
Characterization of a Novel Polerovirus Infecting Maize in China



Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Collection and RNA Isolation
2.2. Small RNA Library Construction, Sequencing, and Data Processing
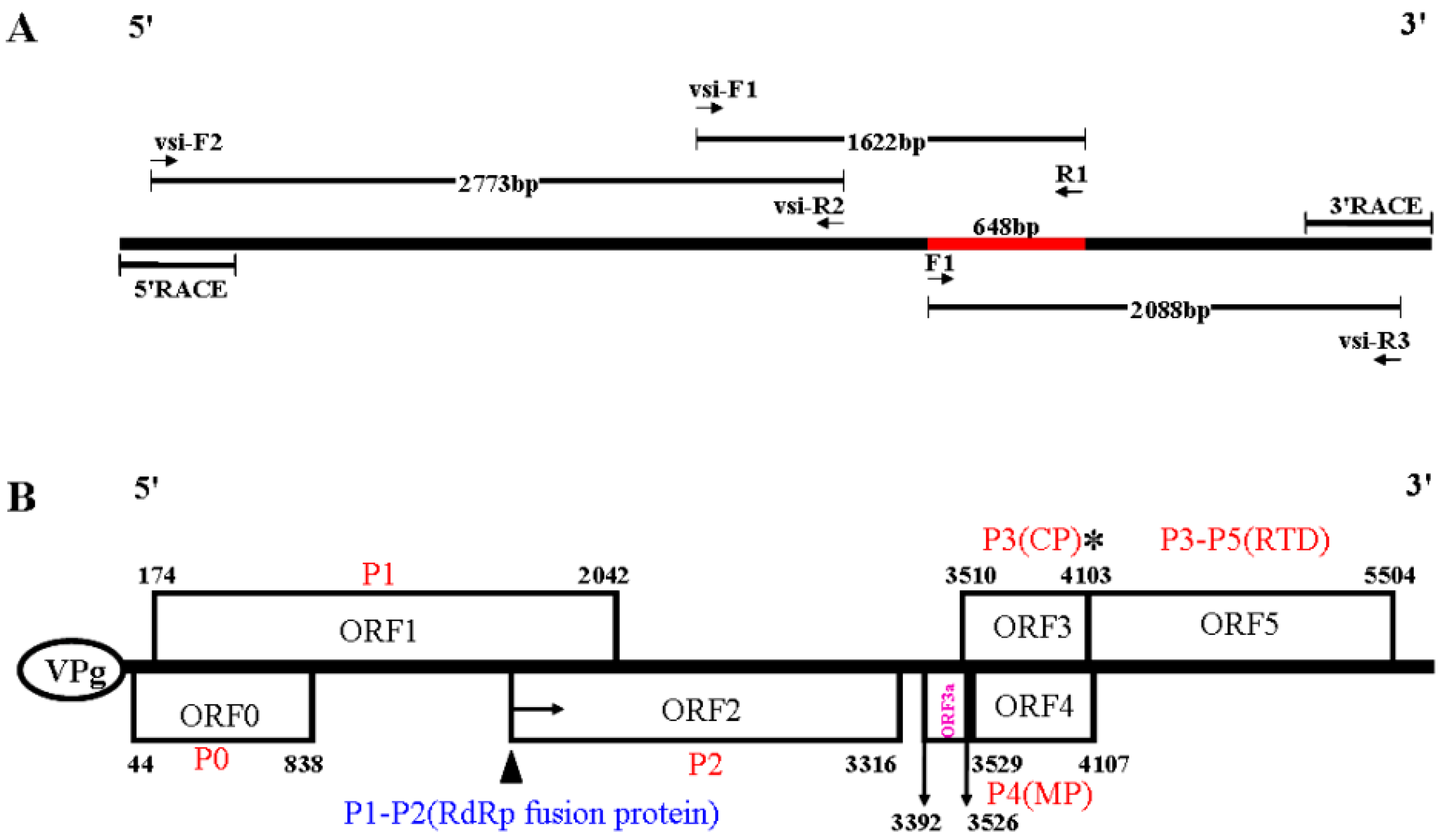
2.3. RT-PCR Validation, Full-Length Genome Amplification, and Sequencing
2.4. Sequence and Molecular Variation Analyses
2.5. Assay of PTGS Suppression Function of P0 Protein Encoded by MaYMV
2.6. Construction of cDNA Clones of MaYMV, Agro-Infiltration, and Detection of Viral RNA
3. Results
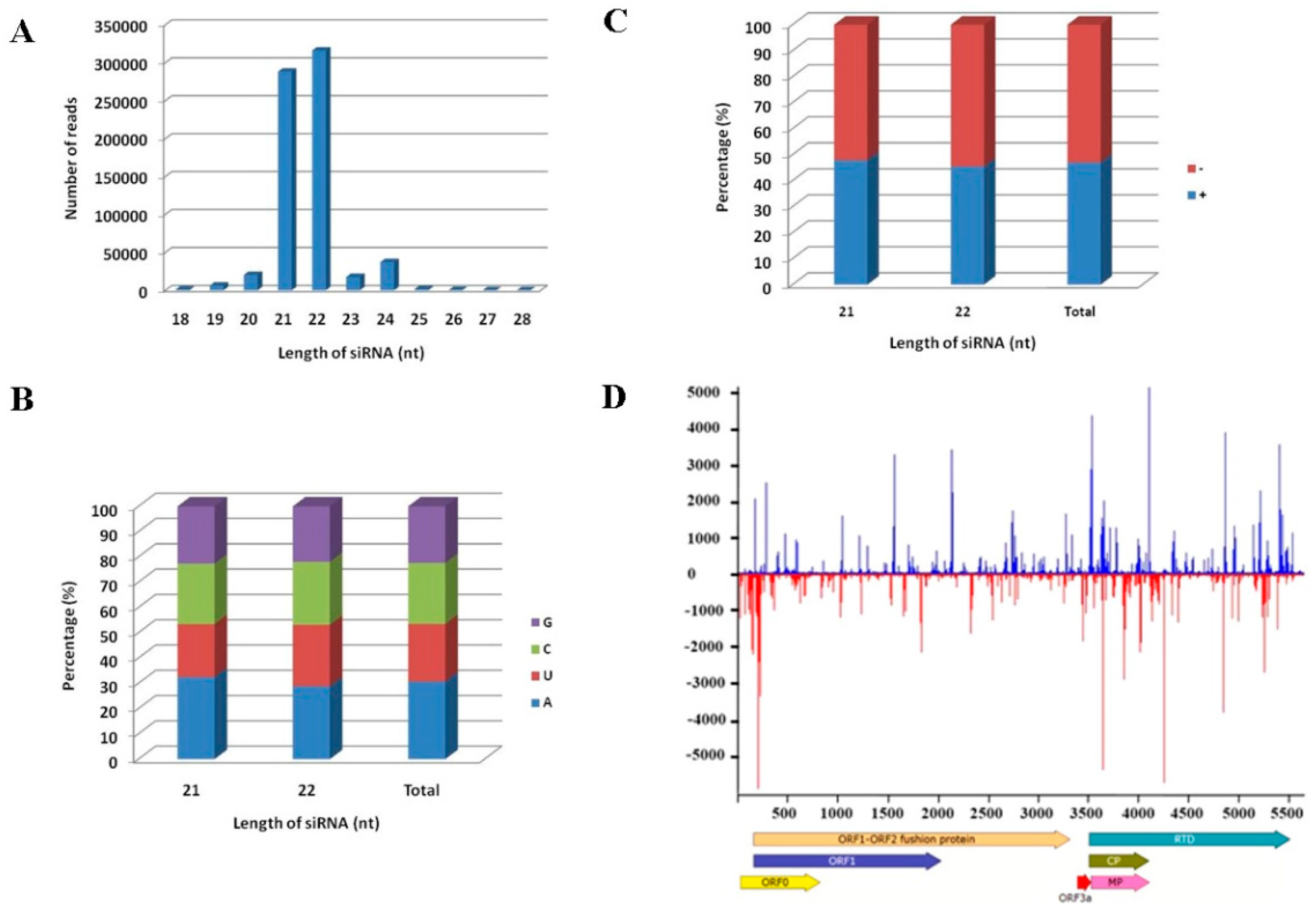
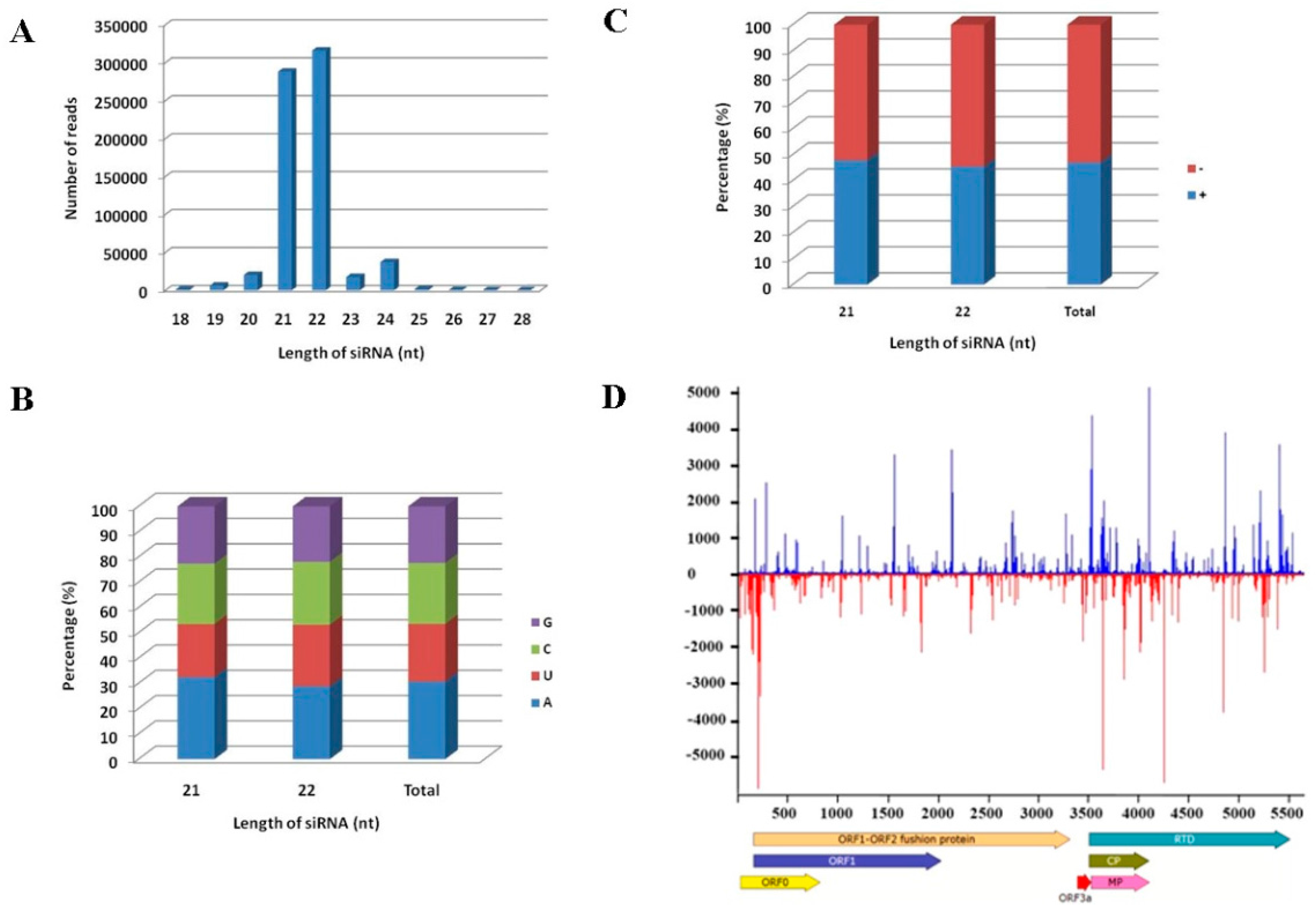
3.1. Analysis of sRNA Library and Identification of Virus in Maize by de Novo Assembly of vsiRNAs
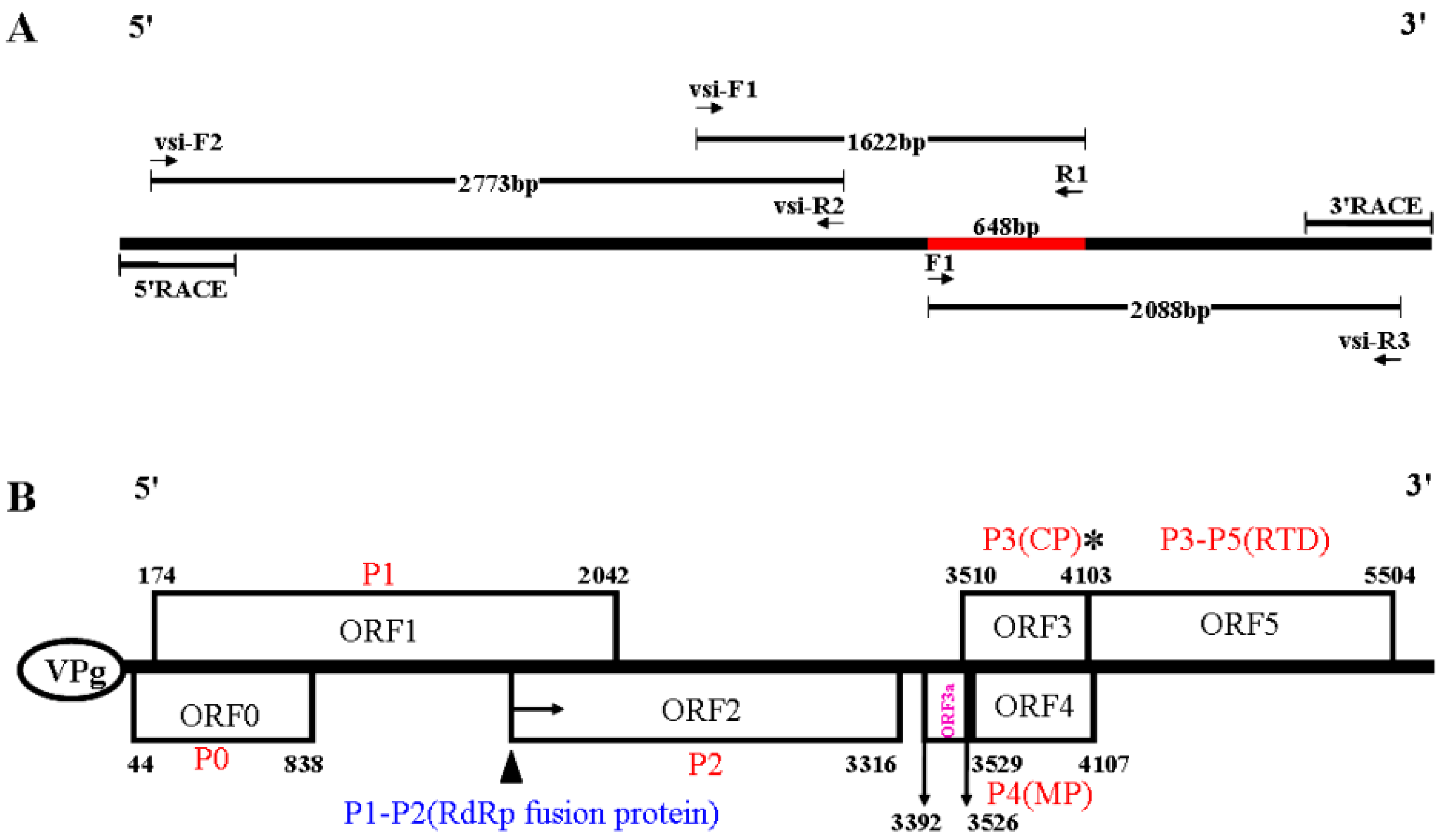
3.2. The Complete Nucleotide Sequence and Genome Organization of a Novel RNA Virus
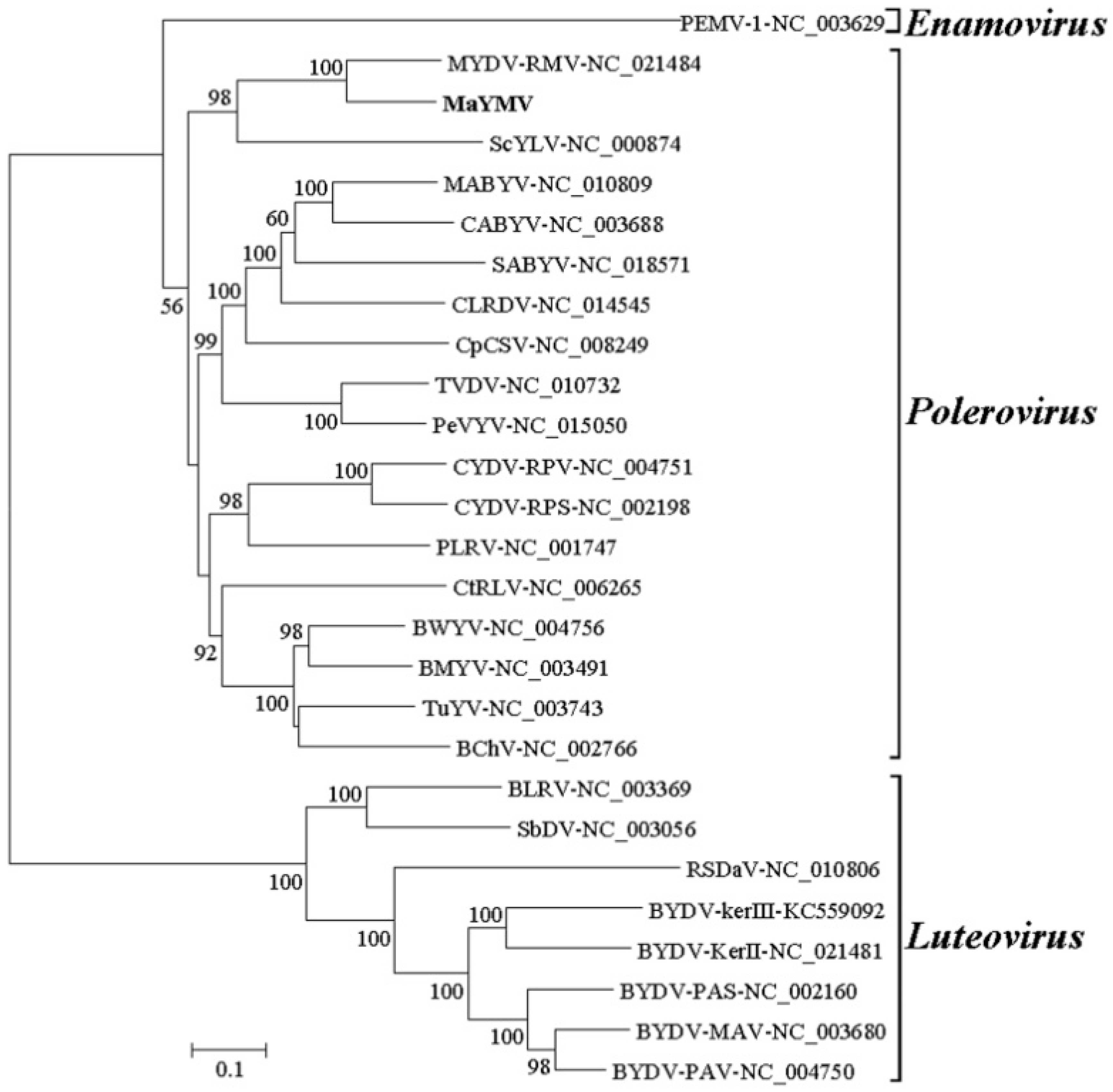
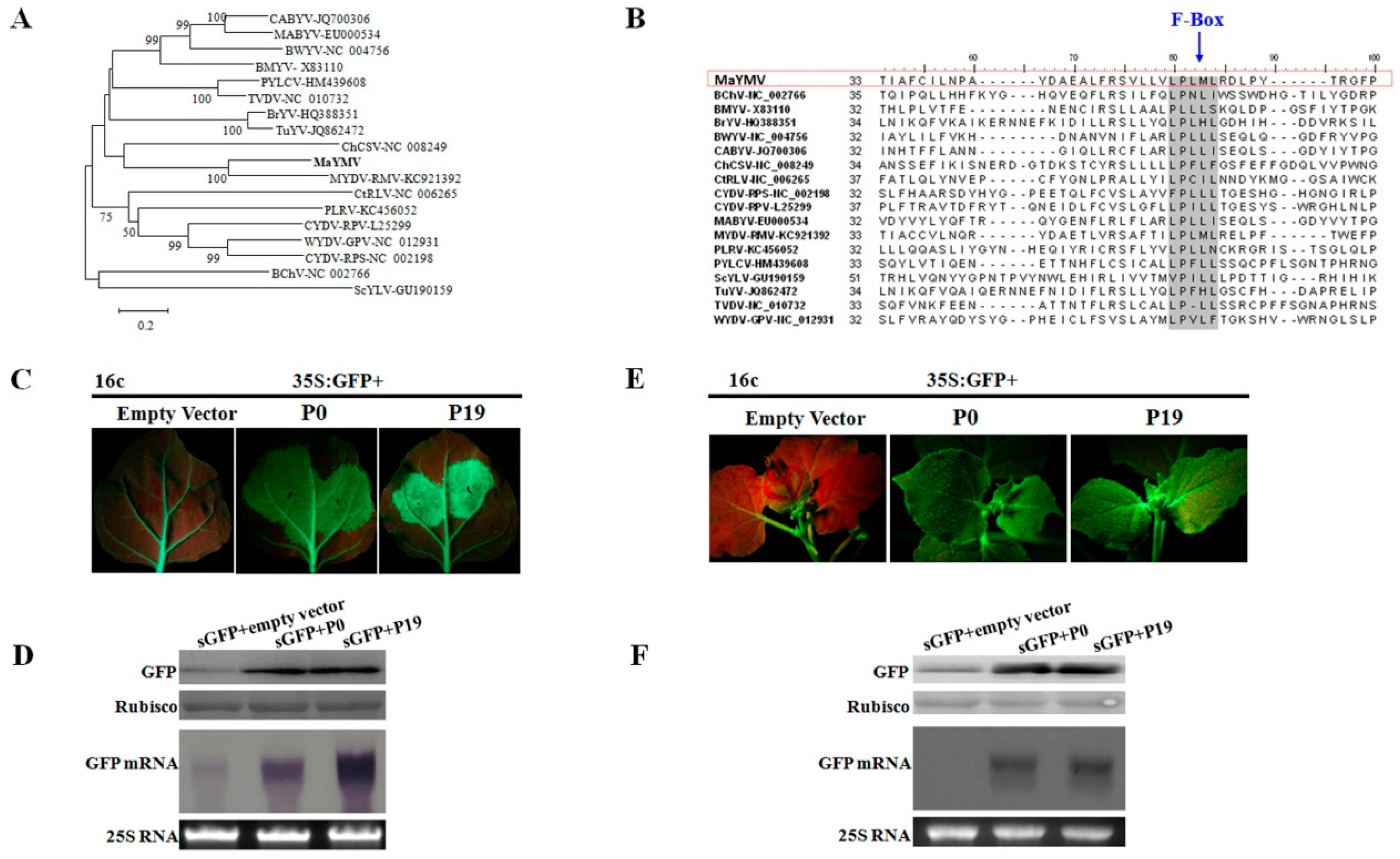
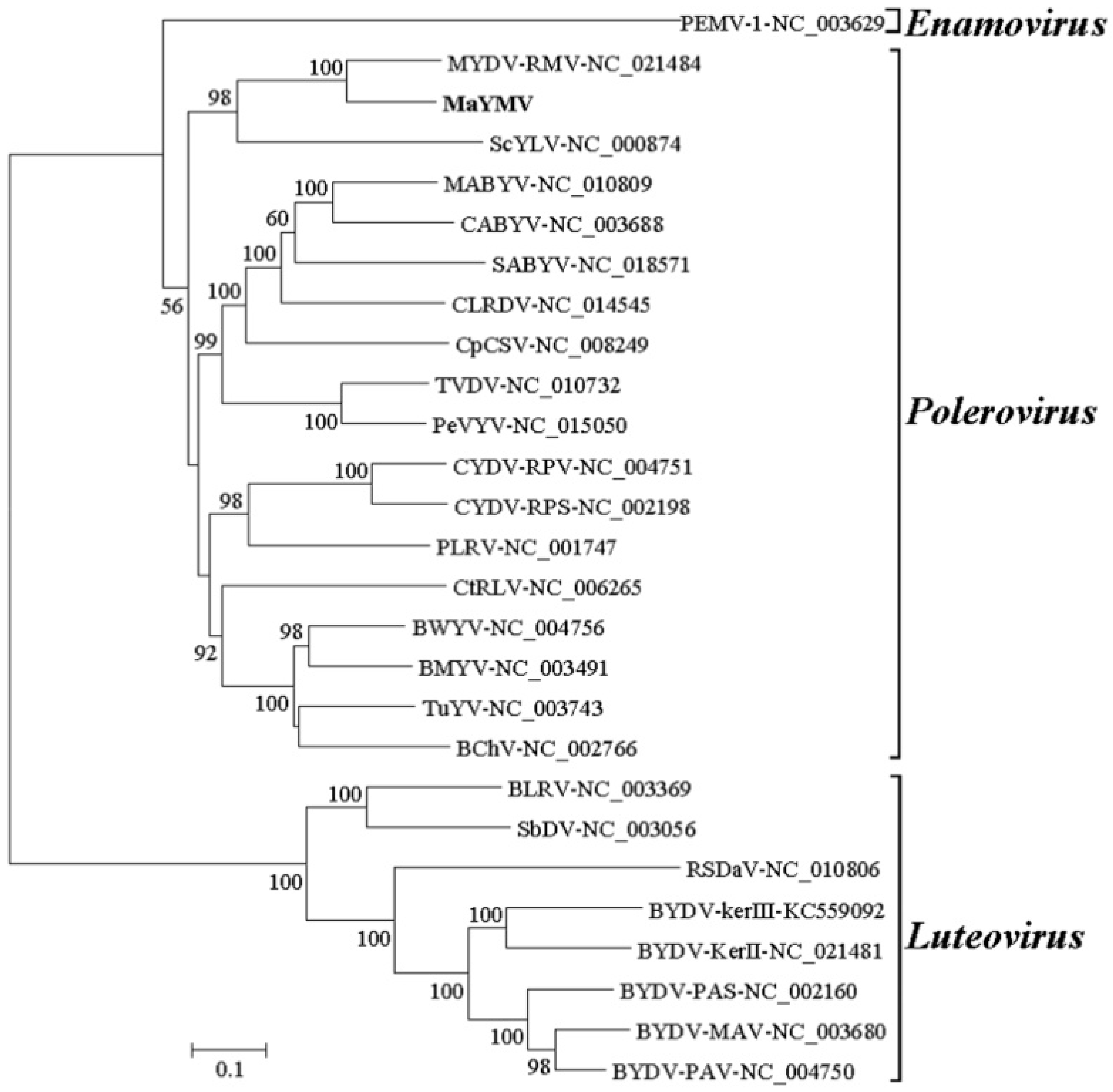
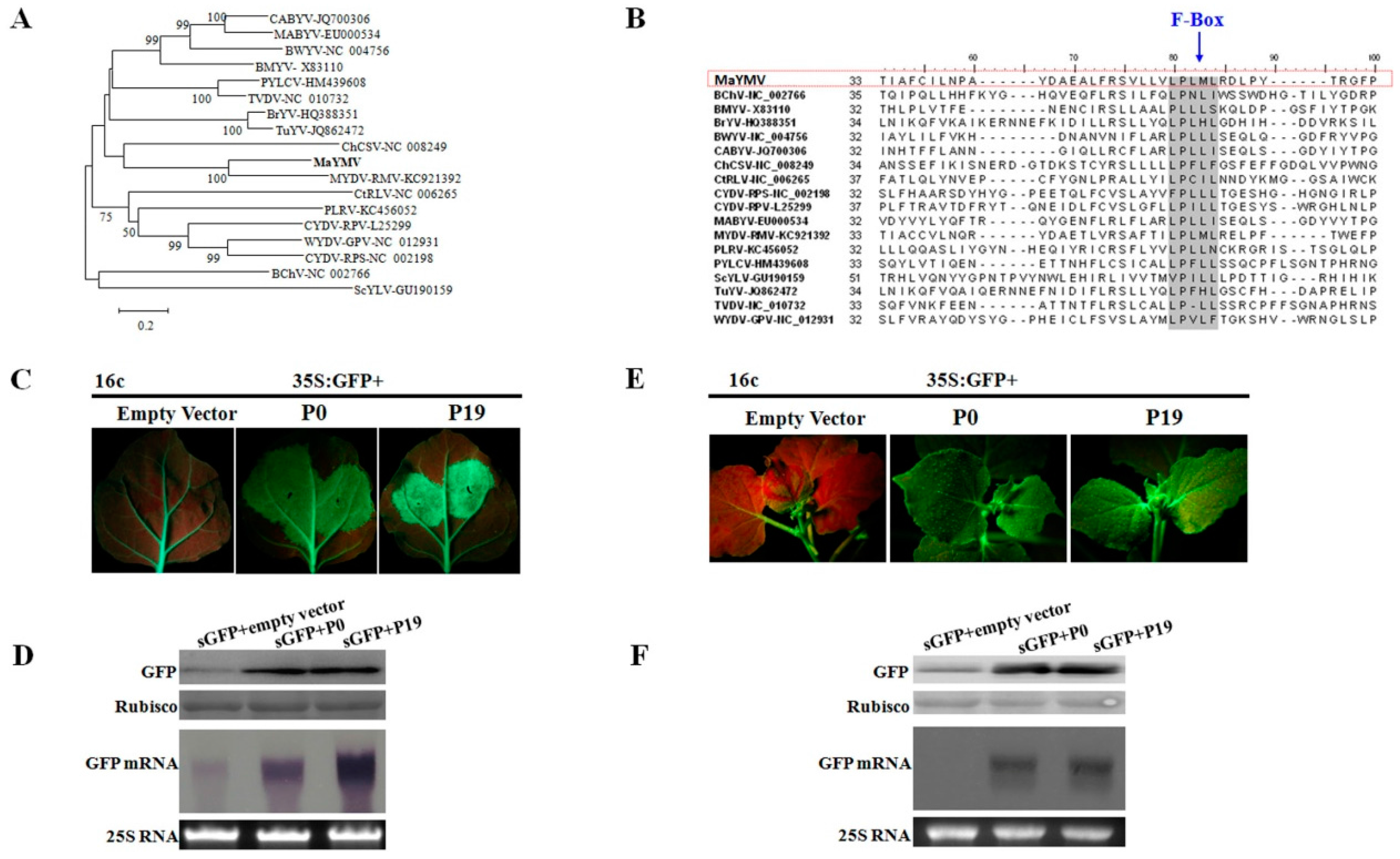
3.3. Phylogenetic Analysis of the MaYMV Genome
3.4. Distribution of vsiRNAs Along the MCMV Genome
3.5. The P0 Protein Encoded by MaYMV Inhibits Local and Systemic RNA Silencing
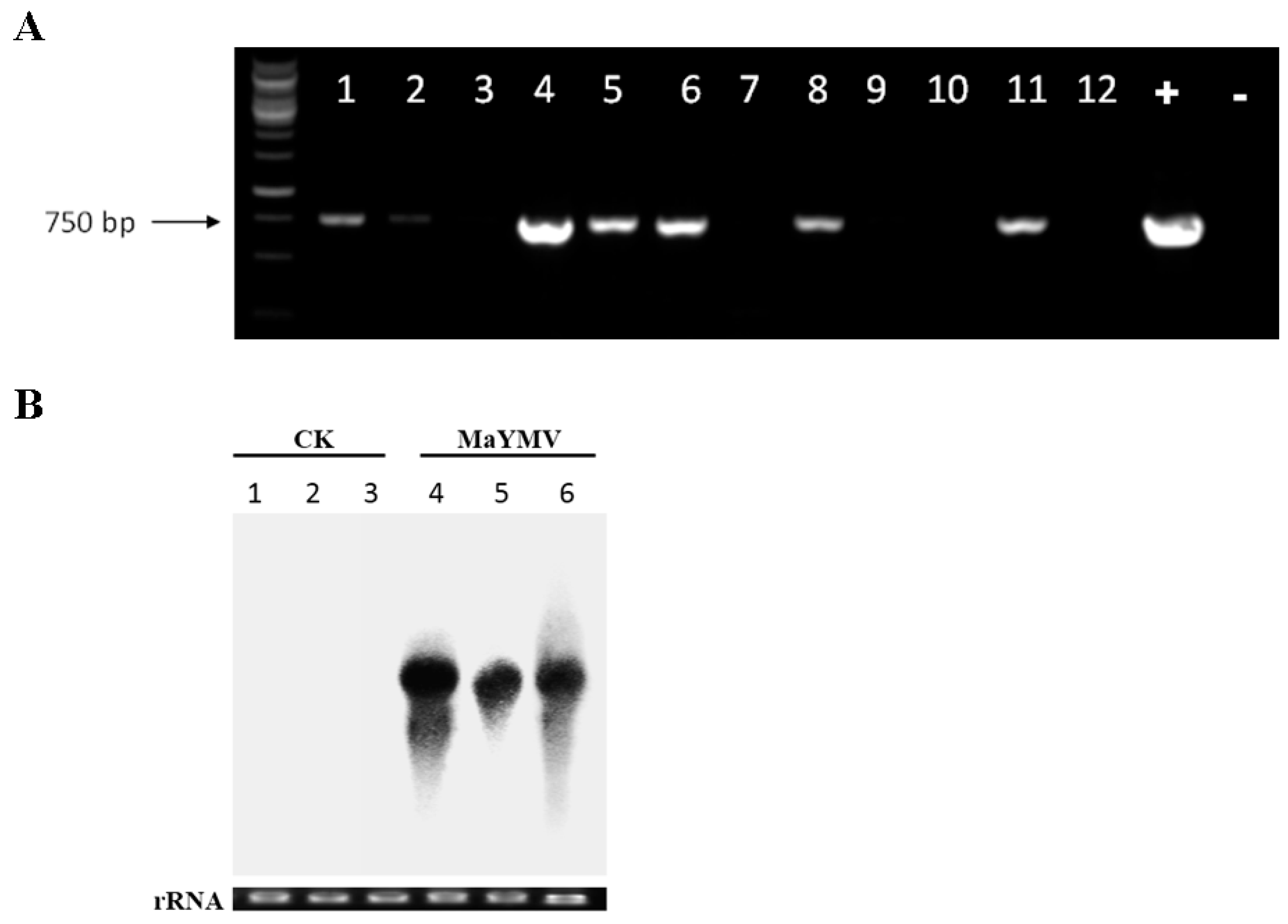
3.6. Infectivity Assays in N. Benthamiana Plants
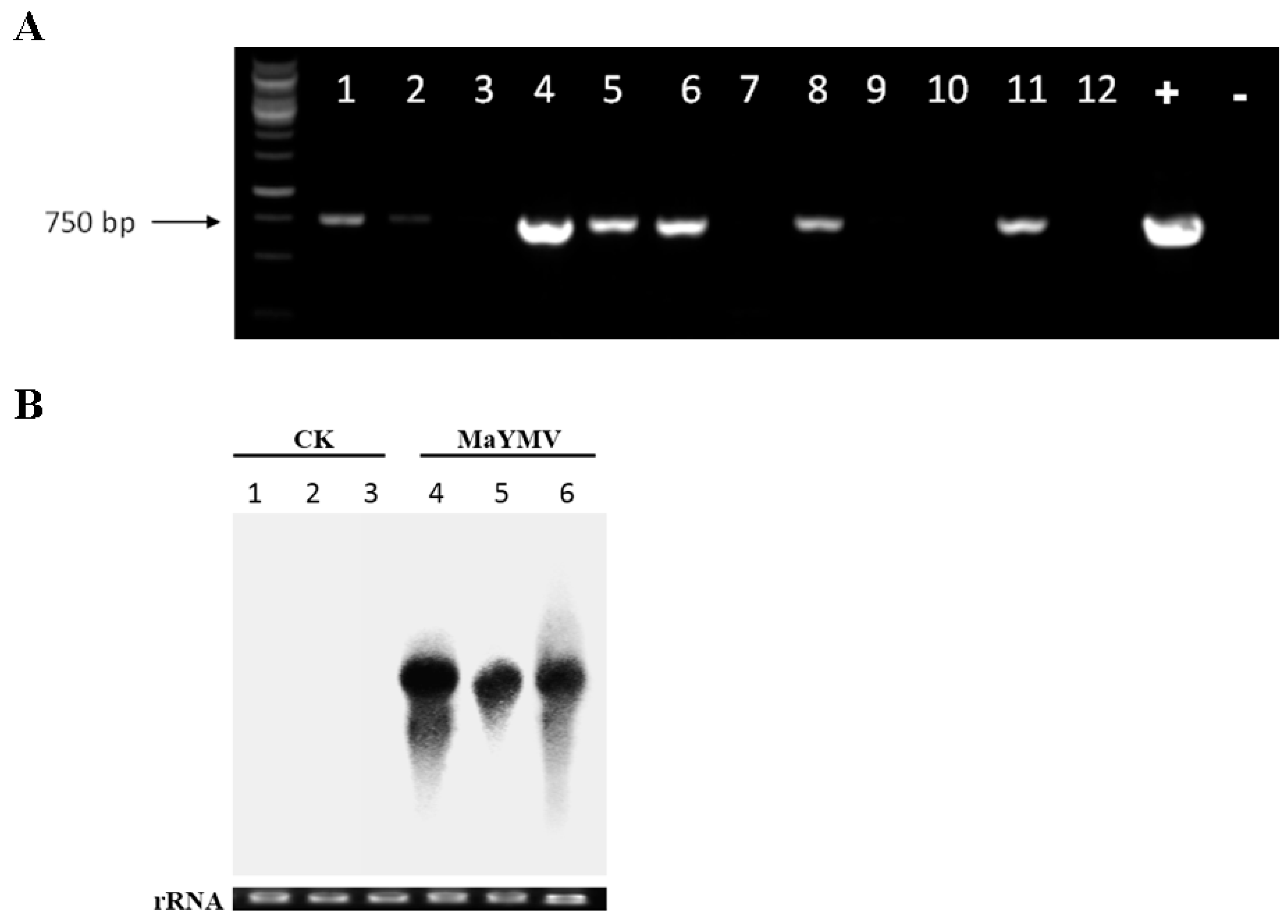
3.7. Field Detection of MaYMV
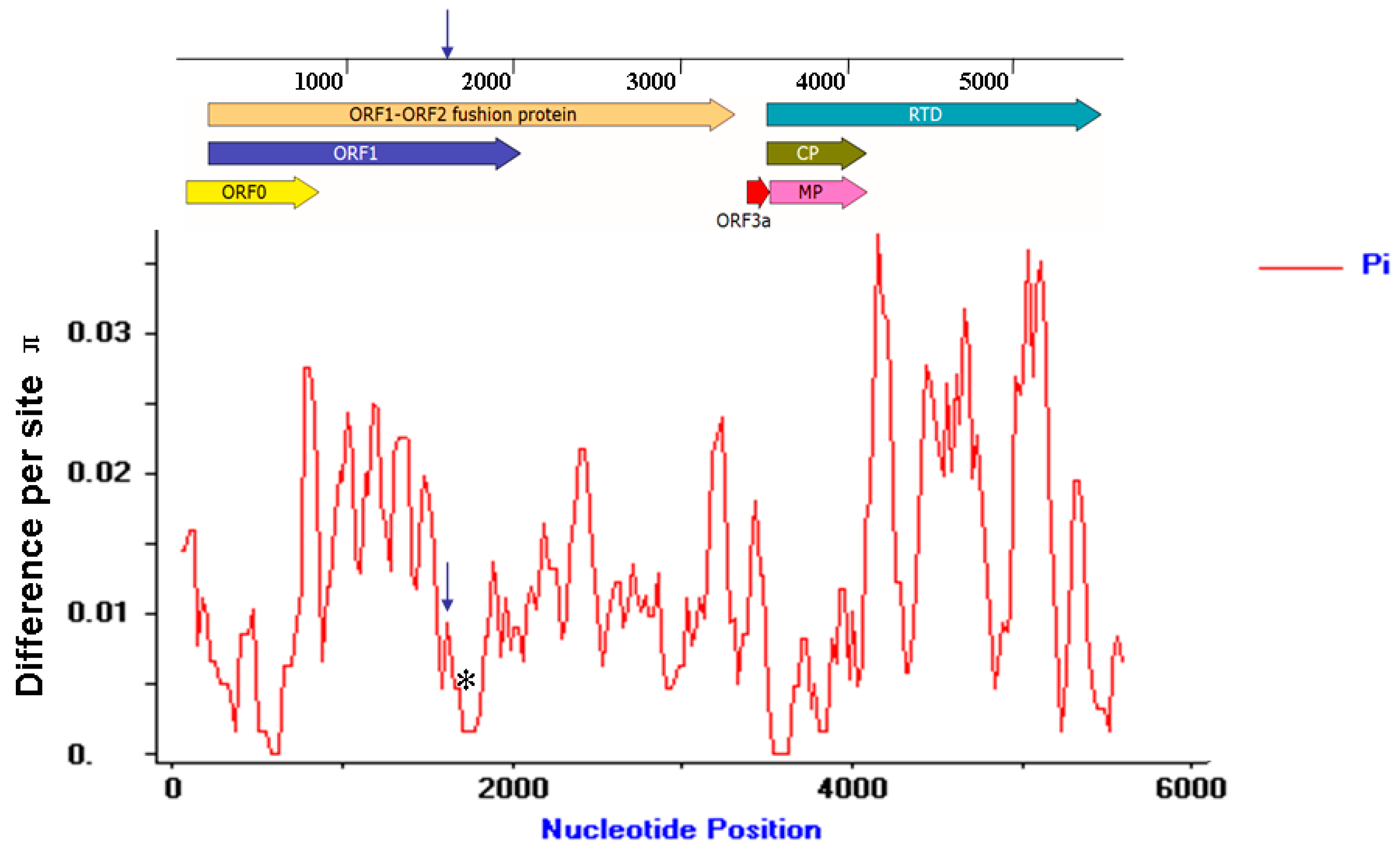
3.8. Molecular Variation of the MaYMV Genome
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Koonin, E.V.; Wolf, Y.I.; Nagasaki, K.; Dolja, V.V. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat. Rev. Microbiol. 2008, 6, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Andrew, M.Q.K.; Michael, J.A.; Eric, B.C.; Elliot, J.L. Virus taxonomy. In Virus Taxonomy: Ninth Report of the International Committee on the Taxonomy of Viruses; Elsevier Academic Press Inc.: San Diego, CA, USA, 2012; pp. 1045–1053. [Google Scholar]
- Moonan, F.; Molina, J.; Mirkov, T.E. Sugarcane yellow leaf virus: An emerging virus that has evolved by recombination between luteoviral and poleroviral ancestors. Virology 2000, 269, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Krueger, E.N.; Beckett, R.J.; Gray, S.M.; Miller, W.A. The complete nucleotide sequence of the genome of Barley yellow dwarf virus-RMV reveals it to be a new Polerovirus distantly related to other yellow dwarf viruses. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Guilley, H.; Wipf-Scheibel, C.; Richards, K.; Lecoq, H.; Jonard, G. Nucleotide sequence of cucurbit aphid-borne yellows luteovirus. Virology 1994, 202, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.H.; Chen, Z.B.; Chen, J.P. Complete nucleotide sequence and genome organization of a Chinese isolate of Tobacco vein distorting virus. Virus Genes 2010, 41, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Kozlowska-Makulska, A.; Guilley, H.; Szyndel, M.S.; Beuve, M.; Lemaire, O.; Herrbach, E.; Bouzoubaa, S. P0 proteins of European beet-infecting poleroviruses display variable RNA silencing suppression activity. J. Gen. Virol. 2010, 91, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Mangwende, T.; Wang, M.L.; Borth, W.; Hu, J.; Moore, P.H.; Mirkov, T.E.; Albert, H.H. The P0 gene of Sugarcane yellow leaf virus encodes an RNA silencing suppressor with unique activities. Virology 2009, 384, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dunoyer, P.; Heim, F.; Richards, K.E.; Jonard, G.; Ziegler-Graff, V. P0 of beet Western yellows virus is a suppressor of posttranscriptional gene silencing. J. Virol. 2002, 76, 6815–6824. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.A.; Dinesh-Kumar, S.P.; Paul, C.P. Luteovirus gene expression. Crit. Rev. Plant Sci. 1995, 14, 179–211. [Google Scholar] [CrossRef]
- Guilley, H.; Richards, K.E.; Jonard, G. Nucleotide sequence of beet mild yellowing virus RNA. Arch. Virol. 1995, 140, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.Y.; Shang, Q.X.; Han, C.G.; Li, D.W.; Yu, J.L. Complete sequence analysis reveals two distinct Poleroviruses infecting cucurbits in China. Arch. Virol. 2008, 153, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.W.; Cheng, Z.M.; Xu, L.; Wu, M.S.; Waterhouse, P.; Zhou, G.H.; Li, S.F. The complete nucleotide sequence of the barley yellow dwarf GPV isolate from China shows that it is a new member of the genus Polerovirus. Arch. Virol. 2009, 154, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Palukaitis, P.; Gray, S.M. Host-dependent requirement for the Potato leafroll virus 17-kda protein in virus movement. Mol. Plant Microbe Interact. 2002, 15, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Brault, W.; Périgon, S.; Reinbold, C.; Erdinger, M.; Scheidecker, D.; Herrbach, E.; Richards, K.; Ziegler-Graff, V. The Polerovirus minor capsid protein determines vector specificity and intestinal tropism in the aphid. J. Virol. 2005, 79, 9685–9693. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Firth, A.E.; Miller, W.A.; Scheidecker, D.; Brault, V.; Reinbold, C.; Rakotondrafara, A.M.; Chung, B.Y.W.; Ziegler-Graff, V. Discovery of a small non-AUG-initiated ORF in poleroviruses and luteoviruses that is required for long-distance movement. PLoS Pathog. 2015, 11, e1004868. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses. Available online: http://www.ictvonline.org/ (accessed on 14 July 2014).
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, V.; Saldarelli, P.; Miozzi, L.; Giampetruzzi, A.; Gisel, A.; Moxon, S.; Dalmaye, T.; Bisztrayf, G.; Burgyan, J. Deep sequencing analysis of viral short RNAs from an infected Pinot Noir grapevine. Virology 2010, 408, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Seguin, J.; Rajeswaran, R.; Malpica-Lopez, N.; Martin, R.R.; Kasschau, K.; Dolja, V.V.; Otten, P.; Farinelli, L.; Pooggin, M.M. De novo reconstruction of consensus master genomes of plant RNA and DNA viruses from siRNAs. PLoS ONE 2014, 9, e88513. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Fu, S.; Qian, Y.J.; Xu, Y.; Zhou, X.P. Identification of Hop stunt viroid infecting Citrus limon in China using small RNAs deep sequencing approach. Virol. J. 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Cheng, X.F.; Wu, X.X.; Wang, A.M.; Wu, X.Y. Characterization of complete genome and small RNA profile of pagoda yellow mosaic associated virus, a novel badnavirus in China. Virus Res. 2014, 188, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.F.; Luo, Y.J.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.X.; Ding, S.W. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Baulcombe, D. RNA silencing in plants. Nature 2004, 431, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.W.; Voinnet, O. Antiviral immunity directed by small RNAs. Cell 2007, 130, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Molnár, A.; Csorba, T.; Lakatos, L.; Várallyay, É.; Lacomme, C.; Burgyán, J. Plant virus-derived small interfering RNAs originate predominantly from highly structured single-stranded viral RNAs. J. Virol. 2005, 79, 7812–7818. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.M.; Wang, M.B.; Lough, T. Gene silencing as an adaptive defence against viruses. Nature 2001, 411, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Pallett, D.W.; Ho, T.; Cooper, I.; Wang, H. Detection of Cereal yellow dwarf virus using small interfering RNAs and enhanced infection rate with Cocksfoot streak virus in wild cocksfoot grass Dactylis glomerata. J. Virol. Methods 2010, 168, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.L.; Wang, Y.; Guo, W.; Xie, Y.; Xie, Q.; Fan, L.J.; Zhou, X.P. Characterization of small interfering RNAs derived from the geminivirus/betasatellite complex using deep sequencing. PLoS ONE 2011, 6, e16928. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.Y.; Wu, J.X.; Zhou, Y.J.; Zhou, X.P. Characterization and subcellular localization of an RNA silencing suppressor encoded by Rice stripe tenuivirus. Virology 2009, 387, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genetics 1983, 105, 437–460. [Google Scholar] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Boil. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Mozo, T.; Hooykaas, P.J. Electroporation of megaplasmids into Agrobacterium. Plant Mol. Biol. 1991, 16, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Dang, M.Q.; Hou, H.W.; Mei, Y.Z.; Qian, Y.J.; Zhou, X.P. Identification of an RNA silencing suppressor encoded by a mastrevirus. J. Gen. Virol. 2014, 95, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.F.; Li, G.X.; Wang, D.W.; Hu, D.W.; Zhou, X.P. A begomovirus DNAβ-encoded protein binds DNA, functions as a suppressor of RNA silencing, and targets the cell nucleus. J. Virol. 2005, 79, 10764–10775. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.D.; Menzel, W.; Lesemann, D.E.; Varrelmann, M.; Vetten, H.J. Chickpea chlorotic stunt virus: A new Polerovirus infecting cool-season food legumes in Ethiopia. Phytopathology 2006, 96, 437–446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Duffus, J.E. Economic significance of Beet western yellows (Radish yellows) on Sugar Beet. Phytopathology 1961, 51, 605–607. [Google Scholar]
- Ellis, M.H.; Silva, T.F.; Stiller, W.N.; Wilson, L.J.; Vaslin, M.F.S.; Sharman, M.; Llewellyn, D.J. Identification of a new Polerovirus (family Luteoviridae) associated with cotton bunchy top disease in Australia. Australas. Plant Pathol. 2013, 42, 261–269. [Google Scholar] [CrossRef]
- Miller, W.A.; Jackson, J.; Feng, Y. Cis-and trans-regulation of luteovirus gene expression by the 3′ end of the viral genome. Virus Res. 2015, 206, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Nakashima, N.; Hinomoto, N.; Kawano, S.; Toyosato, T. The genome sequence of pepper vein yellows virus family Luteoviridae, genus Polerovirus. Arch. Virol. 2011, 156, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, Q.Q.; Wu, L.Q.; Qian, Y.J. Identification and characterization of a maize-associated mastrevirus in China by deep sequencing small RNA populations. Virol. J. 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Donaire, L.; Wang, Y.; Gonzalez-Ibeas, D.; Mayer, K.F.; Aranda, M.A.; Llave, C. Deep-sequencing of plant viral small RNAs reveals effective and widespread targeting of viral genomes. Virology 2009, 392, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Cheng, C.P.; Chang, B.C.H.; Wang, W.C.; Huang, Y.W.; Lee, Y.S.; Huang, H.D.; Hsu, Y.H.; Lin, N.S. Global analyses of small interfering RNAs derived from Bamboo mosaic virus and its associated satellite RNAs in different plants. PLoS ONE 2010, 5, e11928. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.J.; Cai, T.; Hu, Y.G.; Chen, Y.; Hodges, E.; Ni, F.R.; Wu, L.; Li, S.; Zhou, H.; Long, C.Z.; et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Iwasaki, S.; Watanabe, T.; Utsumi, M.; Watanabe, Y. The mechanism selecting the guide strand from small RNA duplexes is different among argonaute proteins. Plant Cell Physiol. 2008, 49, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Miozzi, L.; Gambino, G.; Burgyan, J.; Pantaleo, V. Genome-wide identification of viral and host transcripts targeted by viral siRNAs in Vitis vinifera. Mol. Plant Mothol. 2013, 14, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Llave, C. Virus-derived small interfering RNAs at the core of plant-virus interactions. Trends Plant Sci. 2010, 15, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Silhavy, D.; Burgyán, J. Effects and side-effects of viral RNA silencing suppressors on short RNAs. Trends Plant Sci. 2004, 9, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Induction and suppression of RNA silencing: Insights from viral infections. Nat. Rev. Genet. 2005, 6, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Pazhouhandeh, M.; Dieterle, M.; Marrocco, K.; Lechner, E.; Berry, B.; Brault, V.; Hemmer, O.; Kretsch, T.; Richards, K.E.; Genschik, P.; et al. F-box-like domain in the Polerovirus protein P0 is required for silencing suppressor function. Proc. Natl. Acad. Sci. USA 2006, 103, 1994–1999. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.D.; Empleo, R.; Nguyen, T.T.V.; Moffett, P.; Sacco, M.A. Elicitation of hypersensitive responses in Nicotiana glutinosa by the suppressor of RNA silencing protein P0 from poleroviruses. Mol. Plant Pathol. 2015, 16, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Leiser, R.M.; Ziegler-Graff, V.; Reutenauer, A.; Herrbach, E.; Lemaire, O.; Guilley, H.; Richards, K.; Jonard, G. Agroinfection as an alternative to insects for infecting plants with beet western yellows luteovirus. Proc. Natl. Acad. Sci. USA 1992, 89, 9136–9140. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.Y.; Choi, S.K.; Palukaitis, P.; Gray, S.M. Agrobacterium-mediated infection of whole plants by yellow dwarf viruses. Virus Res. 2011, 160, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Dong, S.W.; Xiang, H.Y.; Chen, X.R.; Li, D.W.; Yu, J.L.; Han, C.G. Development of three full-length infectious cDNA clones of distinct brassica yellows virus genotypes for Agrobacterium-mediated inoculation. Virus Res. 2015, 197, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.; Maiss, E. Biological properties of Beet mild yellowing virus derived from a full-length cDNA clone. J. Gen. Virol. 2006, 87, 445–449. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Year | Sample No. | GenBank No. | Virus | |||
|---|---|---|---|---|---|---|---|
| MaYMV | MCMV | SCMV | SRBSDV | ||||
| Yuanjiang, Yunnan | 2013 | YJ-1 | / | + | + | + | − |
| YJ-3 | KU179221 | + | − | + | + | ||
| YJ-4 | KU248490 | + | − | + | + | ||
| YJ-5 | KU291099 | + | + | + | − | ||
| YJ-6 | KU291100 | + | + | + | − | ||
| YJ-7 | KU291101 | + | − | + | − | ||
| Yuanmou, Yunnan | 2014 | YM-1 | KU291102 | + | + | − | − |
| YM-2 | KU291104 | + | + | + | − | ||
| YM-3 | / | + | + | − | − | ||
| YM-4 | / | + | + | + | − | ||
| YM-5 | / | + | + | + | − | ||
| YM-6 | KU291106 | + | + | − | − | ||
| YM-7 | / | + | + | + | − | ||
| YM-8 | / | + | − | − | − | ||
| YM-9 | / | + | − | − | − | ||
| YM-11 | KU291103 | + | + | − | − | ||
| YM-12 | / | + | + | − | − | ||
| YM-14 | / | + | + | − | − | ||
| YM-15 | KU248489 | + | − | − | − | ||
| YM-16 | / | + | − | − | − | ||
| YM-18 | / | + | − | − | − | ||
| YM-19 | / | + | + | − | − | ||
| YM-20 | KU291105 | + | + | − | − | ||
| YM-21 | / | + | + | − | − | ||
| YM-22 | / | + | + | − | − | ||
| Changshun, Guizhou | 2014 | Csh-1 | KU291107 | + | − | − | − |
| Csh-2 | / | + | − | − | − | ||
| Csh-3 | KU291108 | + | − | − | − | ||
| Csh-5 | / | + | − | − | − | ||
| Csh-7 | / | + | − | − | − | ||
| Csh-8 | / | + | − | − | − | ||
| Csh-9 | / | + | − | − | − | ||
| Csh-11 | / | + | − | − | − | ||
| Csh-12 | / | + | − | − | − | ||
| Csh-14 | / | + | − | − | − | ||
| Dushan, Guizhou | 2014 | Dsh-4 | / | + | − | − | − |
| Dsh-5 | / | + | − | − | − | ||
| Genus | Virus Abbreviation-GenBank Number | Genome Size(nt) | Nucleotide Identity (%) | Amino Acid Similarity (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| P0 | P1 | RdRp (P1–P2) | CP (P3) | ORF3a (P3a) | MP (P4) | RTD (P3–P5) | ||||
| Luteovirus | BLRV-NC_003369 | 5964 | 38.0 | NA | 6.8 | 9.3 | 49.2 | 34.9 | 46.5 | 30.7 |
| Luteovirus | SbDV-NC_003056 | 5853 | 36.8 | NA | 7.5 | 9.9 | 53.9 | 44.4 | 42.7 | 30.5 |
| Luteovirus | RSDaV-NC_010806 | 5808 | 33.7 | NA | 6.0 | 8.3 | 36.0 | 48.9 | 25.4 | 28.8 |
| Luteovirus | BYDV-KerII-NC_021481 | 5736 | 36.2 | NA | 8.2 | 8.6 | 45.0 | 35.6 | 28.9 | 31.5 |
| Luteovirus | BYDV-PAS-NC_002160 | 5695 | 36.2 | NA | 11.1 | 8.1 | 42.8 | 35.6 | 27.5 | 33.4 |
| Luteovirus | BYDV-kerIII-KC559092 | 4625 | 38.1 | NA | 10.9 | 10.1 | 41.7 | 37.8 | 23.0 | 31.7 |
| Luteovirus | BYDV-PAV-NC_004750 | 5677 | 37.2 | NA | 8.5 | 8.4 | 43.8 | 35.6 | 26.1 | 33.1 |
| Luteovirus | BYDV-MAV-NC_003680 | 5273 | 37.4 | NA | 9.1 | 8.4 | 44.6 | 33.3 | 25.3 | 32.4 |
| Polerovirus | BMYV-NC_003491 | 5722 | 50.3 | 20.1 | 33.2 | 50.8 | 59.9 | 65.6 | 42.8 | 34.8 |
| Polerovirus | BWYV-NC_004756 | 5666 | 50.0 | 20.0 | 32.6 | 49.0 | 60.2 | 53.3 | 39.9 | 32.9 |
| Polerovirus | BChV-NC_002766 | 5776 | 48.8 | 16.9 | 25.7 | 43.6 | 61.7 | 65.6 | 38.2 | 33.0 |
| Polerovirus | SABYV-NC_018571 | 5843 | 49.5 | 19.8 | 26.0 | 41.2 | 59.2 | 55.6 | 42.9 | 39.3 |
| Polerovirus | CLRDV-NC_014545 | 5866 | 50.3 | 18.5 | 30.0 | 48.4 | 62.2 | 55.6 | 44.8 | 38.5 |
| Polerovirus | CABYV-NC_003688 | 5669 | 51.1 | 19.7 | 32.7 | 48.3 | 59.5 | 55.6 | 43.2 | 40.5 |
| Polerovirus | MABYV-NC_010809 | 5674 | 51.6 | 21.5 | 33.7 | 49.4 | 57.9 | 53.3 | 45.5 | 38.5 |
| Polerovirus | CpCSV-NC_008249 | 5900 | 53.0 | 17.4 | 29.4 | 45.2 | 61.7 | 46.7 | 37.0 | 39.9 |
| Polerovirus | PeVYV-NC_015050 | 6244 | 49.1 | 21.0 | 29.7 | 47.7 | 54.8 | 48.9 | 31.6 | 36.6 |
| Polerovirus | TVDV-NC_010732 | 5920 | 47.8 | 20.9 | 31.9 | 49.1 | 53.8 | 48.9 | 31.8 | 32.7 |
| Polerovirus | CtRLV-NC_006265 | 5723 | 47.4 | 13.6 | 29.6 | 46.9 | 44.4 | 51.1 | 27.4 | 29.0 |
| Polerovirus | PLRV-NC_001747 | 5987 | 49.6 | 15.2 | 27.6 | 45.4 | 57.7 | 53.3 | 44.9 | 35.1 |
| Polerovirus | TuYV-NC_003743 | 5641 | 51.3 | 20.1 | 33.2 | 49.9 | 60.9 | 44.4 | 42.2 | 34.9 |
| Polerovirus | CYDV-RPS-NC_002198 | 5662 | 48.5 | 15.9 | 27.8 | 42.9 | 58.7 | 42.2 | 43.5 | 34.1 |
| Polerovirus | CYDV-RPV-NC_004751 | 5723 | 49.1 | 16.9 | 28.6 | 43.0 | 60.7 | 40.0 | 40.1 | 36.4 |
| Polerovirus | ScYLV-NC_000874 | 5899 | 56.3 | 10.2 | 29.7 | 44.4 | 41.0 | 56.8 | 32.0 | 16.7 |
| Polerovirus | MYDV-RMV-NC_021484 | 5612 | 73.0 | 48.3 | 68.3 | 78.8 | 77.7 | 33.3 | 62.5 | 60.8 |
| Enamovirus | PEMV1-NC_003629 | 5706 | 40.3 | 10.6 | 16.7 | 27.8 | 32.4 | NA | NA | 42.7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Jiang, G.; Wu, J.; Liu, Y.; Qian, Y.; Zhou, X. Characterization of a Novel Polerovirus Infecting Maize in China. Viruses 2016, 8, 120. https://doi.org/10.3390/v8050120
Chen S, Jiang G, Wu J, Liu Y, Qian Y, Zhou X. Characterization of a Novel Polerovirus Infecting Maize in China. Viruses. 2016; 8(5):120. https://doi.org/10.3390/v8050120
Chicago/Turabian StyleChen, Sha, Guangzhuang Jiang, Jianxiang Wu, Yong Liu, Yajuan Qian, and Xueping Zhou. 2016. "Characterization of a Novel Polerovirus Infecting Maize in China" Viruses 8, no. 5: 120. https://doi.org/10.3390/v8050120
APA StyleChen, S., Jiang, G., Wu, J., Liu, Y., Qian, Y., & Zhou, X. (2016). Characterization of a Novel Polerovirus Infecting Maize in China. Viruses, 8(5), 120. https://doi.org/10.3390/v8050120