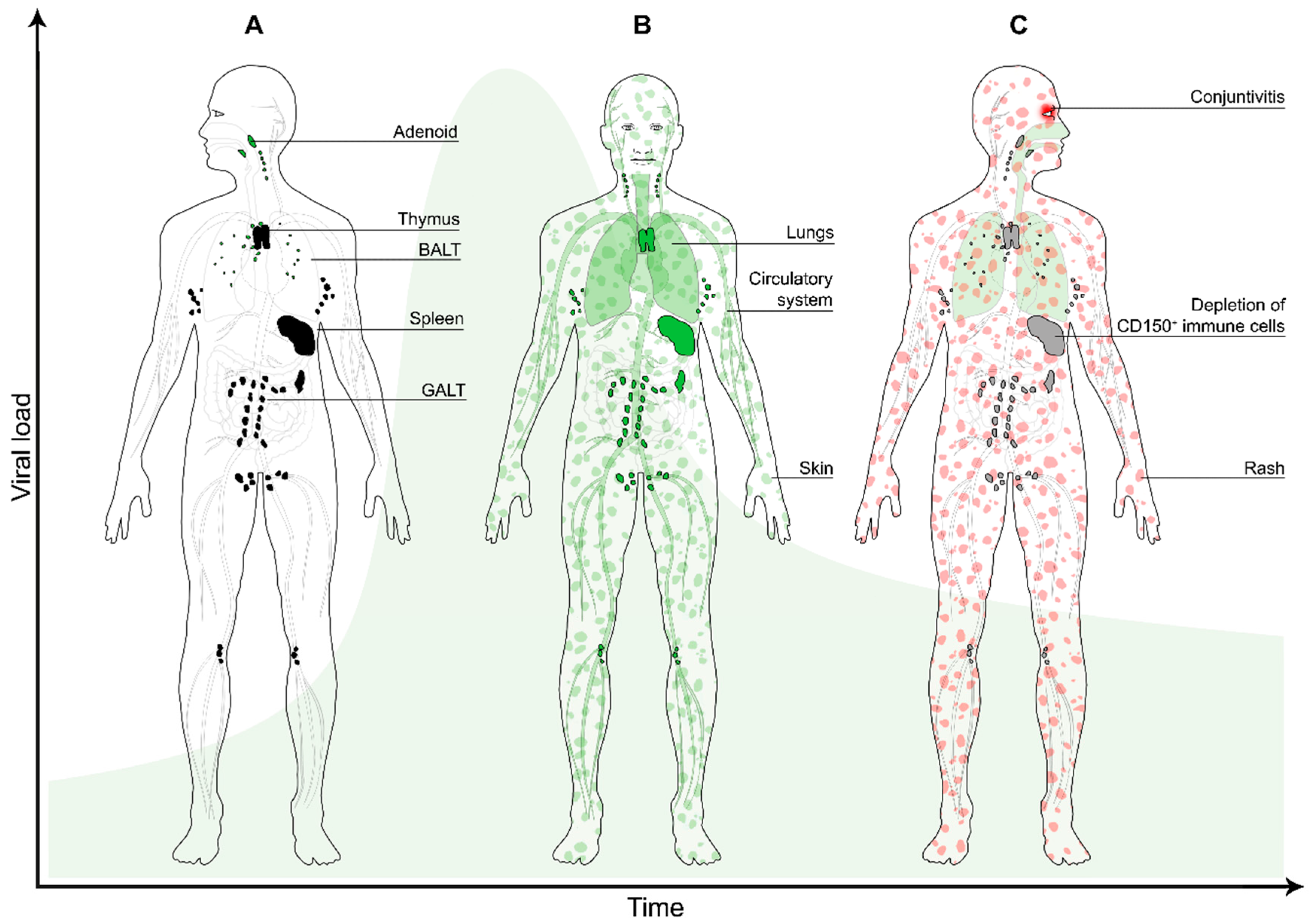
3. Dissemination
4. Transmission
5. Immune Suppression
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| MV | measles virus |
| NHP | non-human primate |
| CNS | central nervous system |
| ADEM | acute disseminated encephalomyelitis |
| MIBE | measles inclusion body encephalitis |
| SSPE | subacute sclerosing panencephalitis |
| WHO | World Health Organisation |
| SLAMF1 | signalling lymphocyte activation molecule family member 1 |
| DCs | dendritic cells |
| HSC | haematopoietic stem cells |
| rMV | recombinant measles virus |
| DC-SIGN | dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin |
| EGFP | enhanced green fluorescent protein |
| HRSV | human respiratory syncytial virus |
| BALT | bronchus-associated lymphoid tissues |
| EBV | Epstein-Barr virus |
| RNP | ribonucleoprotein |
| URT | upper respiratory tract |
| GALT | gut-associated lymphoid tissues |
References
- De Vries, R.D.; Duprex, W.P.; de Swart, R.L. Morbillivirus infections: An introduction. Viruses 2015, 7, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, Y.; Takeda, M.; Ohno, S. Measles virus: Cellular receptors, tropism and pathogenesis. J. Gen. Virol. 2006, 87, 2767–2779. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. Measles virus. In Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Mina, M.J.; Metcalf, C.J.; de Swart, R.L.; Osterhaus, A.D.; Grenfell, B.T. Long-term measles-induced immunomodulation increases overall childhood infectious disease mortality. Science 2015, 348, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.T.; Murray, J.S.; Gacic-Dobo, M.; Dabbagh, A.; Mulders, M.N.; Strebel, P.M.; Okwo-Bele, J.M.; Rota, P.A.; Goodson, J.L. Progress towards regional measles elimination, worldwide, 2000–2014. Wkly. Epidemiol. Rec. 2015, 90, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Tatsuo, H.; Ono, N.; Tanaka, K.; Yanagi, Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature 2000, 406, 893–897. [Google Scholar] [PubMed]
- Schwartzberg, P.L.; Mueller, K.L.; Qi, H.; Cannons, J.L. SLAM receptors and SAP influence lymphocyte interactions, development and function. Nat. Rev. Immunol. 2009, 9, 39–46. [Google Scholar] [CrossRef] [PubMed]
- De Swart, R.L.; Ludlow, M.; de Witte, L.; Yanagi, Y.; van Amerongen, G.; McQuaid, S.; Yuksel, S.; Geijtenbeek, T.B.; Duprex, W.P.; Osterhaus, A.D. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog. 2007, 3, e178. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.T.; Sisson, G.; Tsao, M.S.; Richardson, C.D. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.D.; Mateo, M.; Sinn, P.L.; Prufer, S.; Uhlig, K.M.; Leonard, V.H.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 2011, 480, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Mollo, M.R.; Antonini, D.; Mitchell, K.; Fortugno, P.; Costanzo, A.; Dixon, J.; Brancati, F.; Missero, C. p63-dependent and independent mechanisms of nectin-1 and nectin-4 regulation in the epidermis. Exp. Dermatol 2015, 24, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Fortugno, P.; Bottillo, I.; Lopez, M.; Josselin, E.; Boudghene-Stambouli, O.; Agolini, E.; Bernardini, L.; Bellacchio, E.; Iannicelli, M.; et al. Mutations in PVRL4, encoding cell adhesion molecule nectin-4, cause ectodermal dysplasia-syndactyly syndrome. Am. J. Hum. Genet. 2010, 87, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, H.; Brankin, B.; Brady, C.; Cosby, S.L. Wild-type measles virus infection upregulates poliovirus receptor-related 4 and causes apoptosis in brain endothelial cells by induction of tumor necrosis factor-related apoptosis-inducing ligand. J. Neuropathol. Exp. Neurol. 2013, 72, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Tosaka, K.; Nakao, T. Measles rash. I. Light and electron microscopic study of skin eruptions. Arch. Virol. 1975, 47, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Umino, Y.; Sato, T.A.; Kohama, T.; Ikeda, Y.; Iijima, M.; Fujisawa, R. Detection and comparison of viral antigens in measles and rubella rashes. Clin. Infect. Dis. 1996, 22, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Dörig, R.E.; Marcil, A.; Chopra, A.; Richardson, C.D. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 1993, 75, 295–305. [Google Scholar] [CrossRef]
- Buckland, R.; Wild, T.F. Is CD46 the cellular receptor for measles virus? Virus Res. 1997, 48, 1–9. [Google Scholar] [CrossRef]
- De Witte, L.; de Vries, R.D.; van der Vlist, M.; Yuksel, S.; Litjens, M.; de Swart, R.L.; Geijtenbeek, T.B. DC-SIGN and CD150 have distinct roles in transmission of measles virus from dendritic cells to T-lymphocytes. PLoS Pathog. 2008, 4, e1000049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Vlist, M.; de Witte, L.; de Vries, R.D.; Litjens, M.; de Jong, M.A.; Fluitsma, D.; de Swart, R.L.; Geijtenbeek, T.B. Human Langerhans cells capture measles virus through Langerin and present viral antigens to CD4+ T cells but are incapable of cross-presentation. Eur. J. Immunol. 2011, 41, 2619–2631. [Google Scholar] [CrossRef] [PubMed]
- Leonard, V.H.; Sinn, P.L.; Hodge, G.; Miest, T.; Devaux, P.; Oezguen, N.; Braun, W.; McCray, P.B., Jr.; McChesney, M.B.; Cattaneo, R. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J. Clin. Investig. 2008, 118, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Leonard, V.H.; Hodge, G.; Reyes-Del Valle, J.; McChesney, M.B.; Cattaneo, R. Measles virus selectively blind to signaling lymphocytic activation molecule (SLAM; CD150) is attenuated and induces strong adaptive immune responses in rhesus monkeys. J. Virol. 2010, 84, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; Lemon, K.; Ludlow, M.; McQuaid, S.; Yuksel, S.; van Amerongen, G.; Rennick, L.J.; Rima, B.K.; Osterhaus, A.D.; de Swart, R.L.; et al. In vivo tropism of attenuated and pathogenic measles virus expressing green fluorescent protein in macaques. J. Virol. 2010, 84, 4714–4724. [Google Scholar] [CrossRef] [PubMed]
- Mesman, A.W.; de Vries, R.D.; McQuaid, S.; Duprex, W.P.; de Swart, R.L.; Geijtenbeek, T.B.H. A prominent role for DC-SIGN+ dendritic cells in initiation and dissemination of measles virus infection in non-human primates. PLoS ONE 2012, 7, e49573. [Google Scholar] [CrossRef] [PubMed]
- Lemon, K.; de Vries, R.D.; Mesman, A.W.; McQuaid, S.; van Amerongen, G.; Yuksel, S.; Ludlow, M.; Rennick, L.J.; Kuiken, T.; Rima, B.K.; et al. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog. 2011, 7, e1001263. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, M.; McQuaid, S.; Milner, D.; de Swart, R.L.; Duprex, W.P. Pathological consequences of systemic measles virus infection. J. Pathol. 2015, 235, 253–265. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; Mesman, A.W.; Geijtenbeek, T.B.; Duprex, W.P.; de Swart, R.L. The pathogenesis of measles. Curr. Opin. Virol. 2012, 2, 248–255. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; McQuaid, S.; van Amerongen, G.; Yuksel, S.; Verburgh, R.J.; Osterhaus, A.D.; Duprex, W.P.; de Swart, R.L. Measles immune suppression: Lessons from the macaque model. PLoS Pathog. 2012, 8, e1002885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, K.Y.; Han, S.J.; Cha, H.R.; Seo, S.U.; Song, J.H.; Chung, S.H.; Kweon, M.N. Eye mucosa: An efficient vaccine delivery route for inducing protective immunity. J. Immunol. 2010, 185, 3610–3619. [Google Scholar] [CrossRef] [PubMed]
- Papp, K. Contagion des virus a travers une conjonctive intacte; rougeole, oreillons, rubéole. Rev. Immunol. Ther. Antimicrob. 1954, 18, 380–390. [Google Scholar] [PubMed]
- Ludlow, M.; Rennick, L.J.; Sarlang, S.; Skibinski, G.; McQuaid, S.; Moore, T.; de Swart, R.L.; Duprex, W.P. Wild-type measles virus infection of primary epithelial cells occurs via the basolateral surface without syncytium formation or release of infectious virus. J. Gen. Virol. 2010, 91, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Papp, K. Expériences prouvant que la voie d’infection de la rougeole est la contamination de la muqueuse conjonctivale. Rev. Immunol. Ther. Antimicrob. 1956, 20, 27–36. [Google Scholar] [PubMed]
- Singh, B.K.; Hornick, A.L.; Krishnamurthy, S.; Locke, A.C.; Mendoza, C.A.; Mateo, M.; Miller-Hunt, C.L.; Cattaneo, R.; Sinn, P.L. The nectin-4/afadin protein complex and intercellular membrane pores contribute to rapid spread of measles virus in primary human airway epithelia. J. Virol. 2015, 89, 7089–7096. [Google Scholar] [CrossRef] [PubMed]
- Message, S.D.; Johnston, S.L. The immunology of virus infection in asthma. Eur. Respir. J. 2001, 18, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Yoshikawa, Y.; Yamanouchi, K.; Sata, T.; Nagashima, K.; Takeda, K. Growth of measles virus in epithelial and lymphoid tissues of cynomolgus monkeys. Microb. Immunol. 1986, 30, 1067–1073. [Google Scholar] [CrossRef]
- Hall, W.C.; Kovatch, R.M.; Herman, P.H.; Fox, J.G. Pathology of measles in rhesus monkeys. Vet. Pathol. 1971, 8, 307–319. [Google Scholar] [PubMed]
- McChesney, M.B.; Miller, C.J.; Rota, P.A.; Zhu, Y.D.; Antipa, L.; Lerche, N.W.; Ahmed, R.; Bellini, W.J. Experimental measles. I. Pathogenesis in the normal and the immunized host. Virology 1997, 233, 74–84. [Google Scholar] [CrossRef] [PubMed]
- White, R.G.; Boyd, J.F. The effect of measles on the thymus and other lymphoid tissues. Clin. Exp. Immunol. 1973, 13, 343–357. [Google Scholar] [PubMed]
- Warthin, A.S. Occurence of numerous large giant cells in the tonsils and pharyngeal mucosa in the prodormal stage of measles. Arch. Pathol. 1931, 11, 864–874. [Google Scholar]
- Finkeldey, W. Über Riesenzellbefunde in den Gaumenmandeln, zugleich ein Beitrag zur Histopathologie der Mandelveränderungen im Maserninkubationsstadium. Virchows Arch. 1931, 281, 323–329. [Google Scholar] [CrossRef]
- Stryker, W.A. Disseminated giant cell reaction: A possible prodrome of measles. Am. J. Dis. Child. 1940, 59, 468–478. [Google Scholar] [CrossRef]
- Koethe, S.; Avota, E.; Schneider-Schaulies, S. Measles virus transmission from dendritic cells to T cells: Formation of synapse-like interfaces concentrating viral and cellular components. J. Virol. 2012, 86, 9773–9781. [Google Scholar] [CrossRef] [PubMed]
- Mateo, M.; Generous, A.; Sinn, P.L.; Cattaneo, R. Connections matter—How viruses use cell-cell adhesion components. J. Cell. Sci 2015, 128, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, M.; Lemon, K.; de Vries, R.D.; McQuaid, S.; Millar, E.L.; van Amerongen, G.; Yuksel, S.; Verburgh, R.J.; Osterhaus, A.D.; de Swart, R.L.; et al. Measles virus infection of epithelial cells in the macaque upper respiratory tract is mediated by subepithelial immune cells. J. Virol. 2013, 87, 4033–4042. [Google Scholar] [CrossRef] [PubMed]
- Frenzke, M.; Sawatsky, B.; Wong, X.X.; Delpeut, S.; Mateo, M.; Cattaneo, R.; von Messling, V. Nectin-4-dependent measles virus spread to the cynomolgus monkey tracheal epithelium: Role of infected immune cells infiltrating the lamina propria. J. Virol. 2013, 87, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Mateo, M.; Navaratnarajah, C.K.; Willenbring, R.C.; Maroun, J.W.; Iankov, I.; Lopez, M.; Sinn, P.L.; Cattaneo, R. Different roles of the three loops forming the adhesive interface of nectin-4 in measles virus binding and cell entry, nectin-4 homodimerization, and heterodimerization with nectin-1. J. Virol. 2014, 88, 14161–14171. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, M.; Allen, I.; Schneider-Schaulies, J. Systemic spread of measles virus: Overcoming the epithelial and endothelial barriers. Thromb. Haemost 2009, 102, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, S.; Harms, H.; Runkler, N.; Maisner, A.; Kim, K.S.; Schneider-Schaulies, J. Measles virus-induced block of transendothelial migration of T lymphocytes and infection-mediated virus spread across endothelial cell barriers. J. Virol. 2008, 82, 11273–11282. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.M. Infection of endothelial cells by common human viruses. Rev. Infect. Dis. 1989, 11, S700–S704. [Google Scholar] [CrossRef] [PubMed]
- Moench, T.R.; Griffin, D.E.; Obriecht, C.R.; Vaisberg, A.J.; Johnson, R.T. Acute measles in patients with and without neurological involvement: Distribution of measles virus antigen and RNA. J. Infect. Dis. 1988, 158, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Ono, N.; Tatsuo, H.; Minagawa, H.; Takeda, M.; Takeuchi, K.; Yanagi, Y. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J. Virol. 2002, 76, 6743–6749. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Overholtzer, M. In-cell infection: Bringing uninvited guests. Cell. Res. 2015, 25, 647–648. [Google Scholar] [CrossRef] [PubMed]
- Suringa, D.W.; Bank, L.J.; Ackerman, A.B. Role of measles virus in skin lesions and Koplik‘s spots. N. Engl. J. Med. 1970, 283, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- De Swart, R.L.; Wertheim-van Dillen, P.M.; van Binnendijk, R.S.; Muller, C.P.; Frenkel, J.; Osterhaus, A.D. Measles in a Dutch hospital introduced by an immuno-compromised infant from Indonesia infected with a new virus genotype. Lancet 2000, 355, 201–202. [Google Scholar] [CrossRef]
- Griffin, D.E. Measles virus and the nervous system. Handb. Clin. Neurol. 2014, 123, 577–590. [Google Scholar] [PubMed]
- Fujinami, R.S.; Oldstone, M.B.; Wroblewska, Z.; Frankel, M.E.; Koprowski, H. Molecular mimicry in virus infection: Crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad. Sci. USA 1983, 80, 2346–2350. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.; Bonthius, D.J. Measles virus and associated central nervous system sequelae. Semin. Pediatr. Neurol. 2012, 19, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.F.; Jacobsohn, D.A.; Shulman, S.T.; Bellini, W.J.; Jaggi, P.; de Leon, G.; Keating, G.F.; Kim, F.; Pachman, L.M.; Kletzel, M.; et al. A new complication of stem cell transplantation: Measles inclusion body encephalitis. Pediatrics 2004, 114, e657–e660. [Google Scholar] [CrossRef] [PubMed]
- Bitnun, A.; Shannon, P.; Durward, A.; Rota, P.A.; Bellini, W.J.; Graham, C.; Wang, E.; Ford-Jones, E.L.; Cox, P.; Becker, L.; et al. Measles inclusion-body encephalitis caused by the vaccine strain of measles virus. Clin. Infect. Dis. 1999, 29, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.T.; Halsey, N.A. The clinical significance of measles: A review. J. Infect. Dis. 2004, 189, S4–S16. [Google Scholar] [CrossRef] [PubMed]
- Ehrengruber, M.U.; Ehler, E.; Billeter, M.A.; Naim, H.Y. Measles virus spreads in rat hippocampal neurons by cell-to-cell contact and in a polarized fashion. J. Virol. 2002, 76, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Duprex, W.P.; McQuaid, S.; Roscic-Mrkic, B.; Cattaneo, R.; McCallister, C.; Rima, B.K. In vitro and in vivo infection of neural cells by a recombinant measles virus expressing enchanced green fluorescent protein. J. Virol. 2002, 74, 7972–7979. [Google Scholar] [CrossRef]
- Lawrence, D.M.P.; Patterson, C.E.; Gales, T.L.; D‘orazio, J.L.; Vaughn, M.M.; Rall, G.F. Measles virus spread between neurons requires cell contact but not CD46 expression, syncytium formation, or extracellular virus production. J. Virol. 2000, 74, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Makhortova, N.R.; Askovich, P.; Patterson, C.E.; Gechman, L.A.; Gerard, N.P.; Rall, G.F. Neurokinin-1 enables measles virus trans-synaptic spread in neurons. Virology 2007, 362, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Shirogane, Y.; Suzuki, S.O.; Ikegame, S.; Koga, R.; Yanagi, Y. Mutant fusion proteins with enhanced fusion activity promote measles virus spread in human neuronal cells and brains of suckling hamsters. J. Virol. 2013, 87, 2648–2659. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, I.; Galea, I.; Perry, V.H. What is the blood-brain barrier (not)? Trends Immunol. 2007, 28, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Moss, W.J.; Griffin, D.E. Global measles elimination. Nat. Rev. Microb. 2006, 4, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Racaniello, V. Virology. An exit strategy for measles virus. Science 2011, 334, 1650–1651. [Google Scholar] [CrossRef] [PubMed]
- Christensen, P.E.; Schmidt, H.; Jensen, O.; Bang, H.O.; Andersen, V.; Jordal, B. An epidemic of measles in Southern Greenland, 1951. I. Measles in virgin soil. Acta Med. Scand. 1953, 144, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Smith, J.O.; Schreiber, S.J.; Kopp, P.E.; Getz, W.M. Superspreading and the effect of individual variation on disease emergence. Nature 2005, 438, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Bloch, A.B.; Orenstein, W.A.; Ewing, W.M.; Spain, W.H.; Mallison, G.F.; Herrmann, K.L.; Hinman, A.R. Measles outbreak in a pediatric practice: Airborne transmission in an office setting. Pediatrics 1985, 75, 676–683. [Google Scholar] [PubMed]
- Ehresmann, K.R.; Hedberg, C.W.; Grimm, M.B.; Norton, C.A.; Macdonald, K.L.; Osterholm, M.T. An outbreak of measles at an international sporting event with airborne transmission in a domed stadium. J. Infect. Dis. 1995, 171, 679–683. [Google Scholar] [CrossRef] [PubMed]
- De Jong, J.G.; Winkler, K.C. Survival of measles virus in air. Nature 1964, 201, 1054–1055. [Google Scholar] [CrossRef] [PubMed]
- Van Binnendijk, R.S.; van der Heijden, R.W.; van Amerongen, G.; UytdeHaag, F.G.; Osterhaus, A.D. Viral replication and development of specific immunity in macaques after infection with different measles virus strains. J. Infect. Dis. 1994, 170, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.Y.; Phipps, S. Regulation of inducible BALT formation and contribution to immunity and pathology. Mucosal Immunol. 2010, 3, 537–544. [Google Scholar] [CrossRef] [PubMed]
- GeurtsvanKessel, C.H.; Willart, M.A.; Bergen, I.M.; van Rijt, L.S.; Muskens, F.; Elewaut, D.; Osterhaus, A.D.; Hendriks, R.; Rimmelzwaan, G.F.; Lambrecht, B.N. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J. Exp. Med. 2009, 206, 2339–2349. [Google Scholar] [CrossRef] [PubMed]
- Ryon, J.J.; Moss, W.J.; Monze, M.; Griffin, D.E. Functional and phenotypic changes in circulating lymphocytes from hospitalized zambian children with measles. Clin. Diagn. Lab. Immunol. 2002, 9, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; Yuksel, S.; Osterhaus, A.D.; de Swart, R.L. Specific CD8+ T-lymphocytes control dissemination of measles virus. Eur. J. Immunol. 2010, 40, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E.; Ward, B.J.; Jauregui, E.; Johnson, R.T.; Vaisberg, A. Immune activation in measles. N. Engl. J. Med. 1989, 320, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. Measles virus-induced suppression of immune responses. Immunol. Rev. 2010, 236, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.J.; Griffin, D.E. Changes in cytokine production after measles virus vaccination: Predominant production of IL-4 suggests induction of a Th2 response. Clin. Immunol. Immunopathol. 1993, 67, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Hoffman, S.J.; Moss, W.J.; Griffin, D.E. Altered synthesis of interleukin-12 and type 1 and type 2 cytokinesin rhesus macaques during measles and atypical measles. J. Infect. Dis. 2002, 185, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Atabani, S.F.; Byrnes, A.A.; Jaye, A.; Kidd, I.M.; Magnusen, A.F.; Whittle, H.; Karp, C.L. Natural measles causes prolonged suppression of interleukin-12 production. J. Infect. Dis. 2001, 184, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Karp, C.L.; Wysocka, M.; Wahl, L.M.; Ahearn, J.M.; Cuomo, P.J.; Sherry, B.; Trinchieri, G.; Griffin, D.E. Mechanism of suppression of cell-mediated immunity by measles virus. Science 1996, 273, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Manchester, M.; Smith, K.A.; Eto, D.S.; Perkin, H.B.; Torbett, B.E. Targeting and hematopoietic suppression of human CD34+ cells by measles virus. J. Virol. 2002, 76, 6636–6642. [Google Scholar] [CrossRef] [PubMed]
- Boussaad, I.; Varagnolo, L.; Hornich, V.; Rieger, L.; Krockenberger, M.; Stuehmer, T.; Kranzfelder, D.; Mueller, A.M.; Schneider-Schaulies, S. Wild-type measles virus interferes with short-term engraftment of human CD34+ hematopoietic progenitor cells. J. Virol. 2011, 85, 7710–7718. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; de Swart, R.L. Measles immune suppression: Functional impairment or numbers game? PLoS Pathog. 2014, 10, e1004482. [Google Scholar] [CrossRef] [PubMed]
- Nambulli, S.; Sharp, C.R.; Acciardo, A.S.; Drexler, J.F.; Duprex, W.P. Mapping the evolutionary trajectories of morbilliviruses: What, where and whither. Curr. Opin. Virol. 2016, 16, 95–105. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laksono, B.M.; De Vries, R.D.; McQuaid, S.; Duprex, W.P.; De Swart, R.L. Measles Virus Host Invasion and Pathogenesis. Viruses 2016, 8, 210. https://doi.org/10.3390/v8080210
Laksono BM, De Vries RD, McQuaid S, Duprex WP, De Swart RL. Measles Virus Host Invasion and Pathogenesis. Viruses. 2016; 8(8):210. https://doi.org/10.3390/v8080210
Chicago/Turabian StyleLaksono, Brigitta M., Rory D. De Vries, Stephen McQuaid, W. Paul Duprex, and Rik L. De Swart. 2016. "Measles Virus Host Invasion and Pathogenesis" Viruses 8, no. 8: 210. https://doi.org/10.3390/v8080210
APA StyleLaksono, B. M., De Vries, R. D., McQuaid, S., Duprex, W. P., & De Swart, R. L. (2016). Measles Virus Host Invasion and Pathogenesis. Viruses, 8(8), 210. https://doi.org/10.3390/v8080210