
Mouse Systems to Model Hepatitis C Virus Treatment and Associated Resistance


Abstract
:
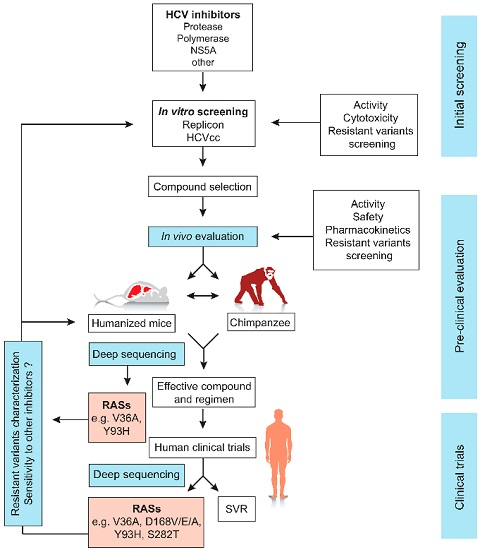{kind=link}
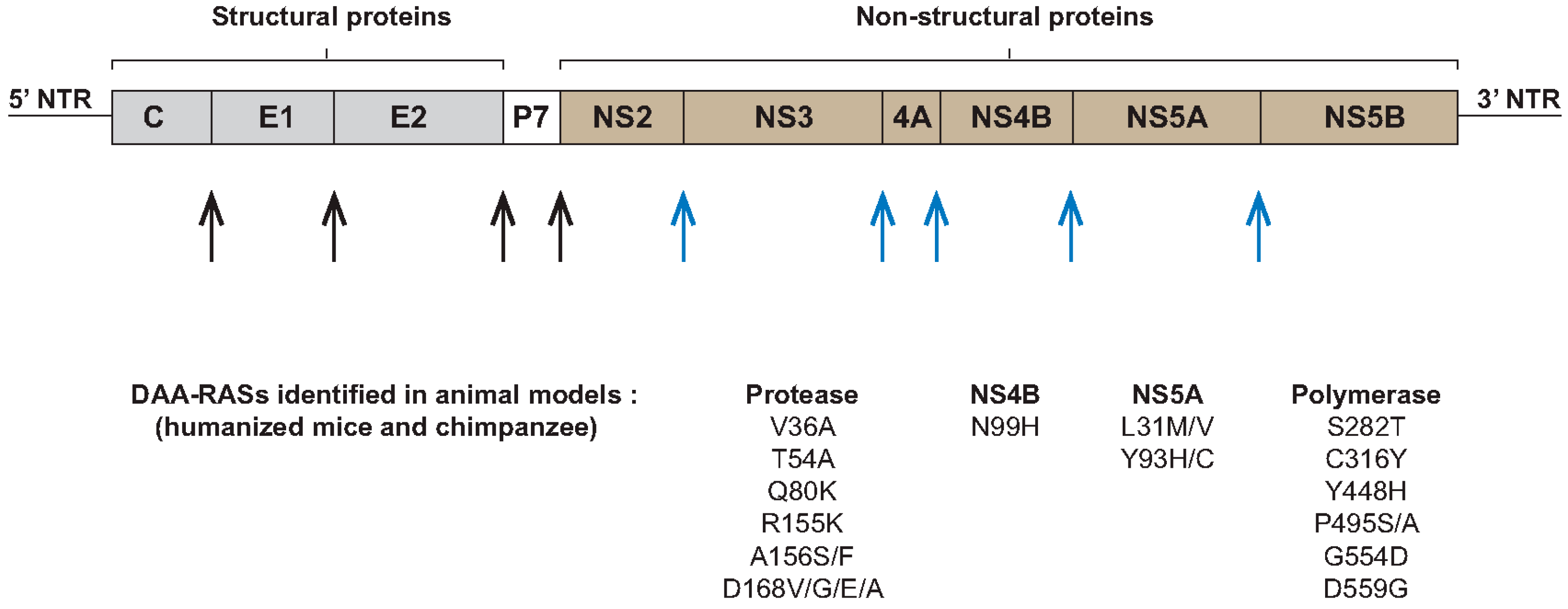{kind=link}
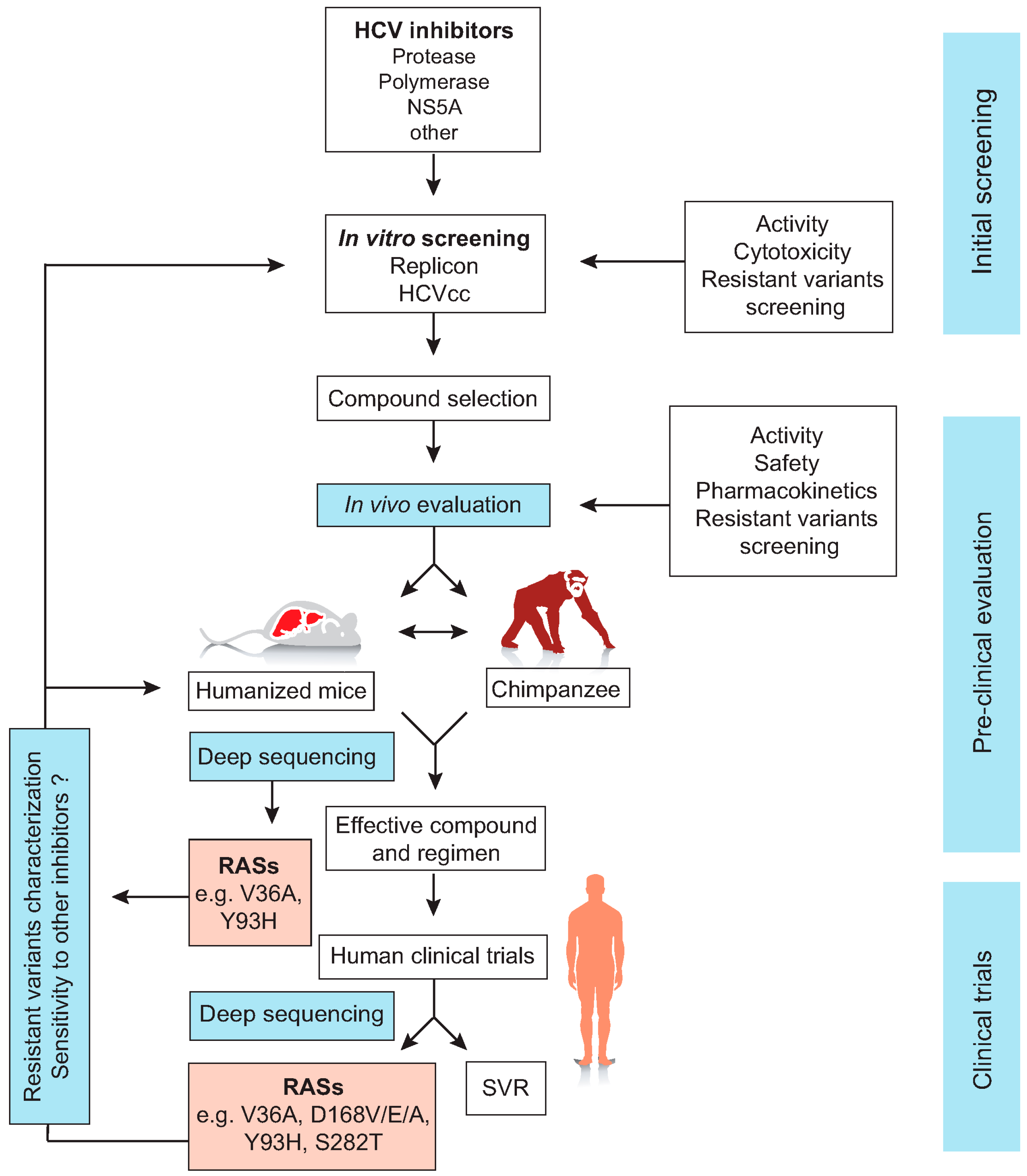{kind=link}
1. Introduction
2. Pre-Clinical in Vivo Models for HCV Treatment
2.1. Non-Rodent HCV Models
2.2. Rodent HCV Models
3. Principles of HCV Resistance
4. HCV Therapy Studies with Resistance Profiling in Human-Liver Mice
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral-hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and natural history of HCV infection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Scull, M.A.; Friling, T.; Horwitz, J.A.; Donovan, B.M.; Dorner, M.; Gerold, G.; Labitt, R.N.; Rice, C.M.; Ploss, A. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology 2013, 444, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.; Horwitz, J.A.; Robbins, J.B.; Barry, W.T.; Feng, Q.; Mu, K.; Jones, C.T.; Schoggins, J.W.; Catanese, M.T.; Burton, D.R.; et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011, 474, U208–U246. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.; Horwitz, J.A.; Donovan, B.M.; Labitt, R.N.; Budell, W.C.; Friling, T.; Vogt, A.; Catanese, M.T.; Satoh, T.; Kawai, T.; et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 2013, 501, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Michta, M.L.; Hopcraft, S.E.; Narbus, C.M.; Kratovac, Z.; Israelow, B.; Sourisseau, M.; Evans, M.J. Species-specific regions of occludin required by hepatitis C virus for cell entry. J. Virol. 2010, 84, 11696–11708. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.N.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Shirasago, Y.; Shimizu, Y.; Tanida, I.; Suzuki, T.; Suzuki, R.; Sugiyama, K.; Wakita, T.; Hanada, K.; Yagi, K.; Kondoh, M.; et al. Occludin-knockout human hepatic huh7.5.1-8-derived cells are completely resistant to hepatitis C virus infection. Biol. Pharm. Bull. 2016, 39, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Bartenschlager, R. Structural and functional properties of the hepatitis C virus p7 viroporin. Viruses 2015, 7, 4461–4481. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; Andre, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.U.; Lam, N.P.; Dahari, H.; Gretch, D.R.; Wiley, T.E.; Layden, T.J.; Perelson, A.S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Gonzalez-Candelas, F.; Moya, A.; Sanjuan, R. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J. Virol. 2009, 83, 5760–5764. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Bukh, J.; Verhoye, L.; Farhoudi, A.; Vanwolleghem, T.; Wang, R.Y.; Desombere, I.; Alter, H.; Purcell, R.H.; Leroux-Roels, G. In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology 2011, 53, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Perin, P.M.; Haid, S.; Brown, R.J.P.; Doerrbecker, J.; Schulze, K.; Zeilinger, C.; von Schaewen, M.; Heller, B.; Vercauteren, K.; Luxenburger, E.; et al. Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 2016, 63, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Calland, N.; Albecka, A.; Belouzard, S.; Wychowski, C.; Duverlie, G.; Descamps, V.; Hober, D.; Dubuisson, J.; Rouille, Y.; Seron, K. (−)-epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 2012, 55, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Ciesek, S.; von Hahn, T.; Colpitts, C.C.; Schang, L.M.; Friesland, M.; Steinmann, J.; Manns, M.P.; Ott, M.; Wedemeyer, H.; Meuleman, P.; et al. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology 2011, 54, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Calland, N.; Sahuc, M.E.; Belouzard, S.; Pene, V.; Bonnafous, P.; Mesalam, A.A.; Deloison, G.; Descamps, V.; Sahpaz, S.; Wychowski, C.; et al. Polyphenols inhibit hepatitis C virus entry by a new mechanism of action. J. Virol. 2015, 89, 10053–10063. [Google Scholar] [CrossRef] [PubMed]
- Haid, S.; Novodomska, A.; Gentzsch, J.; Grethe, C.; Geuenich, S.; Bankwitz, D.; Chhatwal, P.; Jannack, B.; Hennebelle, T.; Bailleul, F.; et al. A plant-derived flavonoid inhibits entry of all HCV genotypes into human hepatocytes. Gastroenterology 2012, 143, U213–U772. [Google Scholar] [CrossRef] [PubMed]
- Anggakusuma; Colpitts, C.C.; Schang, L.M.; Rachmawati, H.; Frentzen, A.; Pfaender, S.; Behrendt, P.; Brown, R.J.P.; Bankwitz, D.; Steinmann, J.; et al. Turmeric curcumin inhibits entry of all hepatitis C virus genotypes into human liver cells. Gut 2014, 63, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Hesselgesser, J.; Paulson, M.; Vanwolleghem, T.; Desollibere, I.; Reiser, I.; Leroux-Roels, G. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 2008, 48, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Catanese, M.T.; Verhoye, L.; Desombere, I.; Farhoudi, A.; Jones, C.T.; Sheahan, T.; Grzyb, K.; Cortese, R.; Rice, C.M.; et al. A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology 2012, 55, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Mailly, L.; Xiao, F.; Lupberger, J.; Wilson, G.K.; Aubert, P.; Duong, F.H.; Calabrese, D.; Leboeuf, C.; Fofana, I.; Thumann, C.; et al. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat. Biotechnol. 2015, 33, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Ottosen, S.; Parsley, T.B.; Yang, L.; Zeh, K.; van Doorn, L.J.; van der Veer, E.; Raney, A.K.; Hodges, M.R.; Patick, A.K. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob. Agents Chemother. 2015, 59, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Van der Ree, M.H.; van der Meer, A.J.; de Bruijne, J.; Maan, R.; van Vliet, A.; Welzel, T.M.; Zeuzem, S.; Lawitz, E.J.; Rodriguez-Torres, M.; Kupcova, V.; et al. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antivir. Res. 2014, 111, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Daito, T.; Watashi, K.; Sluder, A.; Ohashi, H.; Nakajima, S.; Borroto-Esoda, K.; Fujita, T.; Wakita, T. Cyclophilin inhibitors reduce phosphorylation of RNA-dependent protein kinase to restore expression of IFN-stimulated genes in HCV-infected cells. Gastroenterology 2014, 147, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Sakamoto, N.; Tanabe, Y.; Koyama, T.; Itsui, Y.; Takeda, Y.; Chen, C.H.; Kakinuma, S.; Oooka, S.; Maekawa, S.; et al. Suppression of hepatitis C virus replication by cyclosporin A is mediated by blockade of cyclophilins. Gastroenterology 2005, 129, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Goncales, F.L.; Haussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Ascione, A.; de Luca, M.; Tartaglione, M.T.; Lampasi, F.; di Costanzo, G.G.; Lanza, A.G.; Picciotto, F.P.; Marino-Marsilia, G.; Fontanella, L.; Leandro, G. Peginterferon alfa-2a plus ribavirin is more effective than peginterferon alfa-2b plus ribavirin for treating chronic hepatitis C virus infection. Gastroenterology 2010, 138, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Andreone, P.; Pol, S.; Lawitz, E.; Diago, M.; Roberts, S.; Focaccia, R.; Younossi, Z.; Foster, G.R.; Horban, A.; et al. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 2011, 364, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.; Hartung, D.; Rahman, B.; Wasson, N.; Cottrell, E.B.; Fu, R.W. Comparative effectiveness of antiviral treatment for hepatitis C virus infection in adults: A systematic review. Ann. Intern. Med. 2013, 158, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, N.; Imamura, M.; Abe, H.; Hayes, C.N.; Kono, T.; Onishi, M.; Tsuge, M.; Takahashi, S.; Ochi, H.; Iwao, E.; et al. Rapid emergence of telaprevir resistant hepatitis C virus strain from wildtype clone in vivo. Hepatology 2011, 54, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Macartney, M.J.; Irish, D.; Bridge, S.H.; Garcia-Diaz, A.; Booth, C.L.; McCormick, A.L.; Labbett, W.; Smith, C.; Velazquez, C.; Tanwar, S.; et al. Telaprevir or boceprevir based therapy for chronic hepatitis C infection: Development of resistance-associated variants in treatment failure. Antivir. Res. 2014, 105, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.J.; Kuo, G.; Bradley, D.W.; Bonino, F.; Saracco, G.; Lee, C.; Rosenblatt, J.; Choo, Q.L.; Houghton, M. Detection of hepatitis C viral sequences in non-A, non-B hepatitis. Lancet 1990, 335, 1–3. [Google Scholar] [CrossRef]
- Bukh, J. A critical role for the chimpanzee model in the study of hepatitis C. Hepatology 2004, 39, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.J.; Callendret, B.; Zhu, B.G.; Freeman, G.J.; Hasselschwert, D.L.; Satterfield, W.; Sharpe, A.H.; Dustin, L.B.; Rice, C.M.; Grakoui, A.; et al. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc. Natl. Acad. Sci. USA 2013, 110, 15001–15006. [Google Scholar] [CrossRef] [PubMed]
- Muchmore, E.; Popper, H.; Peterson, D.A.; Miller, M.F.; Lieberman, H.M. Non-A, non-B hepatitis-related hepatocellular carcinoma in a chimpanzee. J. Med. Primatol. 1988, 17, 235–246. [Google Scholar] [PubMed]
- Morin, T.J.; Broering, T.J.; Leav, B.A.; Blair, B.M.; Rowley, K.J.; Boucher, E.N.; Wang, Y.; Cheslock, P.S.; Knauber, M.; Olsen, D.B. Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog. 2012, 8, e1002895. [Google Scholar] [CrossRef] [PubMed]
- Coburn, C.A.; Meinke, P.T.; Chang, W.; Fandozzi, C.M.; Graham, D.J.; Hu, B.; Huang, Q.; Kargman, S.; Kozlowski, J.; Liu, R.; et al. Discovery of MK-8742: An HCV NS5A inhibitor with broad genotype activity. Chemmedchem 2013, 8, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.S.; Ludmerer, S.; Handt, L.; Koeplinger, K.; Zhang, N.Y.R.; Graham, D.; Davies, M.E.; MacCoss, M.; Hazuda, D.; Olsen, D.B. Robust antiviral efficacy upon administration of a nucleoside analog to hepatitis C virus-infected chimpanzees. Antimicrob. Agents Chemother. 2009, 53, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; He, Y.; Lu, L.; Lim, H.B.; Tripathi, R.L.; Middleton, T.; Hernandez, L.E.; Beno, D.W.; Long, M.A.; Kati, W.M.; et al. Activity of a potent hepatitis C virus polymerase inhibitor in the chimpanzee model. Antimicrob. Agents Chemother. 2007, 51, 4290–4296. [Google Scholar] [CrossRef] [PubMed]
- Olsen, D.B.; Davies, M.E.; Handt, L.; Koeplinger, K.; Zhang, N.R.; Ludmerer, S.W.; Graham, D.; Liverton, N.; MacCoss, M.; Hazuda, D.; et al. Sustained viral response in a hepatitis C virus-infected chimpanzee via a combination of direct-acting antiviral agents. Antimicrob. Agents Chemother. 2011, 55, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. Nih to End All Support for Chimpanzee Research. Available online: http://www.sciencemag.org/news/2015/11/nih-end-all-support-chimpanzee-research (accessed on 10 December 2015).
- Xie, Z.C.; Riezu-Boj, J.I.; Lasarte, J.J.; Guillen, J.; Su, J.H.; Civeira, M.P.; Prieto, J. Transmission of hepatitis C virus infection to tree shrews. Virology 1998, 244, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, H.; Cao, X.; Ben, K. Efficient infection of tree shrew (Tupaia belangeri) with hepatitis C virus grown in cell culture or from patient plasma. J. Gen. Virol. 2007, 88, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Amako, Y.; Tsukiyama-Kohara, K.; Katsume, A.; Hirata, Y.; Sekiguchi, S.; Tobita, Y.; Hayashi, Y.; Hishima, T.; Funata, N.; Yonekawa, H.; et al. Pathogenesis of hepatitis C virus infection in Tupaia belangeri. J. Virol. 2010, 84, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Mailly, L.; Robinet, E.; Meuleman, P.; Baumert, T.F.; Zeisel, M.B. Hepatitis C virus infection and related liver disease: The quest for the best animal model. Front. Microbiol. 2013, 4, 213. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Yanatori, I.; Ikeda, M.; Kiyokage, E.; Nishina, S.; Tomiyama, Y.; Toida, K.; Kishi, F.; Kato, N.; Imamura, M.; et al. Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. Am. J. Pathol. 2014, 184, 3026–3039. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, C.; Shoenberger, J.M.; Chung, J.; Chang, K.M.; Guidotti, L.G.; Selby, M.; Berger, K.; Lesniewski, R.; Houghton, M.; Chisari, F.V. Hepatitis C virus core and E2 protein expression in transgenic mice. Hepatology 1997, 25, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Frelin, L.; Brenndorfer, E.D.; Ahlen, G.; Weiland, M.; Hultgren, C.; Alheim, M.; Glaumann, H.; Rozell, B.; Milich, D.R.; Bode, J.G.; et al. The hepatitis C virus and immune evasion: Non-structural 3/4A transgenic mice are resistant to lethal tumour necrosis factor alpha mediated liver disease. Gut 2006, 55, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.G.; Moon, H.B.; Kim, J.M.; Hwang, S.B.; Yu, D.Y.; Lee, D.S. Expression of hepatitis C virus nonstructural 4B in transgenic mice. Exp. Mol. Med. 2006, 38, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Steele, R.; Ghosh, A.K.; Zhou, X.Y.; Thornburg, L.; Ray, R.; Phillips, N.J.; Ray, R.B. Expression of hepatitis C virus non-structural 5A protein in the liver of transgenic mice. FEBS Lett. 2003, 555, 528–532. [Google Scholar] [CrossRef]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.Y.; Weinman, S.A.; et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Bitzegeio, J.; Bankwitz, D.; Hueging, K.; Haid, S.; Brohm, C.; Zeisel, M.B.; Herrmann, E.; Iken, M.; Ott, M.; Baumert, T.F.; et al. Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog. 2010. [Google Scholar] [CrossRef] [PubMed]
- Frentzen, A.; Anggakusuma; Gurlevik, E.; Hueging, K.; Knocke, S.; Ginkel, C.; Brown, R.J.P.; Heim, M.; Dill, M.T.; Kroger, A.; et al. Cell entry, efficient RNA replication, and production of infectious hepatitis C virus progeny in mouse liver-derived cells. Hepatology 2014, 59, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Giang, E.; Dorner, M.; Prentoe, J.C.; Dreux, M.; Evans, M.J.; Bukh, J.; Rice, C.M.; Ploss, A.; Burton, D.R.; Law, M. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2012, 109, 6205–6210. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Z.; Zhao, Y.; Zhang, C.; Chen, H.R.; Feng, J.; Chi, X.M.; Pan, Y.; Du, J.; Guo, M.; Cao, H.; et al. Persistent hepatitis C virus infections and hepatopathological manifestations in immune-competent humanized mice. Cell Res. 2014, 24, 1050–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Oei, Y.; Mendel, D.B.; Garrett, E.N.; Patawaran, M.B.; Hollenbach, P.W.; Aukerman, S.L.; Weiner, A.J. Novel robust hepatitis C virus mouse efficacy model. Antimicrob. Agents Chemother. 2006, 50, 3260–3268. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Y.; Konishi, M.; Walton, C.M.; Olive, D.; Hayashi, K.; Wu, C.H. A novel immunocompetent rat model of HCV infection and hepatitis. Gastroenterology 2005, 128, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Ilan, E.; Arazi, J.; Nussbaum, O.; Zauberman, A.; Eren, R.; Lubin, I.; Neville, L.; Ben-Moshe, O.; Kischitzky, A.; Litchi, A.; et al. The hepatitis C virus (HCV)-trimera mouse: A model for evaluation of agents against HCV. J. Infect. Dis. 2002, 185, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Eren, R.; Landstein, D.; Terkieltaub, D.; Nussbaum, O.; Zauberman, A.; Ben-Porath, J.; Gopher, J.; Buchnick, R.; Kovjazin, R.; Rosenthal-Galili, Z. Preclinical evaluation of two neutralizing human monoclonal antibodies against hepatitis C virus (HCV): A potential treatment to prevent HCV reinfection in liver transplant patients. J. Virol. 2006, 80, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.F.; Schiller, D.E.; Elliott, J.F.; Douglas, D.N.; Hao, C.H.; Rinfret, A.; Addison, W.R.; Fischer, K.P.; Churchill, T.A.; Lakey, J.R.T.; et al. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 2001, 7, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Libbrecht, L.; de Vos, R.; de Hemptinne, B.; Gevaert, K.; Vandekerckhove, J.; Roskams, T.; Leroux-Roels, G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 2005, 41, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Bissig, K.D.; Le, T.T.; Woods, N.B.; Verma, I.M. Repopulation of adult and neonatal mice with human hepatocytes: A chimeric animal model. Proc. Natl. Acad. Sci. USA 2007, 104, 20507–20511. [Google Scholar] [CrossRef] [PubMed]
- Bissig, K.D.; Wieland, S.F.; Tran, P.; Isogawa, M.; Le, T.T.; Chisari, F.V.; Verma, I.M. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J. Clin Invest. 2010, 120, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Azuma, H.; Paulk, N.; Ranade, A.; Dorrell, C.; Al-Dhalimy, M.; Ellis, E.; Strom, S.; Kay, M.A.; Finegold, M.; Grompe, M. Robust expansion of human hepatocytes in Fah(−/−)/Rag2(−/−)/Il2rg(−/−) mice. Nat. Biotechnol. 2007, 25, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Sandgren, E.P.; Palmiter, R.D.; Heckel, J.L.; Daugherty, C.C.; Brinster, R.L.; Degen, J.L. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell 1991, 66, 245–256. [Google Scholar] [CrossRef]
- Vercauteren, K.; de Jong, Y.P.; Meuleman, P. HCV animal models and liver disease. J. Hepatol. 2014, 61, S26–S33. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, A.; Stift, J.; Maric, D.; Cui, Q.W.; Dienes, H.P.; Feinstone, S.M. Chimeric mouse model for the infection of hepatitis B and C viruses. PLoS ONE 2013, 8, e77298. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, K.; Hiraga, N.; Imamura, M.; Yoshimi, S.; Murakami, E.; Nakahara, T.; Honda, Y.; Ono, A.; Kawaoka, T.; Tsuge, M.; et al. A novel TK-NOG based humanized mouse model for the study of HBV and HCV infections. Biochem. Biophys. Res. Commun. 2013, 441, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; Mesalam, A.A.; Leroux-Roels, G.; Meuleman, P. Impact of lipids and lipoproteins on hepatitis C virus infection and virus neutralization. WJG 2014, 20, 15975–15991. [Google Scholar] [CrossRef] [PubMed]
- Fauvelle, C.; Felmlee, D.J.; Crouchet, E.; Lee, J.; Heydmann, L.; Lefevre, M.; Magri, A.; Hiet, M.S.; Fofana, I.; Habersetzer, F.; et al. Apolipoprotein E mediates evasion from hepatitis C virus neutralizing antibodies. Gastroenterology 2016, 150, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Granier, C.; Zeisel, M.B.; Guerin, M.; Mancip, J.; Granio, O.; Penin, F.; Lavillette, D.; Bartenschlager, R.; Baumert, T.F.; et al. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J. Biol. Chem. 2012, 287, 31242–31257. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.; Mccaustland, K.; Krawczynski, K.; Spelbring, J.; Humphrey, C.; Cook, E.H. Hepatitis C virus: buoyant density of the factor VIII-derived isolate in sucrose. J. Med. Virol. 1991, 34, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, R.H.G.; Joyce, M.A.; Lund, G.; Lewis, J.; Chen, R.; Barsby, N.; Zhu, L.F.; Tyrrell, D.L.J.; Kneteman, N.M. Lipoprotein profiles in SCID/uPA mice transplanted with human hepatocytes become human-like and correlate with HCV infection success. Am. J. Physiol. Gastrointest Liver Physiol. 2010, 299, G844–G854. [Google Scholar] [CrossRef] [PubMed]
- Prentoe, J.; Verhoye, L.; Velazquez Moctezuma, R.; Buysschaert, C.; Farhoudi, A.; Wang, R.; Alter, H.; Meuleman, P.; Bukh, J. HVR1-mediated antibody evasion of highly infectious in vivo adapted HCV in humanised mice. Gut 2015. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; Van den Eede, N.; Mesalam, A.A.; Belouzard, S.; Catanese, M.T.; Bankwitz, D.; Wong-Staal, F.; Cortese, R.; Dubuisson, J.; Rice, C.M.; et al. Successful anti-scavenger receptor class B type I (SR-BI) monoclonal antibody therapy in humanized mice after challenge with HCV variants with in vitro resistance to SR-BI-targeting agents. Hepatology 2014, 60, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Meuleman, P.; Ploss, A.; Vanwolleghem, T.; Syder, A.J.; McKeating, J.A.; Lanford, R.E.; Feinstone, S.M.; Major, M.E.; Leroux-Roels, G.; et al. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. USA 2006, 103, 3805–3809. [Google Scholar] [CrossRef] [PubMed]
- Calattini, S.; Fusil, F.; Mancip, J.; Thi, V.L.D.; Granier, C.; Gadot, N.; Scoazec, J.Y.; Zeisel, M.B.; Baumert, T.F.; Lavillette, D.; et al. Functional and biochemical characterization of hepatitis C virus (HCV) particles produced in a humanized liver mouse model. J. Biol. Chem. 2015, 290, 23173–23187. [Google Scholar] [CrossRef] [PubMed]
- Strick-Marchand, H.; Dusseaux, M.; Darche, S.; Huntington, N.D.; Legrand, N.; Masse-Ranson, G.; Corcuff, E.; Ahodantin, J.; Weijer, K.; Spits, H.; et al. A novel mouse model for stable engraftment of a human immune system and human hepatocytes. PLoS ONE 2015, 10, e0119820. [Google Scholar]
- Washburn, M.L.; Bility, M.T.; Zhang, L.G.; Kovalev, G.I.; Buntzman, A.; Frelinger, J.A.; Barry, W.; Ploss, A.; Rice, C.M.; Su, L.S. A humanized mouse model to study hepatitis C virus infection, immune response, and liver disease. Gastroenterology 2011, 140, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Gutti, T.L.; Knibbe, J.S.; Makarov, E.; Zhang, J.J.; Yannam, G.R.; Gorantla, S.; Sun, Y.M.; Mercer, D.F.; Suemizu, H.; Wisecarver, J.L.; et al. Human hepatocytes and hematolymphoid dual reconstitution in treosulfan-conditioned uPA-NOG mice. Am. J. Pathol. 2014, 184, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.; Bial, J.; Tarlow, B.; Bial, G.; Jensen, B.; Greiner, D.L.; Brehm, M.A.; Grompe, M. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res. 2014, 13, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Bility, M.T.; Cheng, L.; Zhang, Z.; Luan, Y.; Li, F.; Chi, L.Q.; Zhang, L.G.; Tu, Z.K.; Gao, Y.H.; Fu, Y.X.; et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: Induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, U364–U230. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; Wyles, D.L.; Mena, A.; Pedreira, J.D.; Castro-Iglesias, A.; Cachay, E. Update on hepatitis C virus resistance to direct-acting antiviral agents. Antivir. Res. 2014, 108, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 2016. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Lontok, E.; Harrington, P.; Howe, A.; Kieffer, T.; Lennerstrand, J.; Lenz, O.; McPhee, F.; Mo, H.; Parkin, N.; Pilot-Matias, T.; et al. Hepatitis C virus drug resistance-associated substitutions: State of the art summary. Hepatology 2015, 62, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Welsch, C.; Yamane, D.; McGivern, D.R.; Yi, M.; Zeuzem, S.; Lemon, S.M. Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 2011, 140, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.L.; de Meyer, S.; Bartels, D.J.; Sullivan, J.C.; Zhang, E.Z.; Tigges, A.; Dierynck, I.; Spanks, J.; Dorrian, J.; Jiang, M.; et al. Hepatitis C viral evolution in genotype 1 treatment-naive and treatment-experienced patients receiving telaprevir-based therapy in clinical trials. PLoS ONE 2012, 7, e34372. [Google Scholar] [CrossRef] [PubMed]
- Plaza, Z.; Soriano, V.; Vispo, E.; Gonzalez, M.D.; Barreiro, P.; Seclen, E.; Poveda, E. Prevalence of natural polymorphisms at the HCV NS5A gene associated with resistance to daclatasvir, an NS5A inhibitor. Antivir. Ther. 2012, 17, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Fridell, R.A.; Wang, C.F.; Sun, J.H.; O’Boyle, D.R.; Nower, P.; Valera, L.; Qiu, D.; Roberts, S.; Huang, X.; Kienzle, B.; et al. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: In vitro and in vivo correlations. Hepatology 2011, 54, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- McPhee, F.; Hernandez, D.; Yu, F.; Ueland, J.; Chayama, K.; Toyota, J.; Izumi, N.; Yokosuka, O.; Kawada, N.; Osaki, Y.; et al. A description of virologic escape in HCV genotype 1-infected patients treated with daclatasvir (BMS-790052) in combination with ribavirin and peginterferon alfa-2a or peginterferon alfa-2b. J. Hepatol. 2012, 56, S473–S473. [Google Scholar]
- Nguyen, L.T.; Hall, N.; Sheerin, D.; Carr, M.; de Gascun, C.F.; Irish Hepatitis, C.O.R.N. Naturally occurring HCV NS5A/B inhibitor resistance-associated mutations to direct-acting antivirals. Antivir. Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Paolucci, S.; Fiorina, L.; Mariani, B.; Gulminetti, R.; Novati, S.; Barbarini, G.; Bruno, R.; Baldanti, F. Naturally occurring resistance mutations to inhibitors of HCV NS5A region and NS5B polymerase in DAA treatment-naive patients. Virol. J. 2013. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Steuer, H.M.M.; Niu, C.R.; Zennou, V.; Keilman, M.; Zhu, Y.A.; Lan, S.Y.; Otto, M.J.; et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Harrington, P.R.; O’Rear, J.J.; Naeger, L.K. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology 2015, 61, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Ogert, R.A.; Howe, J.A.; Vierling, J.M.; Kwo, P.Y.; Lawitz, E.J.; McCone, J.; Schiff, E.R.; Pound, D.; Davis, M.N.; Gordon, S.C.; et al. Resistance-associated amino acid variants associated with boceprevir plus pegylated interferon-alpha2b and ribavirin in patients with chronic hepatitis C in the SPRINT-1 trial. Antivir. Ther. 2013, 18, 387–397. [Google Scholar] [PubMed]
- Shepherd, S.J.; Abdelrahman, T.; MacLean, A.R.; Thomson, E.C.; Aitken, C.; Gunson, R.N. Prevalence of HCV NS3 pre-treatment resistance associated amino acid variants within a Scottish cohort. J. Clin. Virol. 2015, 65, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Bartels, D.J.; Zhou, Y.; Zhang, E.Z.; Marcial, M.; Byrn, R.A.; Pfeiffer, T.; Tigges, A.M.; Adiwijaya, B.S.; Lin, C.; Kwong, A.D.; et al. Natural prevalence of hepatitis C virus variants with decreased sensitivity to NS3.4A protease inhibitors in treatment-naive subjects. J. Infect. Dis. 2008, 198, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Kuntzen, T.; Timm, J.; Berical, A.; Lennon, N.; Berlin, A.M.; Young, S.K.; Lee, B.; Heckerman, D.; Carlson, J.; Reyor, L.L.; et al. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naive patients. Hepatology 2008, 48, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Lenz, O.; Verbinnen, T.; Fevery, B.; Tambuyzer, L.; Vijgen, L.; Peeters, M.; Buelens, A.; Ceulemans, H.; Beumont, M.; Picchio, G.; et al. Virology analyses of HCV isolates from genotype 1-infected patients treated with simeprevir plus peginterferon/ribavirin in phase IIb/III studies. J. Hepatol. 2015, 62, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Bagaglio, S.; Andolina, A.; Merli, M.; Uberti-Foppa, C.; Morsica, G. Frequency of natural resistance within NS5A replication complex domain in hepatitis C genotypes 1a, 1b: Possible implication of subtype-specific resistance selection in multiple direct acting antivirals drugs combination treatment. Viruses 2016. [Google Scholar] [CrossRef] [PubMed]
- Mizokami, M.; Yokosuka, O.; Takehara, T.; Sakamoto, N.; Korenaga, M.; Mochizuki, H.; Nakane, K.; Enomoto, H.; Ikeda, F.; Yanase, M.; et al. Ledipasvir and sofosbuvir fixed-dose combination with and without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C: An open-label, randomised, phase 3 trial. Lancet Infect. Dis. 2015, 15, 645–653. [Google Scholar] [CrossRef]
- Chayama, K.; Notsumata, K.; Kurosaki, M.; Sato, K.; Rodrigues, L.; Setze, C.; Badri, P.; Pilot-Matias, T.; Vilchez, R.A.; Kumada, H. Randomized trial of interferon- and ribavirin-free ombitasvir/paritaprevir/ritonavir in treatment-experienced hepatitis C virus-infected patients. Hepatology 2015, 61, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Kowdley, K.V.; Coakley, E.; Sigal, S.; Nelson, D.R.; Crawford, D.; Weiland, O.; Aguilar, H.; Xiong, J.; Pilot-Matias, T.; et al. Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N. Engl. J. Med. 2014, 370, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; O’Boyle, D.R., 2nd; Fridell, R.A.; Langley, D.R.; Wang, C.; Roberts, S.B.; Nower, P.; Johnson, B.M.; Moulin, F.; Nophsker, M.J.; et al. Resensitizing daclatasvir-resistant hepatitis C variants by allosteric modulation of NS5A. Nature 2015, 527, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kneteman, N.M.; Weiner, A.J.; O’Connell, J.; Collett, M.; Gao, T.; Aukerman, L.; Kovelsky, R.; Ni, Z.J.; Zhu, Q.; Hashash, A.; et al. Anti-HCV therapies in chimeric SCID-Alb/uPA mice parallel outcomes in human clinical application. Hepatology 2006, 43, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Ohara, E.; Hiraga, N.; Imamura, M.; Iwao, E.; Kamiya, N.; Yamada, I.; Kono, T.; Onishi, M.; Hirata, D.; Mitsui, F.; et al. Elimination of hepatitis C virus by short term NS3-4A and NS5B inhibitor combination therapy in human hepatocyte chimeric mice. J. Hepatol. 2011, 54, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Hayes, C.N.; Hiraga, N.; Imamura, M.; Tsuge, M.; Miki, D.; Takahashi, S.; Ochi, H.; Chayama, K. A translational study of resistance emergence using sequential direct-acting antiviral agents for hepatitis C using ultra-deep sequencing. Am. J. Gastroenterol. 2013, 108, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Kai, Y.; Hikita, H.; Tatsumi, T.; Nakabori, T.; Saito, Y.; Morishita, N.; Tanaka, S.; Nawa, T.; Oze, T.; Sakamori, R.; et al. Emergence of hepatitis C virus NS5A L31V plus Y93H variant upon treatment failure of daclatasvir and asunaprevir is relatively resistant to ledipasvir and NS5B polymerase nucleotide inhibitor GS-558093 in human hepatocyte chimeric mice. J. Gastroenterol. 2015, 50, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, J.J.; Thomson, M.; Xie, M.; Horton, J.; Johnson, J.; Krull, D.; Mathis, A.; Morikawa, Y.; Parks, D.; Peterson, R.; et al. Preclinical characterization and in vivo efficacy of GSK8853, a small molecule inhibitor of the hepatitis C virus NS4B protein. Antimicrob. Agents Chemother. 2015, 59, 6539–6550. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Hiraga, N.; Imamura, M.; Hayes, C.N.; Zhang, Y.Z.; Kosaka, K.; Okazaki, A.; Murakami, E.; Tsuge, M.; Abe, H.; et al. Combination therapies with NS5A, NS3 and NS5B inhibitors on different genotypes of hepatitis C virus in human hepatocyte chimeric mice. Gut 2013, 62, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, N.; Abe, H.; Imamura, M.; Tsuge, M.; Takahashi, S.; Hayes, C.N.; Ochi, H.; Tateno, C.; Yoshizato, K.; Nakamura, Y.; et al. Impact of viral amino acid substitutions and host interleukin-28b polymorphism on replication and susceptibility to interferon of hepatitis C virus. Hepatology 2011, 54, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Kurbanov, F.; Tanaka, Y.; Matsuura, K.; Sugauchi, F.; Elkady, A.; Khan, A.; Hasegawa, I.; Ohno, T.; Tokuda, H.; Mizokami, M. Positive selection of core 70Q variant genotype 1b hepatitis C virus strains induced by pegylated interferon and ribavirin. J. Infect. Dis. 2010, 201, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Dallas, A.; Ilves, H.; Shorenstein, J.; MacLachlan, I.; Klumpp, K.; Johnston, B.H. Formulated minimal-length synthetic small hairpin RNAs are potent inhibitors of hepatitis C virus in mice with humanized livers. Gastroenterology 2014, 146, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Dallas, A.; Ilves, H.; Ma, H.; Chin, D.J.; MacLachlan, I.; Klumpp, K.; Johnston, B.H. Inhibition of hepatitis C virus in chimeric mice by short synthetic hairpin RNAs: Sequence analysis of surviving virus shows added selective pressure of combination therapy. J. Virol. 2014, 88, 4647–4656. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; Brown, R.J.; Mesalam, A.A.; Doerrbecker, J.; Bhuju, S.; Geffers, R.; van den Eede, N.; McClure, C.P.; Troise, F.; Verhoye, L.; et al. Targeting a host-cell entry factor barricades antiviral-resistant HCV variants from on-therapy breakthrough in human-liver mice. Gut 2015. [Google Scholar] [CrossRef]
- Lisboa-Neto, G.; Noble, C.F.; Pinho, J.R.R.; Malta, F.M.; Gomes-Gouvea, M.S.; Alvarado-Mora, M.V.; da Silva, M.H.; Leite, A.G.B.; Piccoli, L.Z.; Rodrigues, F.K.; et al. Resistance mutations are rare among protease inhibitor treatment-naive hepatitis C genotype-1 patients with or without HIV coinfection. Antivir. Ther. 2015, 20, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.; Cortes Martins, H.; Coutinho, R.; Leitao, E.; Silva, R.; Padua, E. Molecular characterization of hepatitis C virus for determination of subtypes and detection of resistance mutations to protease inhibitors in a group of intravenous drug users co-infected with HIV. J. Med. Virol. 2015, 87, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Aissa Larousse, J.; Trimoulet, P.; Recordon-Pinson, P.; Papuchon, J.; Azzouz, M.M.; Ben Mami, N.; Cheikh, I.; Triki, H.; Fleury, H. Natural prevalence of hepatitis C virus (HCV) variants resistant to protease and polymerase inhibitors in patients infected with HCV genotype 1 in Tunisia. J. Med. Virol. 2014, 86, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Franco, S.; Bellido, R.; Aparicio, E.; Canete, N.; Garcia-Retortillo, M.; Sola, R.; Tural, C.; Clotet, B.; Paredes, R.; Martinez, M.A. Natural prevalence of HCV minority variants that are highly resistant to NS3/4A protease inhibitors. J. Viral Hepat. 2011, 18, E578–E582. [Google Scholar] [CrossRef] [PubMed]
- Applegate, T.L.; Gaudieri, S.; Plauzolles, A.; Chopra, A.; Grebely, J.; Lucas, M.; Hellard, M.; Luciani, F.; Dore, G.J.; Matthews, G.V. Naturally occurring dominant drug resistance mutations occur infrequently in the setting of recently acquired hepatitis C. Antivir. Ther. 2015, 20, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, V.C.; Cento, V.; Mirabelli, C.; Artese, A.; Costa, G.; Alcaro, S.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus genetic variability and the presence of NS5B resistance- associated mutations as natural polymorphisms in selected genotypes could affect the response to NS5B inhibitors. Antimicrob. Agents Chemother. 2014, 58, 2781–2797. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Queiroz, A.T.L.; Pessoa, M.G.; da Silva, E.F.; Mazo, D.F.C.; Carrilho, F.J.; Carvalho, R.J.; de Carvalho, I.M.V.G. The presence of resistance mutations to protease and polymerase inhibitors in hepatitis C virus sequences from the Los Alamos databank. J. Viral Hepat. 2013, 20, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Dryer, P.D.; Limketkai, B.N.; Martin, C.M.; Ma, G.; Sherman, K.E.; Taylor, L.E.; Mayer, K.H.; Jamieson, D.J.; Blackard, J.T. Screening for hepatitis C virus non-nucleotide resistance mutations in treatment-naive women. J. Antimicrob. Chemother. 2009, 64, 945–948. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Kanda, T.; Matsumura, H.; Moriyama, M.; Yokosuka, O.; Omata, M. HCV NS5A resistance-associated variants in a group of real-world Japanese patients chronically infected with HCV genotype 1b. Hepatol. Int. 2015, 9, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, S.; Imamura, M.; Murakami, E.; Hiraga, N.; Tsuge, M.; Kawakami, Y.; Aikata, H.; Abe, H.; Hayes, C.N.; Sasaki, T.; et al. Long term persistence of NS5A inhibitor-resistant hepatitis C virus in patients who failed daclatasvir and asunaprevir therapy. J. Med. Virol. 2015, 87, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, K.; Imamura, M.; Hayes, C.N.; Abe, H.; Hiraga, N.; Yoshimi, S.; Murakami, E.; Kawaoka, T.; Tsuge, M.; Aikata, H.; et al. Emergence of resistant variants detected by ultra-deep sequencing after asunaprevir and daclatasvir combination therapy in patients infected with hepatitis C virus genotype 1. J. Viral Hepat. 2015, 22, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Kan, H.; Hiraga, N.; Imamura, M.; Hayes, C.N.; Uchida, T.; Miyaki, E.; Tsuge, M.; Abe, H.; Aikata, H.; Miki, D.; et al. Combination therapies with daclatasvir and asunaprevir on NS3-D168 mutated HCV in human hepatocyte chimeric mice. Antivir. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Hiraga, N.; Imamura, M.; Yoshimi, S.; Kan, H.; Miyaki, E.; Tsuge, M.; Abe, H.; Hayes, C.N.; Aikata, H.; et al. Elimination of HCV via a non-ISG-mediated mechanism by vaniprevir and BMS-788329 combination therapy in human hepatocyte chimeric mice. Virus Res. 2015, 213, 62–68. [Google Scholar] [CrossRef] [PubMed]
- De Jong, Y.P.; Rice, C.M.; Ploss, A. Evaluation of combination therapy against hepatitis C virus infection in human liver chimeric mice. J. Hepatol. 2011, 54, 848–850. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesalam, A.A.; Vercauteren, K.; Meuleman, P. Mouse Systems to Model Hepatitis C Virus Treatment and Associated Resistance. Viruses 2016, 8, 176. https://doi.org/10.3390/v8060176
Mesalam AA, Vercauteren K, Meuleman P. Mouse Systems to Model Hepatitis C Virus Treatment and Associated Resistance. Viruses. 2016; 8(6):176. https://doi.org/10.3390/v8060176
Chicago/Turabian StyleMesalam, Ahmed Atef, Koen Vercauteren, and Philip Meuleman. 2016. "Mouse Systems to Model Hepatitis C Virus Treatment and Associated Resistance" Viruses 8, no. 6: 176. https://doi.org/10.3390/v8060176
APA StyleMesalam, A. A., Vercauteren, K., & Meuleman, P. (2016). Mouse Systems to Model Hepatitis C Virus Treatment and Associated Resistance. Viruses, 8(6), 176. https://doi.org/10.3390/v8060176