
Measles Virus Fusion Protein: Structure, Function and Inhibition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Diseases
3. Cell Entry: The Viral Membrane Fusion Machinery
4. Host Cellular Receptors and Pathogenesis
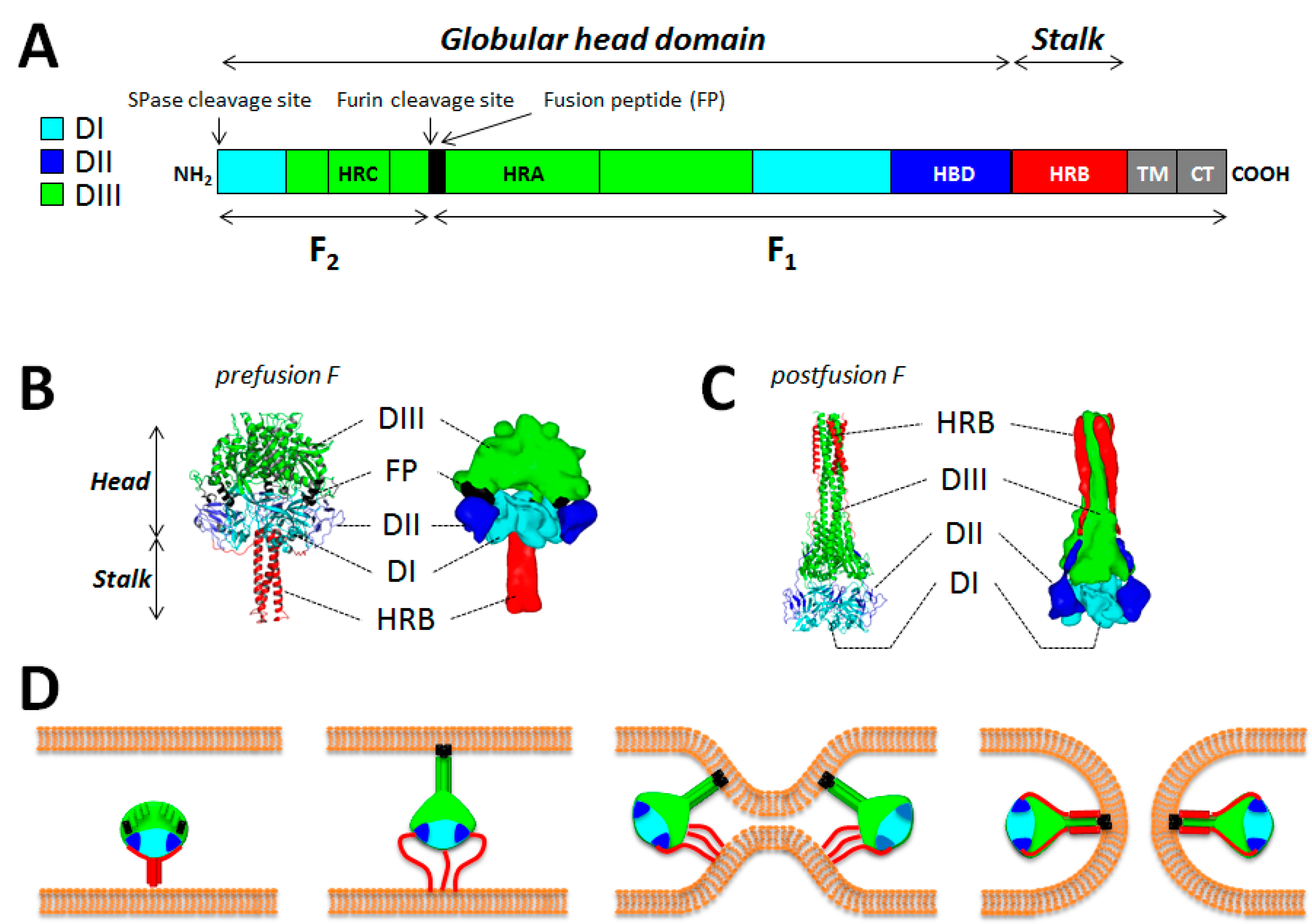
5. The Fusion Protein F
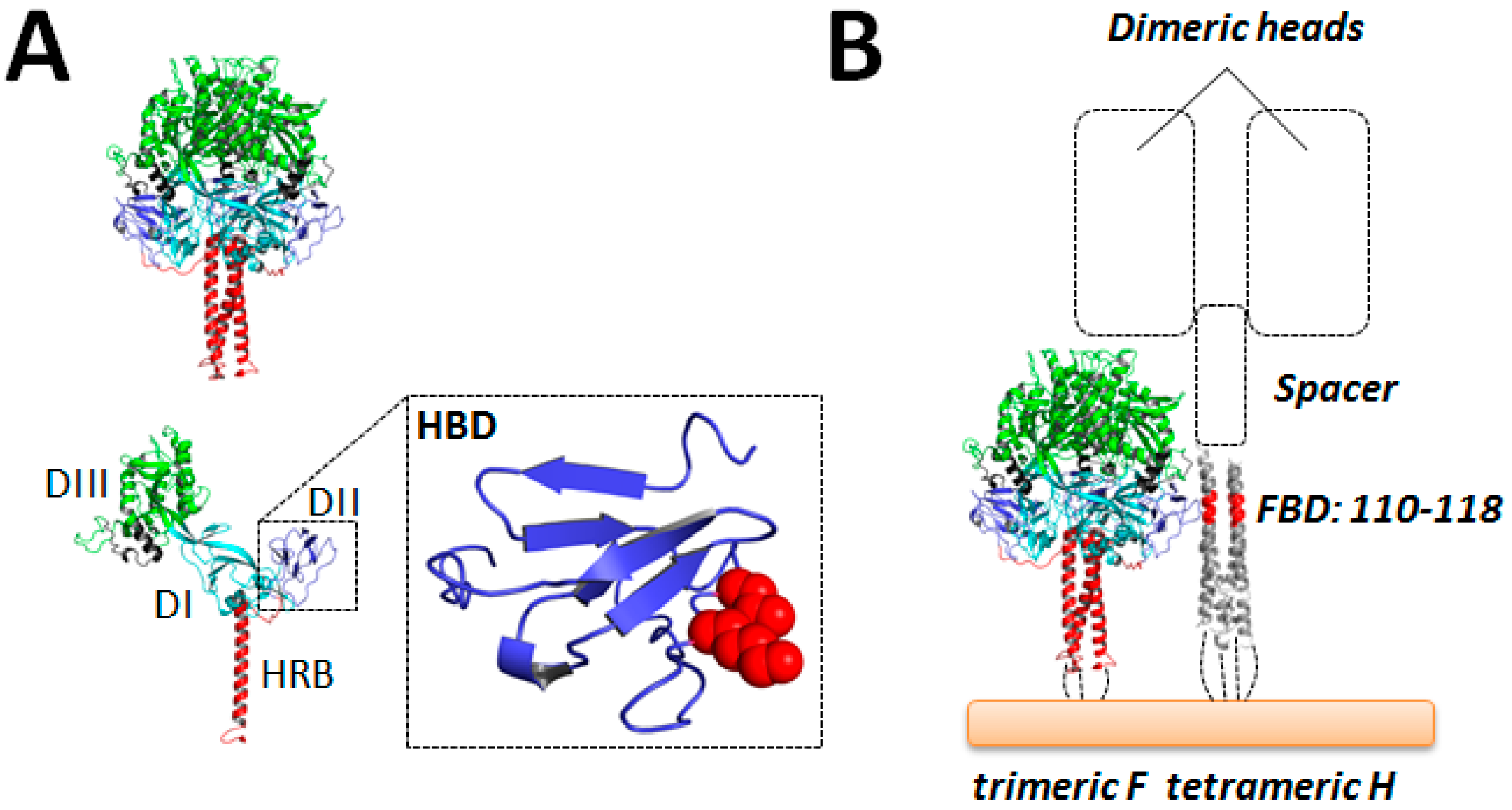
5.1. General Information
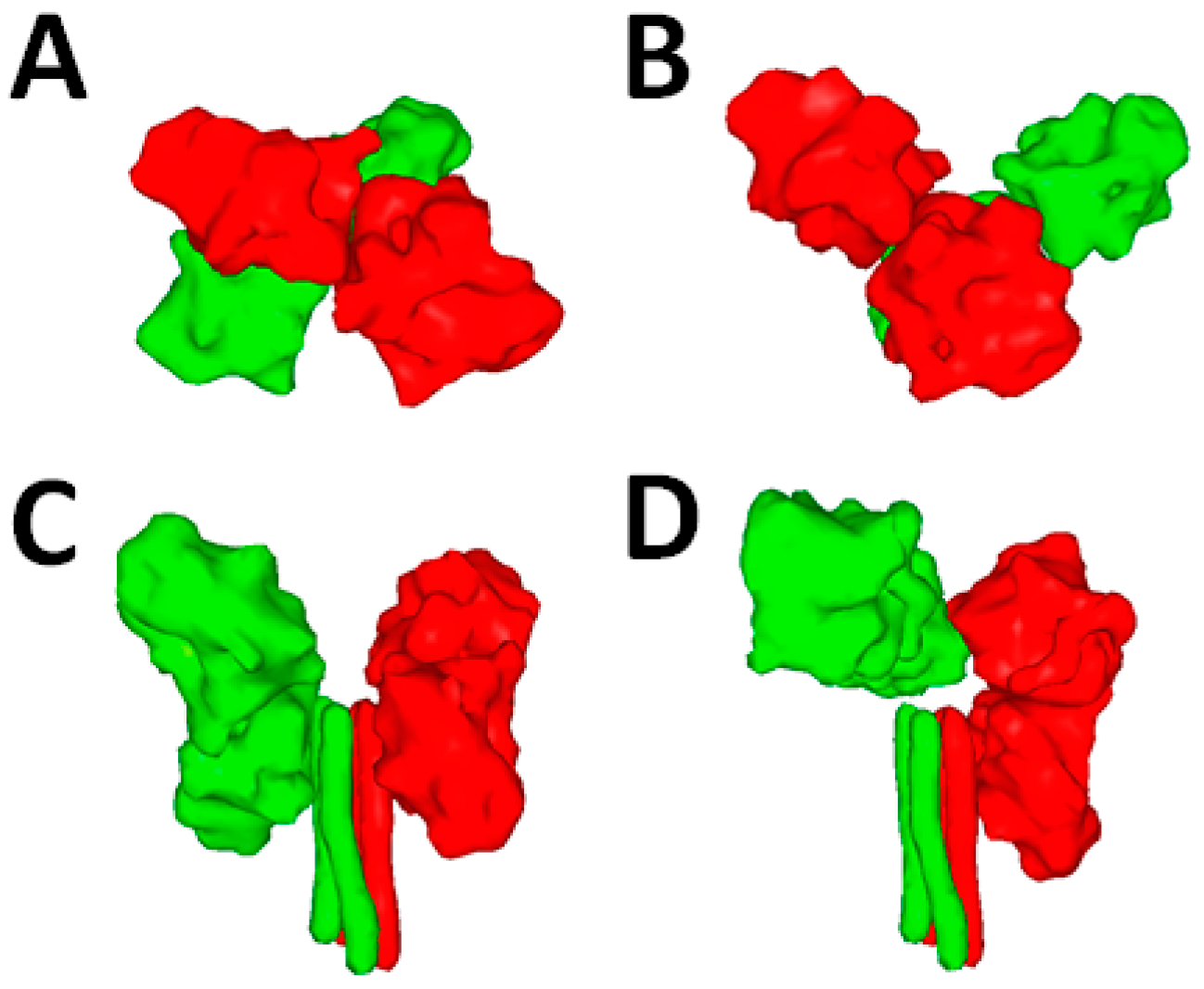
5.2. F Structural Information
5.3. The F-Refolding Cascade
6. The F-Activation Stimulus
6.1. The Receptor-Binding H Protein: General Information
6.2. Structural Information
6.2.1. H-Heads
6.2.2. H-Stalks
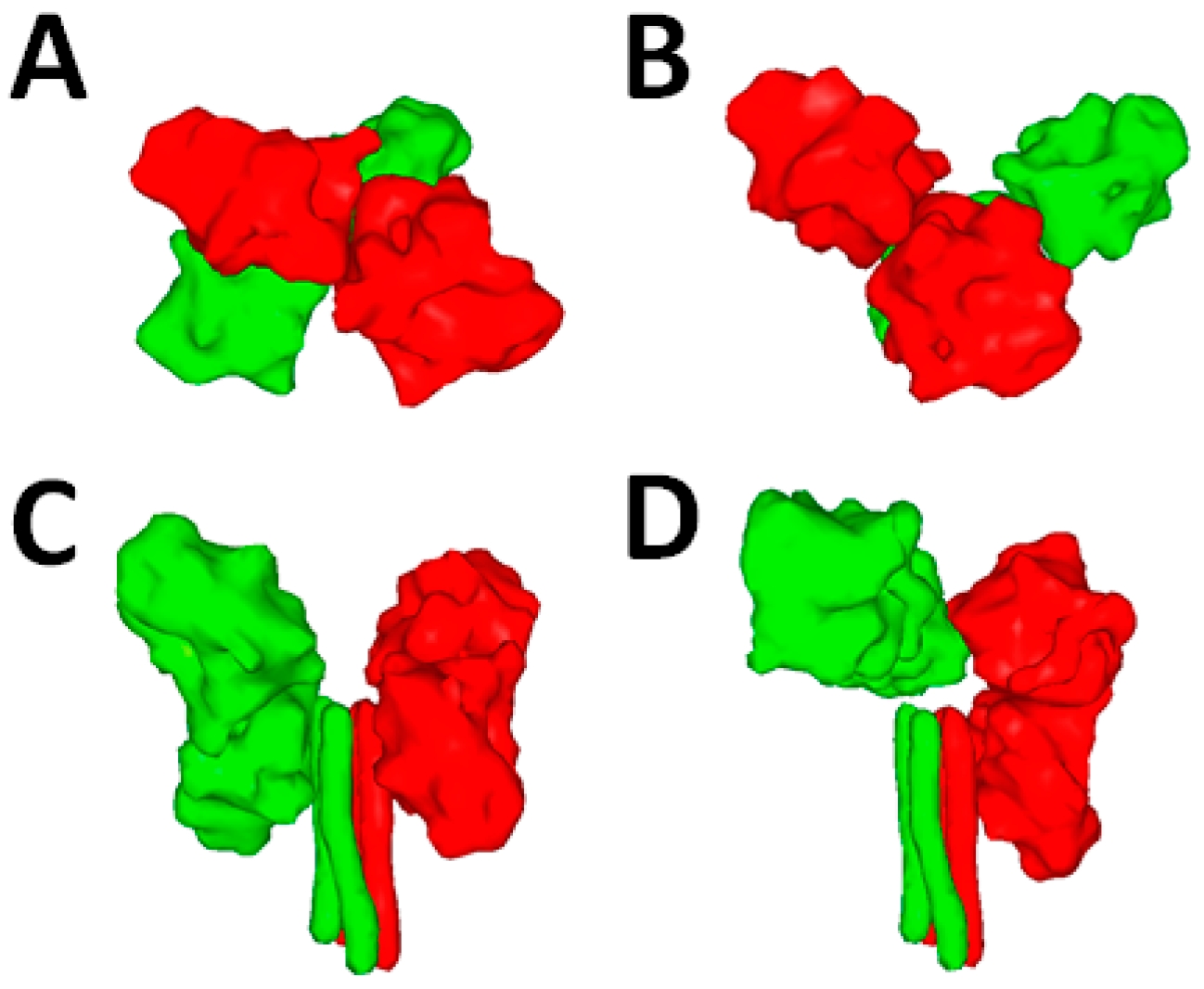
6.2.3. Tetrameric Attachment Protein Structures
MeV H
NDV HN
hPIV5 HN
6.3. Functional Information
7. The Putative F-H Binding Interface
8. Energetics of F Activation
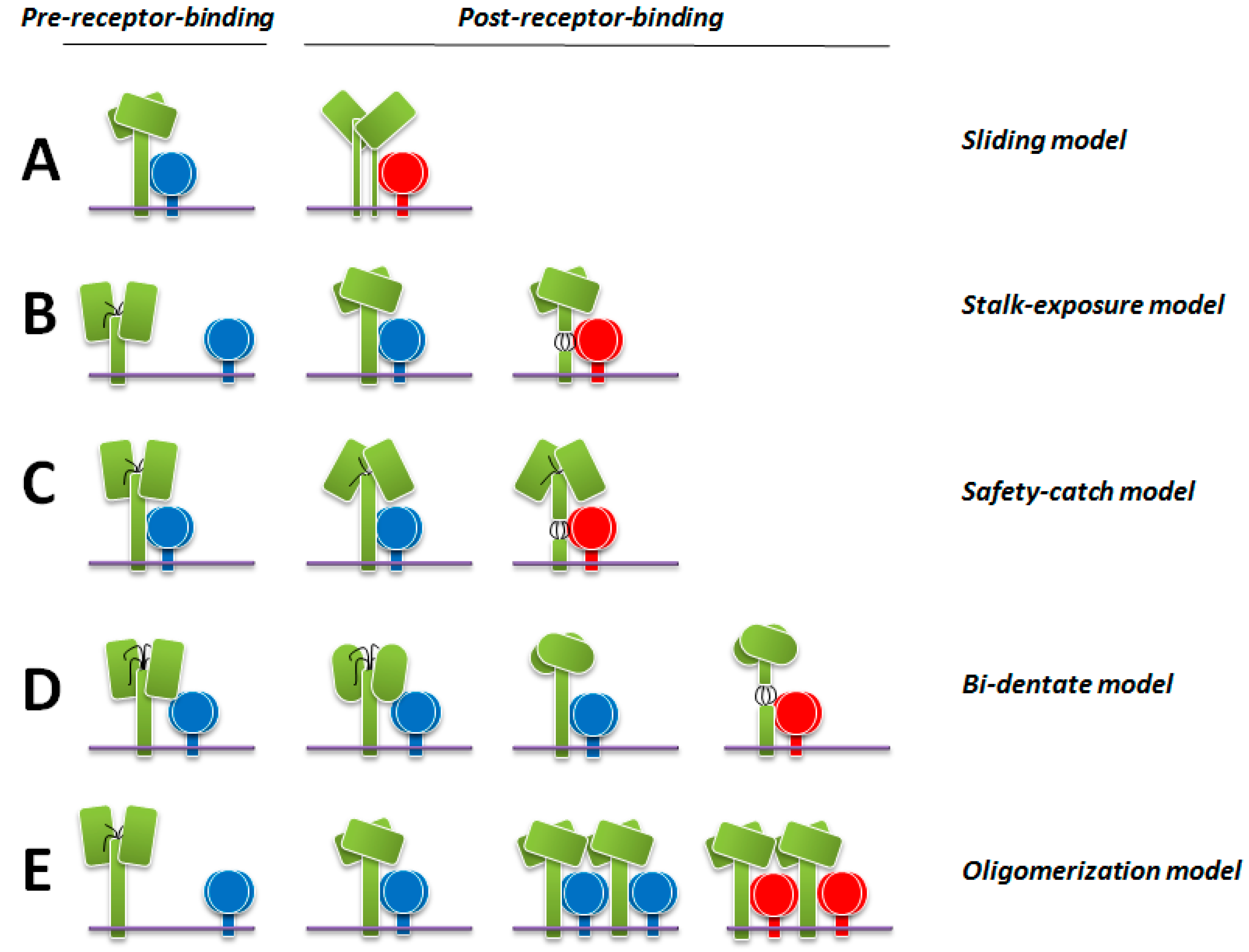
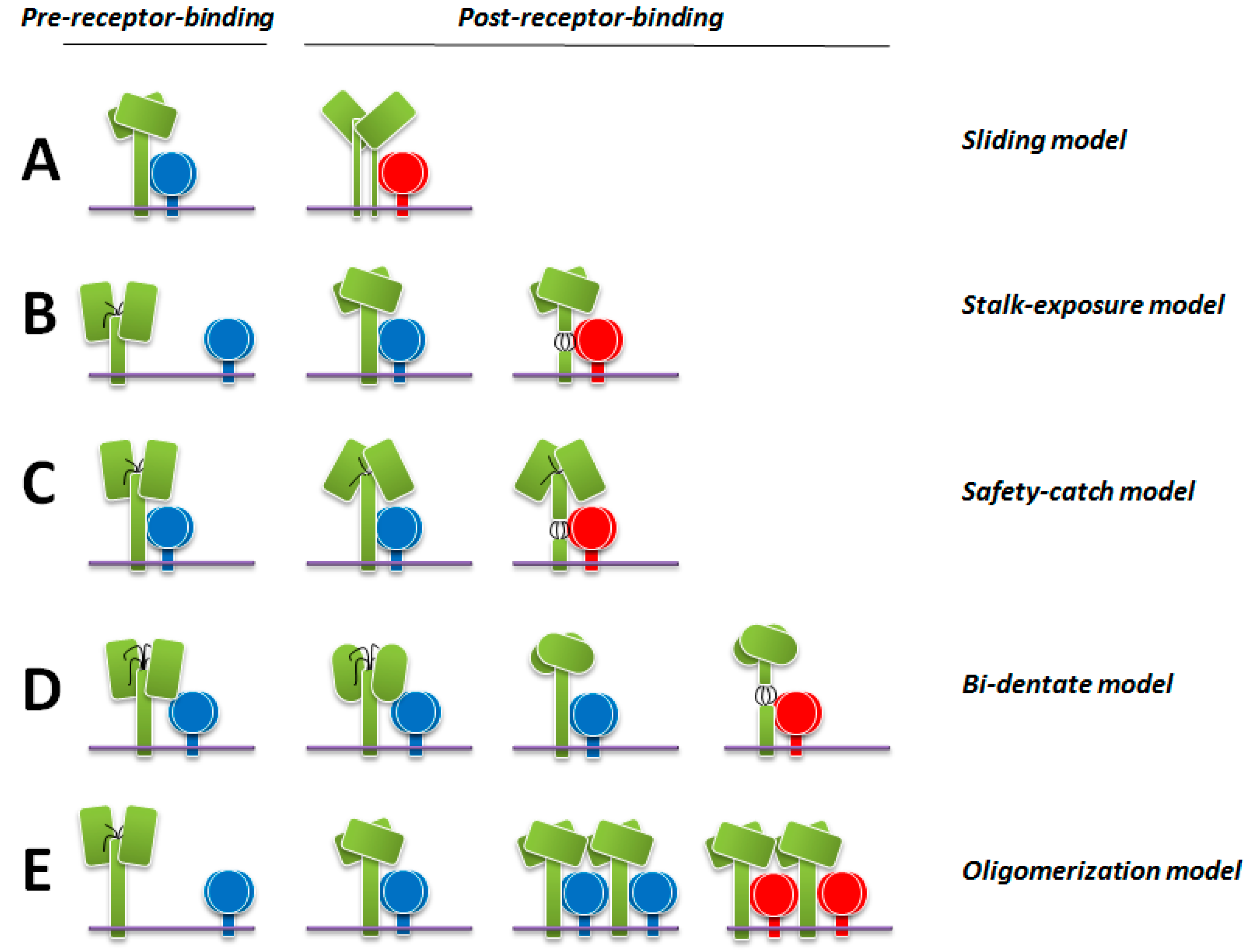
9. Latest Models of F Activation
9.1. The Sliding Model
9.2. The Stalk-Exposure/Induced Fit Model
9.3. The Safety-Catch Model
9.4. The Bi-Dentate Attachment Protein/F Interaction Model
9.5. The Receptor-Induced Oligomerization Model
10. The Concept of the F-Triggering Range
11. Development of MeV F Inhibitors
11.1. Anti-F Peptidic Inhibitors
11.2. Anti-F Inhibitory Antibodies
11.3. Anti-F Small-Molecule Blockers
11.4. Mechanism of Viral Resistance to AS-48 and Chemical Analogs
12. Functional Impact of the F Protein in Brain Disorders: A Highly Regulated or Dysregulated Membrane Fusion Process?
13. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: The viruses and their replication. In Fields’ Virology, 5th ed.; Fileds, B., Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1449–1496. [Google Scholar]
- Simons, E.; Ferrari, M.; Fricks, J.; Wannemuehler, K.; Anand, A.; Burton, A.; Strebel, P. Assessment of the 2010 global measles mortality reduction goal: Results from a model of surveillance data. Lancet 2012, 379, 2173–2178. [Google Scholar] [CrossRef]
- Plemper, R.K.; Hammond, A.L. Synergizing vaccinations with therapeutics for measles eradication. Expert Opin. Drug Discov. 2014, 9, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.; et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Westbury, H.A.; Hooper, P.T.; Selleck, P.W.; Murray, P.K. Equine morbillivirus pneumonia: Susceptibility of laboratory animals to the virus. Aust. Vet. J. 1995, 72, 278–279. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.M.; Hooper, P.T.; Selleck, P.W.; Gleeson, L.J.; Daniels, P.W.; Westbury, H.A.; Murray, P.K. Transmission studies of hendra virus (equine morbilli-virus) in fruit bats, horses and cats. Aust. Vet. J. 1998, 76, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, V.; Contamin, H.; Loth, P.; Grosjean, I.; Courbot, M.C.; Deubel, V.; Buckland, R.; Wild, T.F. Antibody prophylaxis and therapy against nipah virus infection in hamsters. J. Virol. 2006, 80, 1972–1978. [Google Scholar] [CrossRef] [PubMed]
- Roelke-Parker, M.E.; Munson, L.; Packer, C.; Kock, R.; Cleaveland, S.; Carpenter, M.; O’Brien, S.J.; Pospischil, A.; Hofmann-Lehmann, R.; Lutz, H.; et al. A canine distemper virus epidemic in Serengeti lions (Panthera leo). Nature 1996, 379, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Nagata, N.; Ami, Y.; Seki, F.; Suzaki, Y.; Iwata-Yoshikawa, N.; Suzuki, T.; Fukushi, S.; Mizutani, T.; Yoshikawa, T.; et al. Lethal canine distemper virus outbreak in cynomolgus monkeys in japan in 2008. J. Virol. 2013, 87, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Osterhaus, A.D.; Vedder, E.J. Identification of virus causing recent seal deaths. Nature 1988, 335, 20. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.J. Morbilliviruses in marine mammals. Trends Microbiol. 1995, 3, 4–9. [Google Scholar] [CrossRef]
- Kennedy, S. Morbillivirus infections in aquatic mammals. J. Comp. Pathol. 1998, 119, 201–225. [Google Scholar] [CrossRef]
- Origgi, F.C.; Plattet, P.; Sattler, U.; Robert, N.; Casaubon, J.; Mavrot, F.; Pewsner, M.; Wu, N.; Giovannini, S.; Oevermann, A.; et al. Emergence of canine distemper virus strains with modified molecular signature and enhanced neuronal tropism leading to high mortality in wild carnivores. Vet. Pathol. 2012, 49, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Normile, D. Animal science. Rinderpest, deadly for cattle, joins smallpox as a vanquished disease. Science 2010, 330, 435. [Google Scholar] [CrossRef] [PubMed]
- Albina, E.; Kwiatek, O.; Minet, C.; Lancelot, R.; Servan de Almeida, R.; Libeau, G. Peste des petits ruminants, the next eradicated animal disease? Vet. Microbiol. 2013, 165, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Stone, B.M.; Blyde, D.J.; Saliki, J.T.; Blas-Machado, U.; Bingham, J.; Hyatt, A.; Wang, J.; Payne, J.; Crameri, S. Fatal cetacean morbillivirus infection in an australian offshore bottlenose dolphin (Tursiops truncatus). Aust. Vet. J. 2011, 89, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Philip Earle, J.A.; Melia, M.M.; Doherty, N.V.; Nielsen, O.; Cosby, S.L. Phocine distemper virus in seals, east coast, United States, 2006. Emerg. Infect. Dis. 2011, 17, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Moss, W.J.; Griffin, D.E. Global measles elimination. Nat. Rev. Microbiol. 2006, 4, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Butler, D. Measles by the numbers: A race to eradication. Nature 2015, 518, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Kupferschmidt, K. Public health. Europe’s embarrassing problem. Science 2012, 336, 406–407. [Google Scholar] [CrossRef] [PubMed]
- Sammons, J.S. Responding to measles in the postelimination era. Ann. Intern. Med. 2014, 161, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Mina, M.J.; Metcalf, C.J.; de Swart, R.L.; Osterhaus, A.D.; Grenfell, B.T. Long-term measles-induced immunomodulation increases overall childhood infectious disease mortality. Science 2015, 348, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Krumm, S.A.; Yan, D.; Hovingh, E.S.; Evers, T.J.; Enkirch, T.; Reddy, G.P.; Sun, A.; Saindane, M.T.; Arrendale, R.F.; Painter, G.; et al. An orally available, small-molecule polymerase inhibitor shows efficacy against a lethal morbillivirus infection in a large animal model. Sci. Transl. Med. 2014, 6, 232ra252. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Huey, D.; Jurgens, E.; Welsch, J.C.; DeVito, I.; Talekar, A.; Horvat, B.; Niewiesk, S.; Moscona, A.; Porotto, M. Prevention of measles virus infection by intranasal delivery of fusion inhibitor peptides. J. Virol. 2015, 89, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Welsch, J.C.; Talekar, A.; Mathieu, C.; Pessi, A.; Moscona, A.; Horvat, B.; Porotto, M. Fatal measles virus infection prevented by brain-penetrant fusion inhibitors. J. Virol. 2013, 87, 13785–13794. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Ray, W.C.; Peeples, M.E. Structure and function of respiratory syncytial virus surface glycoproteins. Curr. Top. Microbiol. Immunol. 2013, 372, 83–104. [Google Scholar] [PubMed]
- De Vries, R.D.; Mesman, A.W.; Geijtenbeek, T.B.; Duprex, W.P.; de Swart, R.L. The pathogenesis of measles. Curr. Opin. Virol. 2012, 2, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.S.; Frenzke, M.; Leonard, V.H.; Welstead, G.G.; Richardson, C.D.; Cattaneo, R. Measles virus infection of alveolar macrophages and dendritic cells precedes spread to lymphatic organs in transgenic mice expressing human signaling lymphocytic activation molecule (SLAM, CD150). J. Virol. 2010, 84, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Avota, E.; Koethe, S.; Schneider-Schaulies, S. Membrane dynamics and interactions in measles virus dendritic cell infections. Cell. Microbiol. 2013, 15, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Koethe, S.; Avota, E.; Schneider-Schaulies, S. Measles virus transmission from dendritic cells to t cells: Formation of synapse-like interfaces concentrating viral and cellular components. J. Virol. 2012, 86, 9773–9781. [Google Scholar] [CrossRef] [PubMed]
- Lemon, K.; de Vries, R.D.; Mesman, A.W.; McQuaid, S.; van, A.G.; Yuksel, S.; Ludlow, M.; Rennick, L.J.; Kuiken, T.; Rima, B.K.; et al. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog. 2011, 7, e1001263. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, M.; McQuaid, S.; Milner, D.; de Swart, R.L.; Duprex, W.P. Pathological consequences of systemic measles virus infection. J. Pathol. 2015, 235, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.; Bonthius, D.J. Measles virus and associated central nervous system sequelae. Semin. Pediatr. Neurol. 2012, 19, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Vandevelde, M.; Zurbriggen, A. Demyelination in canine distemper virus infection: A review. Acta Neuropathol. 2005, 109, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Norrby, E.; Kristensson, K. Measles virus in the brain. Brain Res. Bull. 1997, 44, 213–220. [Google Scholar] [CrossRef]
- Bonaparte, M.I.; Dimitrov, A.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Dimitrov, D.S.; Wang, L.F.; Eaton, B.T.; et al. Ephrin-b2 ligand is a functional receptor for hendra virus and nipah virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657. [Google Scholar] [CrossRef] [PubMed]
- Negrete, O.A.; Levroney, E.L.; Aguilar, H.C.; Bertolotti-Ciarlet, A.; Nazarian, R.; Tajyar, S.; Lee, B. Ephrinb2 is the entry receptor for nipah virus, an emergent deadly paramyxovirus. Nature 2005, 436, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Negrete, O.A.; Wolf, M.C.; Aguilar, H.C.; Enterlein, S.; Wang, W.; Muhlberger, E.; Su, S.V.; Bertolotti-Ciarlet, A.; Flick, R.; Lee, B. Two key residues in ephrinb3 are critical for its use as an alternative receptor for nipah virus. PLoS Pathog. 2006, 2, e7. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.; Klein, R. Multiple roles of ephrins in morphogenesis, neuronal networking, and brain function. Genes Dev. 2003, 17, 1429–1450. [Google Scholar] [CrossRef] [PubMed]
- Tatsuo, H.; Ono, N.; Tanaka, K.; Yanagi, Y. Slam (CDW150) is a cellular receptor for measles virus. Nature 2000, 406, 893–897. [Google Scholar] [PubMed]
- Tatsuo, H.; Ono, N.; Yanagi, Y. Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J. Virol. 2001, 75, 5842–5850. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.T.; Sisson, G.; Tsao, M.S.; Richardson, C.D. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.D.; Mateo, M.; Sinn, P.L.; Prufer, S.; Uhlig, K.M.; Leonard, V.H.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 2011, 480, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Pratakpiriya, W.; Seki, F.; Otsuki, N.; Sakai, K.; Fukuhara, H.; Katamoto, H.; Hirai, T.; Maenaka, K.; Techangamsuwan, S.; Lan, N.T.; et al. Nectin4 is an epithelial cell receptor for canine distemper virus and involved in neurovirulence. J. Virol. 2012, 86, 10207–10210. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Delpeut, S.; Richardson, C.D. Dog nectin-4 is an epithelial cell receptor for canine distemper virus that facilitates virus entry and syncytia formation. Virology 2013, 436, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Veillette, A. Slam family receptors in normal immunity and immune pathologies. Curr. Opin. Immunol. 2016, 38, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Leonard, V.H.; Hodge, G.; Reyes-Del, V.J.; McChesney, M.B.; Cattaneo, R. Measles virus selectively blind to signaling lymphocytic activation molecule (SLAM; CD150) is attenuated and induces strong adaptive immune responses in rhesus monkeys. J. Virol. 2010, 84, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Leonard, V.H.; Sinn, P.L.; Hodge, G.; Miest, T.; Devaux, P.; Oezguen, N.; Braun, W.; McCray, P.B., Jr.; McChesney, M.B.; Cattaneo, R. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J. Clin. Investig. 2008, 118, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Dorig, R.E.; Marcil, A.; Chopra, A.; Richardson, C.D. The human cd46 molecule is a receptor for measles virus (edmonston strain). Cell 1993, 75, 295–305. [Google Scholar] [CrossRef]
- Erlenhofer, C.; Duprex, W.P.; Rima, B.K.; ter Meulen, V.; Schneider-Schaulies, J. Analysis of receptor (CD46, CD150) usage by measles virus. J. Gen. Virol. 2002, 83, 1431–1436. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beineke, A.; Puff, C.; Seehusen, F.; Baumgartner, W. Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet. Immunol. Immunopathol. 2009, 127, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zurbriggen, A.; Graber, H.U.; Vandevelde, M. Selective spread and reduced virus release leads to canine distemper virus persistence in the nervous system. Vet. Microbiol. 1995, 44, 281–288. [Google Scholar] [CrossRef]
- Wyss-Fluehmann, G.; Zurbriggen, A.; Vandevelde, M.; Plattet, P. Canine distemper virus persistence in demyelinating encephalitis by swift intracellular cell-to-cell spread in astrocytes is controlled by the viral attachment protein. Acta Neuropathol. 2010, 119, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Duprex, W.P.; McQuaid, S.; Hangartner, L.; Billeter, M.A.; Rima, B.K. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J. Virol. 1999, 73, 9568–9575. [Google Scholar] [PubMed]
- Ehrengruber, M.U.; Ehler, E.; Billeter, M.A.; Naim, H.Y. Measles virus spreads in rat hippocampal neurons by cell-to-cell contact and in a polarized fashion. J. Virol. 2002, 76, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.M.; Patterson, C.E.; Gales, T.L.; D’Orazio, J.L.; Vaughn, M.M.; Rall, G.F. Measles virus spread between neurons requires cell contact but not CD46 expression, syncytium formation, or extracellular virus production. J. Virol. 2000, 74, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Makhortova, N.R.; Askovich, P.; Patterson, C.E.; Gechman, L.A.; Gerard, N.P.; Rall, G.F. Neurokinin-1 enables measles virus trans-synaptic spread in neurons. Virology 2007, 362, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Hornick, A.L.; Krishnamurthy, S.; Locke, A.C.; Mendoza, C.A.; Mateo, M.; Miller-Hunt, C.L.; Cattaneo, R.; Sinn, P.L. The nectin-4/afadin protein complex and intercellular membrane pores contribute to rapid spread of measles virus in primary human airway epithelia. J. Virol. 2015, 89, 7089–7096. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.; Khosravi, M.; Avila, M.; Ader-Ebert, N.; Bringolf, F.; Zurbriggen, A.; Vandevelde, M.; Plattet, P. Slam- and nectin-4-independent noncytolytic spread of canine distemper virus in astrocytes. J. Virol. 2015, 89, 5724–5733. [Google Scholar] [CrossRef] [PubMed]
- Klenk, H.D.; Garten, W. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 1994, 2, 39–43. [Google Scholar] [CrossRef]
- Plemper, R.K.; Hammond, A.L.; Cattaneo, R. Measles virus envelope glycoproteins hetero-oligomerize in the endoplasmic reticulum. J. Biol. Chem. 2001, 276, 44239–44246. [Google Scholar] [CrossRef] [PubMed]
- Brindley, M.A.; Chaudhury, S.; Plemper, R.K. Measles virus glycoprotein complexes preassemble intracellularly and relax during transport to the cell surface in preparation for fusion. J. Virol. 2015, 89, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Lescar, J.; Roussel, A.; Wien, M.W.; Navaza, J.; Fuller, S.D.; Wengler, G.; Wengler, G.; Rey, F.A. The fusion glycoprotein shell of semliki forest virus: An icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 2001, 105, 137–148. [Google Scholar] [CrossRef]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.A.; Dutch, R.E.; Lamb, R.A.; Jardetzky, T.S. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell 1999, 3, 309–319. [Google Scholar] [CrossRef]
- Plemper, R.K.; Compans, R.W. Mutations in the putative HR-C region of the measles virus F2 glycoprotein modulate syncytium formation. J. Virol. 2003, 77, 4181–4190. [Google Scholar] [CrossRef] [PubMed]
- Dutch, R.E.; Jardetzky, T.S.; Lamb, R.A. Virus membrane fusion proteins: Biological machines that undergo a metamorphosis. Biosci. Rep. 2000, 20, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Berger, B.; Kim, P.S. LearnCoil-VMF: Computational evidence for coiled-coil-like motifs in many viral membrane-fusion proteins. J. Mol. Biol. 1999, 290, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Hughson, F.M. Enveloped viruses: A common mode of membrane fusion? Curr. Biol. CB 1997, 7, R565–R569. [Google Scholar] [CrossRef]
- Moll, M.; Klenk, H.D.; Maisner, A. Importance of the cytoplasmic tails of the measles virus glycoproteins for fusogenic activity and the generation of recombinant measles viruses. J. Virol. 2002, 76, 7174–7186. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.G.; Russell, C.J.; Lamb, R.A. Fusion protein of the paramyxovirus SV5: Destabilizing and stabilizing mutants of fusion activation. Virology 2000, 270, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Waning, D.L.; Russell, C.J.; Jardetzky, T.S.; Lamb, R.A. Activation of a paramyxovirus fusion protein is modulated by inside-out signaling from the cytoplasmic tail. Proc. Natl. Acad. Sci. USA 2004, 101, 9217–9222. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, H.C.; Matreyek, K.A.; Choi, D.Y.; Filone, C.M.; Young, S.; Lee, B. Polybasic kkr motif in the cytoplasmic tail of nipah virus fusion protein modulates membrane fusion by inside-out signaling. J. Virol. 2007, 81, 4520–4532. [Google Scholar] [CrossRef] [PubMed]
- Runkler, N.; Dietzel, E.; Moll, M.; Klenk, H.D.; Maisner, A. Glycoprotein targeting signals influence the distribution of measles virus envelope proteins and virus spread in lymphocytes. J. Gen. Virol. 2008, 89, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Runkler, N.; Dietzel, E.; Carsillo, M.; Niewiesk, S.; Maisner, A. Sorting signals in the measles virus wild-type glycoproteins differently influence virus spread in polarized epithelia and lymphocytes. J. Gen. Virol. 2009, 90, 2474–2482. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Culler, M.R.; Hellman, L.M.; Fried, M.G.; Creamer, T.P.; Dutch, R.E. Beyond anchoring: The expanding role of the hendra virus fusion protein transmembrane domain in protein folding, stability, and function. J. Virol. 2012, 86, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.D.; Leonard, V.H.; Cattaneo, R. The measles virus fusion protein transmembrane region modulates availability of an active glycoprotein complex and fusion efficiency. J. Virol. 2008, 82, 11437–11445. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Colman, P.M.; Cosgrove, L.J.; Lawrence, M.C.; Lawrence, L.J.; Tulloch, P.A.; Gorman, J.J. Cloning, expression, and crystallization of the fusion protein of newcastle disease virus. Virology 2001, 290, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.S.; Paterson, R.G.; Wen, X.; Lamb, R.A.; Jardetzky, T.S. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9288–9293. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gorman, J.J.; McKimm-Breschkin, J.; Lawrence, L.J.; Tulloch, P.A.; Smith, B.J.; Colman, P.M.; Lawrence, M.C. The structure of the fusion glycoprotein of newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure 2001, 9, 255–266. [Google Scholar] [CrossRef]
- Wong, J.J.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure and stabilization of the hendra virus f glycoprotein in its prefusion form. Proc. Natl. Acad. Sci. USA 2016, 113, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chan, Y.P.; Bradel-Tretheway, B.; Akyol-Ataman, Z.; Zhu, Y.; Dutta, S.; Yan, L.; Feng, Y.; Wang, L.F.; Skiniotis, G.; et al. Crystal structure of the pre-fusion nipah virus fusion glycoprotein reveals a novel hexamer-of-trimers assembly. PLoS Pathog. 2015, 11, e1005322. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 2011, 85, 7788–7796. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of rsv fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.D.; Liu, Y.; Kors, C.A.; Leser, G.P.; Jardetzky, T.S.; Lamb, R.A. Structure of the cleavage-activated prefusion form of the parainfluenza virus 5 fusion protein. Proc. Natl. Acad. Sci. USA 2012, 109, 16672–16677. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.J.; Jardetzky, T.S.; Lamb, R.A. Membrane fusion machines of paramyxoviruses: Capture of intermediates of fusion. EMBO J. 2001, 20, 4024–4034. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L.V.; Kozlov, M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, M.M.; McMahon, H.T.; Chernomordik, L.V. Protein-driven membrane stresses in fusion and fission. Trends Biochem. Sci. 2010, 35, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Brindley, M.A.; Plattet, P.; Plemper, R.K. Efficient replication of a paramyxovirus independent of full zippering of the fusion protein six-helix bundle domain. Proc. Natl. Acad. Sci. USA 2014, 111, E3795–E3804. [Google Scholar] [CrossRef] [PubMed]
- Villar, E.; Barroso, I.M. Role of sialic acid-containing molecules in paramyxovirus entry into the host cell: A minireview. Glycoconj. J. 2006, 23, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Jardetzky, T.S. Structural basis of viral invasion: Lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 2007, 17, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K.; Hammond, A.L.; Cattaneo, R. Characterization of a region of the measles virus hemagglutinin sufficient for its dimerization. J. Virol. 2000, 74, 6485–6493. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Thompson, T.B.; Wurzburg, B.A.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 2005, 13, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.C.; Borg, N.A.; Streltsov, V.A.; Pilling, P.A.; Epa, V.C.; Varghese, J.N.; Kimm-Breschkin, J.L.; Colman, P.M. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J. Mol. Biol. 2004, 335, 1343–1357. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, V.; von, I.M.; Groves, D.; Kiefel, M.; Takimoto, T.; Portner, A.; Taylor, G. Second sialic acid binding site in newcastle disease virus hemagglutinin-neuraminidase: Implications for fusion. J. Virol. 2004, 78, 3733–3741. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Rajashankar, K.R.; Chan, Y.P.; Himanen, J.P.; Broder, C.C.; Nikolov, D.B. Host cell recognition by the henipaviruses: Crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. USA 2008, 105, 9953–9958. [Google Scholar] [CrossRef] [PubMed]
- Bowden, T.A.; Crispin, M.; Harvey, D.J.; Aricescu, A.R.; Grimes, J.M.; Jones, E.Y.; Stuart, D.I. Crystal structure and carbohydrate analysis of nipah virus attachment glycoprotein: A template for antiviral and vaccine design. J. Virol. 2008, 82, 11628–11636. [Google Scholar] [CrossRef] [PubMed]
- Santiago, C.; Celma, M.L.; Stehle, T.; Casasnovas, J.M. Structure of the measles virus hemagglutinin bound to the CD46 receptor. Nat. Struct. Mol. Biol. 2010, 17, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Colf, L.A.; Juo, Z.S.; Garcia, K.C. Structure of the measles virus hemagglutinin. Nat. Struct. Mol. Biol. 2007, 14, 1227–1228. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, T.; Kajikawa, M.; Maita, N.; Takeda, M.; Kuroki, K.; Sasaki, K.; Kohda, D.; Yanagi, Y.; Maenaka, K. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. USA 2007, 104, 19535–19540. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, T.; Ose, T.; Kubota, M.; Maita, N.; Kamishikiryo, J.; Maenaka, K.; Yanagi, Y. Structure of the measles virus hemagglutinin bound to its cellular receptor slam. Nat. Struct. Mol. Biol. 2011, 18, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, G.; Qi, J.; Li, Y.; He, Y.; Xu, X.; Shi, J.; Zhang, C.W.; Yan, J.; Gao, G.F. Structure of measles virus hemagglutinin bound to its epithelial receptor nectin-4. Nat. Struct. Mol. Biol. 2013, 20, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Swanson, K.A.; Leser, G.P.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. USA 2011, 108, 14920–14925. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Paterson, R.G.; Leser, G.P.; Lamb, R.A.; Jardetzky, T.S. Structure of the ulster strain newcastle disease virus hemagglutinin-neuraminidase reveals auto-inhibitory interactions associated with low virulence. PLoS Pathog. 2012, 8, e1002855. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.D.; Yuan, P.; Bose, S.; Kors, C.A.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) ectodomain. PLoS Pathog. 2013, 9, e1003534. [Google Scholar] [CrossRef] [PubMed]
- Ader, N.; Brindley, M.A.; Avila, M.; Origgi, F.C.; Langedijk, J.P.; Orvell, C.; Vandevelde, M.; Zurbriggen, A.; Plemper, R.K.; Plattet, P. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger f protein refolding for membrane fusion. J. Biol. Chem. 2012, 287, 16324–16334. [Google Scholar] [CrossRef] [PubMed]
- Navaratnarajah, C.K.; Negi, S.; Braun, W.; Cattaneo, R. Membrane fusion triggering: Three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J. Biol. Chem. 2012, 287, 38543–38551. [Google Scholar] [CrossRef] [PubMed]
- Brindley, M.A.; Takeda, M.; Plattet, P.; Plemper, R.K. Triggering the measles virus membrane fusion machinery. Proc. Natl. Acad. Sci. USA 2012, 109, E3018–E3027. [Google Scholar] [CrossRef] [PubMed]
- Heminway, B.R.; Yu, Y.; Galinski, M.S. Paramyxovirus mediated cell fusion requires co-expression of both the fusion and hemagglutinin-neuraminidase glycoproteins. Virus Res. 1994, 31, 1–16. [Google Scholar] [CrossRef]
- Hu, X.L.; Ray, R.; Compans, R.W. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J. Virol. 1992, 66, 1528–1534. [Google Scholar] [PubMed]
- Morrison, T.; McQuain, C.; McGinnes, L. Complementation between avirulent newcastle disease virus and a fusion protein gene expressed from a retrovirus vector: Requirements for membrane fusion. J. Virol. 1991, 65, 813–822. [Google Scholar] [PubMed]
- Lee, J.K.; Prussia, A.; Paal, T.; White, L.K.; Snyder, J.P.; Plemper, R.K. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 2008, 283, 16561–16572. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Wang, Z.; Mirza, A.M.; Iorio, R.M. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 1995, 209, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Mirza, A.M.; Mahon, P.J.; Iorio, R.M. Functional chimeric HN glycoproteins derived from newcastle disease virus and human parainfluenza virus-3. Arch. Virol. Suppl. 1997, 13, 115–130. [Google Scholar] [PubMed]
- Tanabayashi, K.; Compans, R.W. Functional interaction of paramyxovirus glycoproteins: Identification of a domain in sendai virus hn which promotes cell fusion. J. Virol. 1996, 70, 6112–6118. [Google Scholar] [PubMed]
- Wang, Z.; Mirza, A.M.; Li, J.; Mahon, P.J.; Iorio, R.M. An oligosaccharide at the C-terminus of the F-specific domain in the stalk of the human parainfluenza virus 3 hemagglutinin-neuraminidase modulates fusion. Virus Res. 2004, 99, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Tsurudome, M.; Kawano, M.; Yuasa, T.; Tabata, N.; Nishio, M.; Komada, H.; Ito, Y. Identification of regions on the hemagglutinin-neuraminidase protein of human parainfluenza virus type 2 important for promoting cell fusion. Virology 1995, 213, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Pte-Sengupta, S.; Navaratnarajah, C.K.; Cattaneo, R. Hydrophobic and charged residues in the central segment of the measles virus hemagglutinin stalk mediate transmission of the fusion-triggering signal. J. Virol. 2013, 87, 10401–10404. [Google Scholar] [CrossRef] [PubMed]
- Navaratnarajah, C.K.; Kumar, S.; Generous, A.; pte-Sengupta, S.; Mateo, M.; Cattaneo, R. The measles virus hemagglutinin stalk: Structures and functions of the central fusion activation and membrane-proximal segments. J. Virol. 2014, 88, 6158–6167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bose, S.; Zokarkar, A.; Welch, B.D.; Leser, G.P.; Jardetzky, T.S.; Lamb, R.A. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc. Natl. Acad. Sci. USA 2012, 109, E2625–E2634. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Song, A.S.; Jardetzky, T.S.; Lamb, R.A. Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J. Virol. 2014, 88, 3925–3941. [Google Scholar] [CrossRef] [PubMed]
- Brindley, M.A.; Suter, R.; Schestak, I.; Kiss, G.; Wright, E.R.; Plemper, R.K. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J. Virol. 2013, 87, 11693–11703. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Stone, J.A.; Bradel-Tretheway, B.; Dabundo, J.; avides Montano, J.A.; Santos-Montanez, J.; Biering, S.B.; Nicola, A.V.; Iorio, R.M.; Lu, X.; et al. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced nipah virus fusion and cell entry. PLoS Pathog. 2013, 9, e1003770. [Google Scholar] [CrossRef] [PubMed]
- Ader-Ebert, N.; Khosravi, M.; Herren, M.; Avila, M.; Alves, L.; Bringolf, F.; Orvell, C.; Langedijk, J.P.; Zurbriggen, A.; Plemper, R.K.; et al. Sequential conformational changes in the morbillivirus attachment protein initiate the membrane fusion process. PLoS Pathog. 2015, 11, e1004880. [Google Scholar] [CrossRef] [PubMed]
- Navaratnarajah, C.K.; Rosemarie, Q.; Cattaneo, R. A structurally unresolved head segment of defined length favors proper measles virus hemagglutinin tetramerization and efficient membrane fusion triggering. J. Virol. 2015, 90, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Jardetzky, T.S.; Lamb, R.A. Timing is everything: Fine-tuned molecular machines orchestrate paramyxovirus entry. Virology 2015, 479–480, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, H.C.; Matreyek, K.A.; Filone, C.M.; Hashimi, S.T.; Levroney, E.L.; Negrete, O.A.; Bertolotti-Ciarlet, A.; Choi, D.Y.; McHardy, I.; Fulcher, J.A.; et al. N-glycans on nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J. Virol. 2006, 80, 4878–4889. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, H.C.; Ataman, Z.A.; Aspericueta, V.; Fang, A.Q.; Stroud, M.; Negrete, O.A.; Kammerer, R.A.; Lee, B. A novel receptor-induced activation site in the nipah virus attachment glycoprotein (G) involved in triggering the fusion glycoprotein (F). J. Biol. Chem. 2009, 284, 1628–1635. [Google Scholar] [CrossRef] [PubMed]
- Apte-Sengupta, S.; Negi, S.; Leonard, V.H.; Oezguen, N.; Navaratnarajah, C.K.; Braun, W.; Cattaneo, R. Base of the measles virus fusion trimer head receives the signal that triggers membrane fusion. J. Biol. Chem. 2012, 287, 33026–33035. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Heath, C.M.; Shah, P.A.; Alayyoubi, M.; Jardetzky, T.S.; Lamb, R.A. Mutations in the parainfluenza virus 5 fusion protein reveal domains important for fusion triggering and metastability. J. Virol. 2013, 87, 13520–13531. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.; Khosravi, M.; Alves, L.; Ader-Ebert, N.; Bringolf, F.; Zurbriggen, A.; Plemper, R.K.; Plattet, P. Canine distemper virus envelope protein interactions modulated by hydrophobic residues in the fusion protein globular head. J. Virol. 2015, 89, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Paal, T.; Brindley, M.A.; St, C.C.; Prussia, A.; Gaus, D.; Krumm, S.A.; Snyder, J.P.; Plemper, R.K. Probing the spatial organization of measles virus fusion complexes. J. Virol. 2009, 83, 10480–10493. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.A.; Leser, G.P.; Jardetzky, T.S.; Lamb, R.A. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: Implications for the mechanism of fusion triggering. J. Virol. 2009, 83, 10857–10868. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.G.; Johnson, M.L.; Lamb, R.A. Paramyxovirus fusion (F) protein and hemagglutinin-neuraminidase (HN) protein interactions: Intracellular retention of f and hn does not affect transport of the homotypic hn or f protein. Virology 1997, 237, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ader, N.; Brindley, M.; Avila, M.; Orvell, C.; Horvat, B.; Hiltensperger, G.; Schneider-Schaulies, J.; Vandevelde, M.; Zurbriggen, A.; Plemper, R.K.; et al. Mechanism for active membrane fusion triggering by morbillivirus attachment protein. J. Virol. 2013, 87, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.P.; Lu, M.; Dutta, S.; Yan, L.; Barr, J.; Flora, M.; Feng, Y.R.; Xu, K.; Nikolov, D.B.; Wang, L.F.; et al. Biochemical, conformational, and immunogenic analysis of soluble trimeric forms of henipavirus fusion glycoproteins. J. Virol. 2012, 86, 11457–11471. [Google Scholar] [CrossRef] [PubMed]
- Talekar, A.; Moscona, A.; Porotto, M. Measles virus fusion machinery activated by sialic acid binding globular domain. J. Virol. 2013, 87, 13619–13627. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, M.; Bringolf, F.; Rothlisberger, S.; Bieringer, M.; Schneider-Schaulies, J.; Zurbriggen, A.; Origgi, F.; Plattet, P. Canine distemper virus fusion activation: Critical role of residue E123 of CD150/SLAM. J. Virol. 2015, 90, 1622–1637. [Google Scholar] [CrossRef] [PubMed]
- Plattet, P.; Langedijk, J.P.; Zipperle, L.; Vandevelde, M.; Orvell, C.; Zurbriggen, A. Conserved leucine residue in the head region of morbillivirus fusion protein regulates the large conformational change during fusion activity. Biochemistry 2009, 48, 9112–9121. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Palmer, S.G.; Palermo, L.M.; Moscona, A. Mechanism of fusion triggering by human parainfluenza virus type III: Communication between viral glycoproteins during entry. J. Biol. Chem. 2012, 287, 778–793. [Google Scholar] [CrossRef] [PubMed]
- Gui, L.; Jurgens, E.M.; Ebner, J.L.; Porotto, M.; Moscona, A.; Lee, K.K. Electron tomography imaging of surface glycoproteins on human parainfluenza virus 3: Association of receptor binding and fusion proteins before receptor engagement. mBio 2015, 6, e02393–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Palmer, S.G.; Porotto, M.; Palermo, L.M.; Niewiesk, S.; Wilson, I.A.; Moscona, A. Interaction between the hemagglutinin-neuraminidase and fusion glycoproteins of human parainfluenza virus type III regulates viral growth in vivo. MBio 2013, 4, e00803–e00813. [Google Scholar] [CrossRef] [PubMed]
- Crennell, S.; Takimoto, T.; Portner, A.; Taylor, G. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol. 2000, 7, 1068–1074. [Google Scholar] [PubMed]
- Lamb, R.A. Paramyxovirus fusion: A hypothesis for changes. Virology 1993, 197, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sergel, T.; McGinnes, L.W.; Peeples, M.E.; Morrison, T.G. The attachment function of the newcastle disease virus hemagglutinin-neuraminidase protein can be separated from fusion promotion by mutation. Virology 1993, 193, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.; Alves, L.; Khosravi, M.; der-Ebert, N.; Origgi, F.; Schneider-Schaulies, J.; Zurbriggen, A.; Plemper, R.K.; Plattet, P. Molecular determinants defining the triggering range of prefusion f complexes of canine distemper virus. J. Virol. 2014, 88, 2951–2966. [Google Scholar] [CrossRef] [PubMed]
- Shirogane, Y.; Watanabe, S.; Yanagi, Y. Cooperation between different RNA virus genomes produces a new phenotype. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Summers, B.A.; Greisen, H.A.; Appel, M.J. Early events in canine distemper demyelinating encephalomyelitis. Acta Neuropathol. 1979, 46, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Summers, B.A.; Greisen, H.A.; Appel, M.J. Canine distemper encephalomyelitis: Variation with virus strain. J. Comp. Pathol. 1984, 94, 65–75. [Google Scholar] [CrossRef]
- Summers, B.A.; Whitaker, J.N.; Appel, M.J. Demyelinating canine distemper encephalomyelitis: Measurement of myelin basic protein in cerebrospinal fluid. J. Neuroimmunol. 1987, 14, 227–233. [Google Scholar] [CrossRef]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar] [CrossRef] [PubMed]
- Bossart, K.N.; Wang, L.F.; Eaton, B.T.; Broder, C.C. Functional expression and membrane fusion tropism of the envelope glycoproteins of hendra virus. Virology 2001, 290, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Bossart, K.N.; Wang, L.F.; Flora, M.N.; Chua, K.B.; Lam, S.K.; Eaton, B.T.; Broder, C.C. Membrane fusion tropism and heterotypic functional activities of the nipah virus and hendra virus envelope glycoproteins. J. Virol. 2002, 76, 11186–11198. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Barney, S.; Lambert, A.L.; Guthrie, K.; Medinas, R.; Davis, D.E.; Bucy, T.; Erickson, J.; Merutka, G.; Petteway, S.R., Jr. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA 1996, 93, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.B.; Dutch, R.E.; Lamb, R.A. A core trimer of the paramyxovirus fusion protein: Parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology 1998, 248, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Young, J.K.; Hicks, R.P.; Wright, G.E.; Morrison, T.G. Analysis of a peptide inhibitor of paramyxovirus (NDV) fusion using biological assays, nmr, and molecular modeling. Virology 1997, 238, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Bossart, K.N.; Mungall, B.A.; Crameri, G.; Wang, L.F.; Eaton, B.T.; Broder, C.C. Inhibition of henipavirus fusion and infection by heptad-derived peptides of the nipah virus fusion glycoprotein. Virol. J. 2005, 2. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Rockx, B.; Yokoyama, C.C.; Talekar, A.; Devito, I.; Palermo, L.M.; Liu, J.; Cortese, R.; Lu, M.; Feldmann, H.; et al. Inhibition of nipah virus infection in vivo: Targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 2010, 6, e1001168. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Pessi, A.; Gui, L.; Santoprete, A.; Talekar, A.; Moscona, A.; Porotto, M. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 2011, 286, 42141–42149. [Google Scholar] [CrossRef] [PubMed]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Malvoisin, E.; Wild, F. Contribution of measles virus fusion protein in protective immunity: Anti-F monoclonal antibodies neutralize virus infectivity and protect mice against challenge. J. Virol. 1990, 64, 5160–5162. [Google Scholar] [PubMed]
- Sheshberadaran, H.; Chen, S.N.; Norrby, E. Monoclonal antibodies against five structural components of measles virus. I. Characterization of antigenic determinants on nine strains of measles virus. Virology 1983, 128, 341–353. [Google Scholar] [CrossRef]
- Young, M.K.; Nimmo, G.R.; Cripps, A.W.; Jones, M.A. Post-exposure passive immunisation for preventing measles. Cochrane Database Syst. Rev. 2014, 4. [Google Scholar] [CrossRef]
- Hampp, C.; Kauf, T.L.; Saidi, A.S.; Winterstein, A.G. Cost-effectiveness of respiratory syncytial virus prophylaxis in various indications. Arch. Pediatr. Adolesc. Med. 2011, 165, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Mahadevia, P.J.; Masaquel, A.S.; Polak, M.J.; Weiner, L.B. Cost utility of palivizumab prophylaxis among pre-term infants in the united states: A national policy perspective. J. Med. Econ. 2012, 15, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K.; Erlandson, K.J.; Lakdawala, A.S.; Sun, A.; Prussia, A.; Boonsombat, J.; ki-Sener, E.; Yalcin, I.; Yildiz, I.; Temiz-Arpaci, O.; et al. A target site for template-based design of measles virus entry inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 5628–5633. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K.; Doyle, J.; Sun, A.; Prussia, A.; Cheng, L.T.; Rota, P.A.; Liotta, D.C.; Snyder, J.P.; Compans, R.W. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob. Agents Chemother. 2005, 49, 3755–3761. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Prussia, A.; Zhan, W.; Murray, E.E.; Doyle, J.; Cheng, L.T.; Yoon, J.J.; Radchenko, E.V.; Palyulin, V.A.; Compans, R.W.; et al. Nonpeptide inhibitors of measles virus entry. J. Med. Chem. 2006, 49, 5080–5092. [Google Scholar] [CrossRef] [PubMed]
- Singethan, K.; Hiltensperger, G.; Kendl, S.; Wohlfahrt, J.; Plattet, P.; Holzgrabe, U.; Schneider-Schaulies, J. N-(3-cyanophenyl)-2-phenylacetamide, an effective inhibitor of morbillivirus-induced membrane fusion with low cytotoxicity. J. Gen. Virol. 2010, 91, 2762–2772. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Yoon, J.J.; Yin, Y.; Prussia, A.; Yang, Y.; Min, J.; Plemper, R.K.; Snyder, J.P. Potent non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase complex. J. Med. Chem. 2008, 51, 3731–3741. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.; Prussia, A.; White, L.K.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Compans, R.W.; Plemper, R.K. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J. Virol. 2006, 80, 1524–1536. [Google Scholar] [CrossRef] [PubMed]
- Prussia, A.J.; Plemper, R.K.; Snyder, J.P. Measles virus entry inhibitors: A structural proposal for mechanism of action and the development of resistance. Biochemistry 2008, 47, 13573–13583. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Lee, S.; Thakkar, V.D.; Luo, M.; Moore, M.L.; Plemper, R.K. Cross-resistance mechanism of respiratory syncytial virus against structurally diverse entry inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E3441–E3449. [Google Scholar] [CrossRef] [PubMed]
- Cianci, C.; Yu, K.L.; Combrink, K.; Sin, N.; Pearce, B.; Wang, A.; Civiello, R.; Voss, S.; Luo, G.; Kadow, K.; et al. Orally active fusion inhibitor of respiratory syncytial virus. Antimicrob. Agents Chemother. 2004, 48, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Moeremans, M.; Gevers, T.; Willebrords, R.; Sommen, C.; Lacrampe, J.; Janssens, F.; Wyde, P.R. Substituted benzimidazoles with nanomolar activity against respiratory syncytial virus. Antivir. Res. 2003, 60, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Bonfanti, J.F.; Meyer, C.; Doublet, F.; Fortin, J.; Muller, P.; Queguiner, L.; Gevers, T.; Janssens, P.; Szel, H.; Willebrords, R.; et al. Selection of a respiratory syncytial virus fusion inhibitor clinical candidate. 2. Discovery of a morpholinopropylaminobenzimidazole derivative (TMC353121). J. Med. Chem. 2008, 51, 875–896. [Google Scholar] [CrossRef] [PubMed]
- Battles, M.B.; Langedijk, J.P.; Furmanova-Hollenstein, P.; Chaiwatpongsakorn, S.; Costello, H.M.; Kwanten, L.; Vranckx, L.; Vink, P.; Jaensch, S.; Jonckers, T.H.; et al. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat. Chem. Biol. 2016, 12, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Poor, T.A.; Jones, L.M.; Sood, A.; Leser, G.P.; Plasencia, M.D.; Rempel, D.L.; Jardetzky, T.S.; Woods, R.J.; Gross, M.L.; Lamb, R.A. Probing the paramyxovirus fusion (F) protein-refolding event from pre- to postfusion by oxidative footprinting. Proc. Natl. Acad.Sci. USA 2014, 111, E2596–E2605. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, F.A.; Gibbs, E.L.; Carpenter, P.R.; Spies, H.W. Electroencephalographic abnormality in “uncomplicated” childhood diseases. J. Am. Med. Assoc. 1959, 171, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Pampiglione, G. Prodromal phase of measles: Some neurophysiological studies. Br. Med. J. 1964, 2, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Budka, H.; Urbanits, S.; Liberski, P.P.; Eichinger, S.; Popow-Kraupp, T. Subacute measles virus encephalitis: A new and fatal opportunistic infection in a patient with aids. Neurology 1996, 46, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.M.; Weitman, S.D.; Winick, N.J.; Bellini, W.J.; Timmons, C.F.; Siegel, J.D. Subacute measles encephalitis in the young immunocompromised host: Report of two cases diagnosed by polymerase chain reaction and treated with ribavirin and review of the literature. Clin. Infect. Dis. 1993, 16, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, R.; Schmid, A.; Rebmann, G.; Baczko, K.; ter, M.V.; Bellini, W.J.; Rozenblatt, S.; Billeter, M.A. Accumulated measles virus mutations in a case of subacute sclerosing panencephalitis: Interrupted matrix protein reading frame and transcription alteration. Virology 1986, 154, 97–107. [Google Scholar] [CrossRef]
- Cattaneo, R.; Schmid, A.; Eschle, D.; Baczko, K.; ter, M.V.; Billeter, M.A. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell 1988, 55, 255–265. [Google Scholar] [CrossRef]
- Wong, T.C.; Ayata, M.; Ueda, S.; Hirano, A. Role of biased hypermutation in evolution of subacute sclerosing panencephalitis virus from progenitor acute measles virus. J. Virol. 1991, 65, 2191–2199. [Google Scholar] [PubMed]
- Baczko, K.; Lampe, J.; Liebert, U.G.; Brinckmann, U.; ter Meulen, V.; Pardowitz, I.; Budka, H.; Cosby, S.L.; Isserte, S.; Rima, B.K. Clonal expansion of hypermutated measles virus in a sspe brain. Virology 1993, 197, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.; Spielhofer, P.; Cattaneo, R.; Baczko, K.; ter Meulen, V.; Billeter, M.A. Subacute sclerosing panencephalitis is typically characterized by alterations in the fusion protein cytoplasmic domain of the persisting measles virus. Virology 1992, 188, 910–915. [Google Scholar] [CrossRef]
- Watanabe, S.; Shirogane, Y.; Suzuki, S.O.; Ikegame, S.; Koga, R.; Yanagi, Y. Mutant fusion proteins with enhanced fusion activity promote measles virus spread in human neuronal cells and brains of suckling hamsters. J. Virol. 2013, 87, 2648–2659. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ohno, S.; Shirogane, Y.; Suzuki, S.O.; Koga, R.; Yanagi, Y. Measles virus mutants possessing the fusion protein with enhanced fusion activity spread effectively in neuronal cells, but not in other cells, without causing strong cytopathology. J. Virol. 2015, 89, 2710–2717. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.R.; Albertyn, C.; Heckmann, J.M.; Smuts, H.E. Molecular characterisation of virus in the brains of patients with measles inclusion body encephalitis (MIBE). Virol. J. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, E.M.; Mathieu, C.; Palermo, L.M.; Hardie, D.; Horvat, B.; Moscona, A.; Porotto, M. Measles fusion machinery is dysregulated in neuropathogenic variants. mBio 2015, 6, e02528–14. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8, 112. https://doi.org/10.3390/v8040112
Plattet P, Alves L, Herren M, Aguilar HC. Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses. 2016; 8(4):112. https://doi.org/10.3390/v8040112
Chicago/Turabian StylePlattet, Philippe, Lisa Alves, Michael Herren, and Hector C. Aguilar. 2016. "Measles Virus Fusion Protein: Structure, Function and Inhibition" Viruses 8, no. 4: 112. https://doi.org/10.3390/v8040112
APA StylePlattet, P., Alves, L., Herren, M., & Aguilar, H. C. (2016). Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses, 8(4), 112. https://doi.org/10.3390/v8040112