
Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes


Abstract
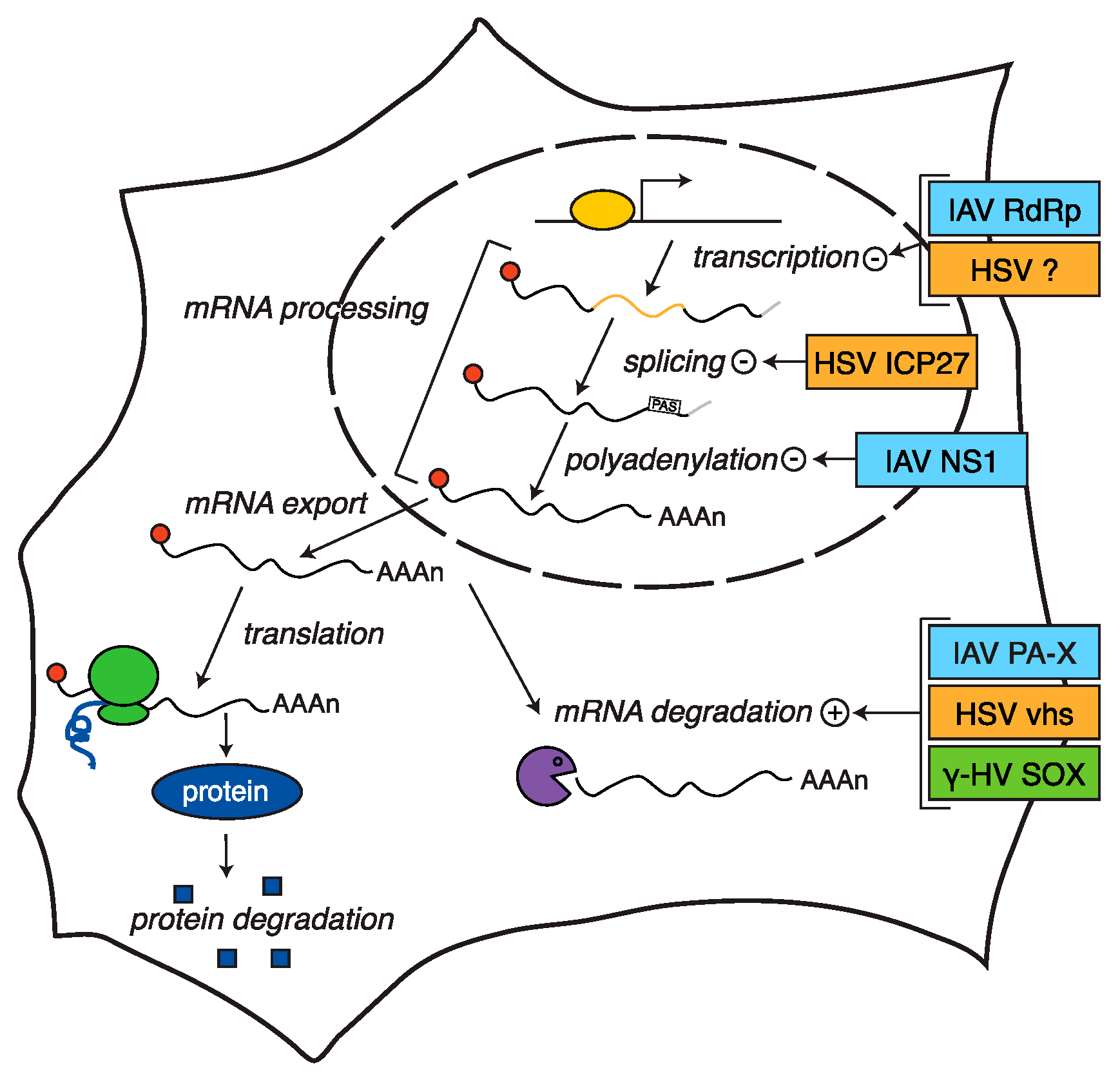
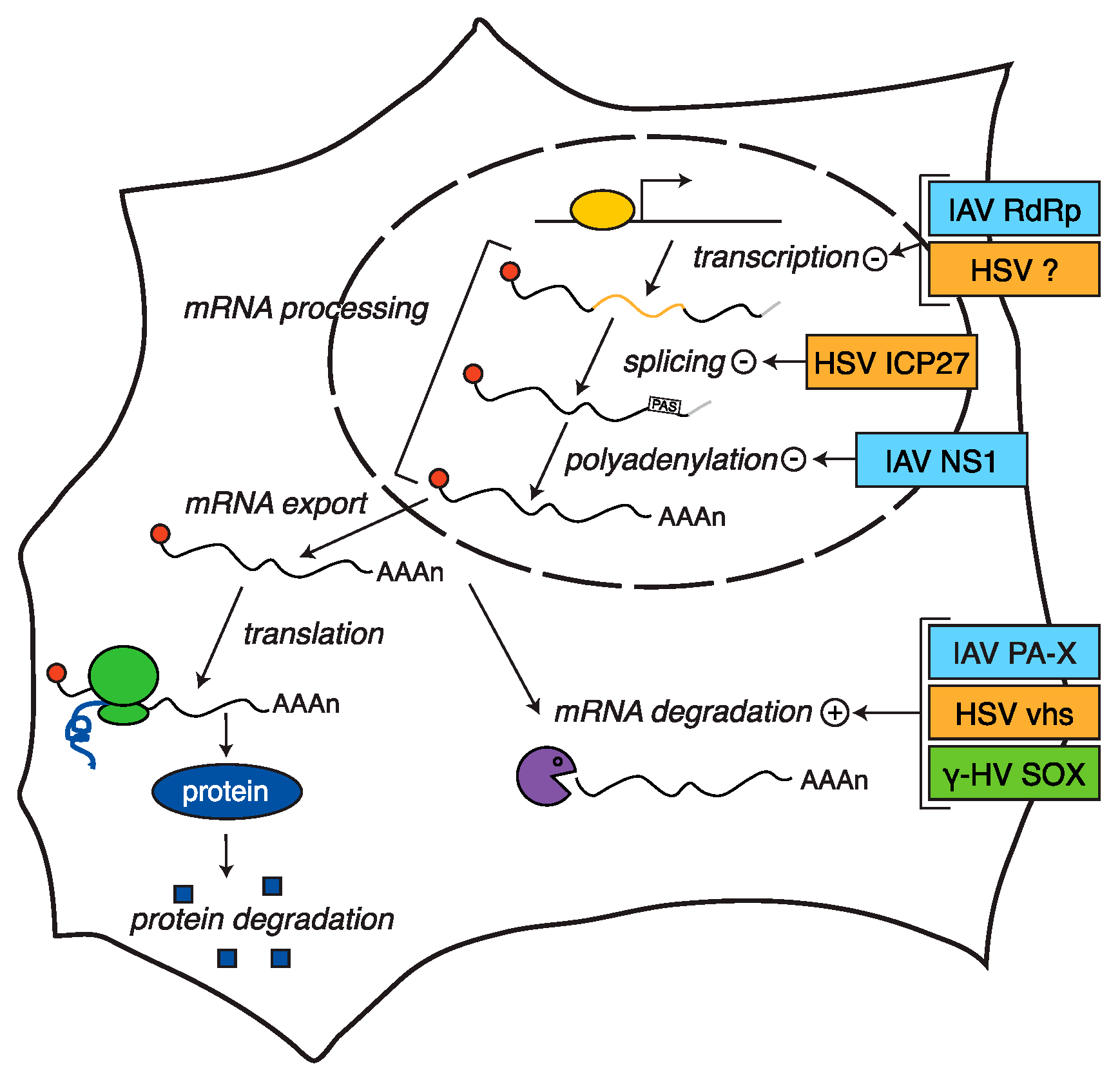
:1. Introduction
2. Host Shutoff in Herpesviruses
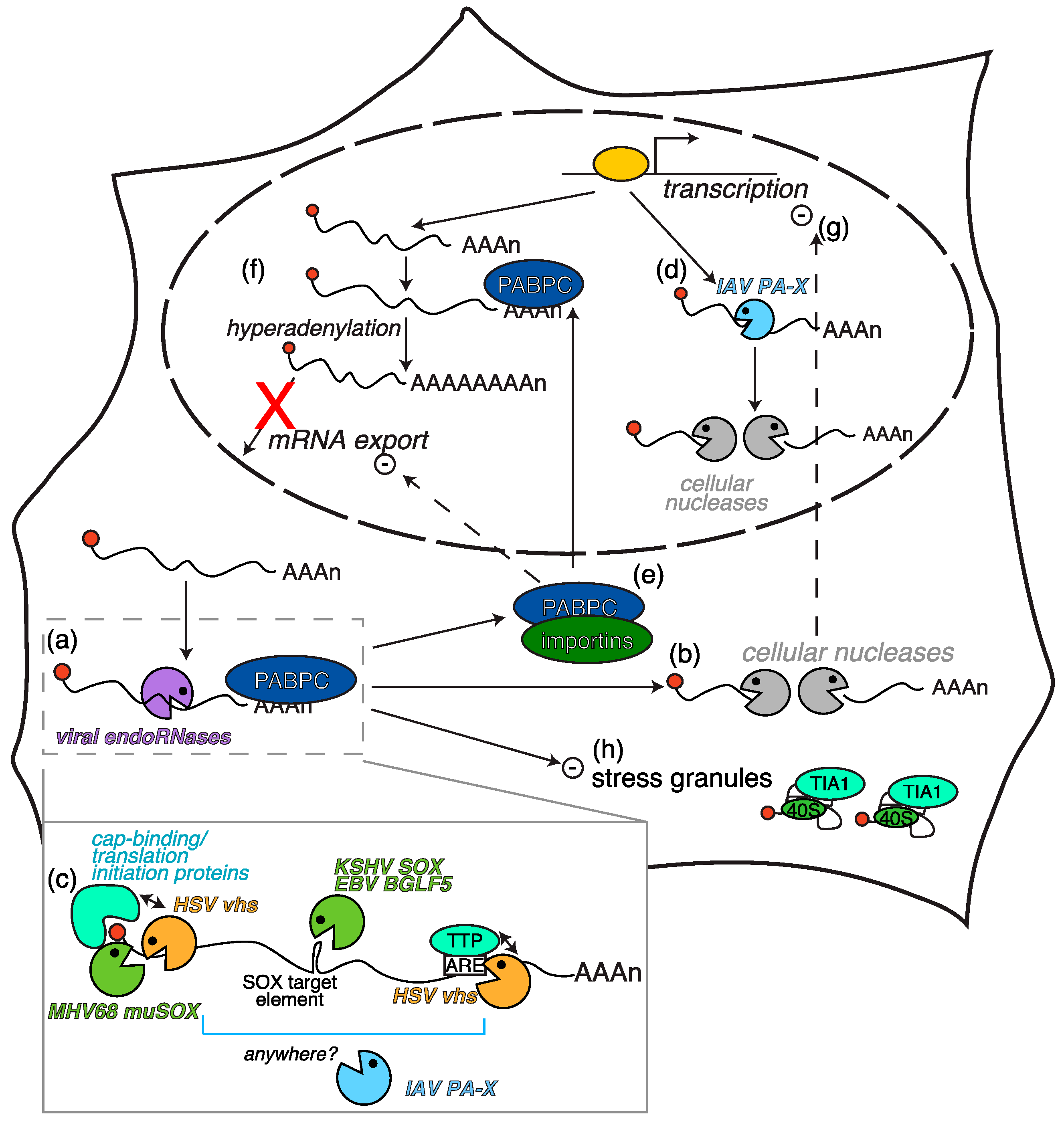
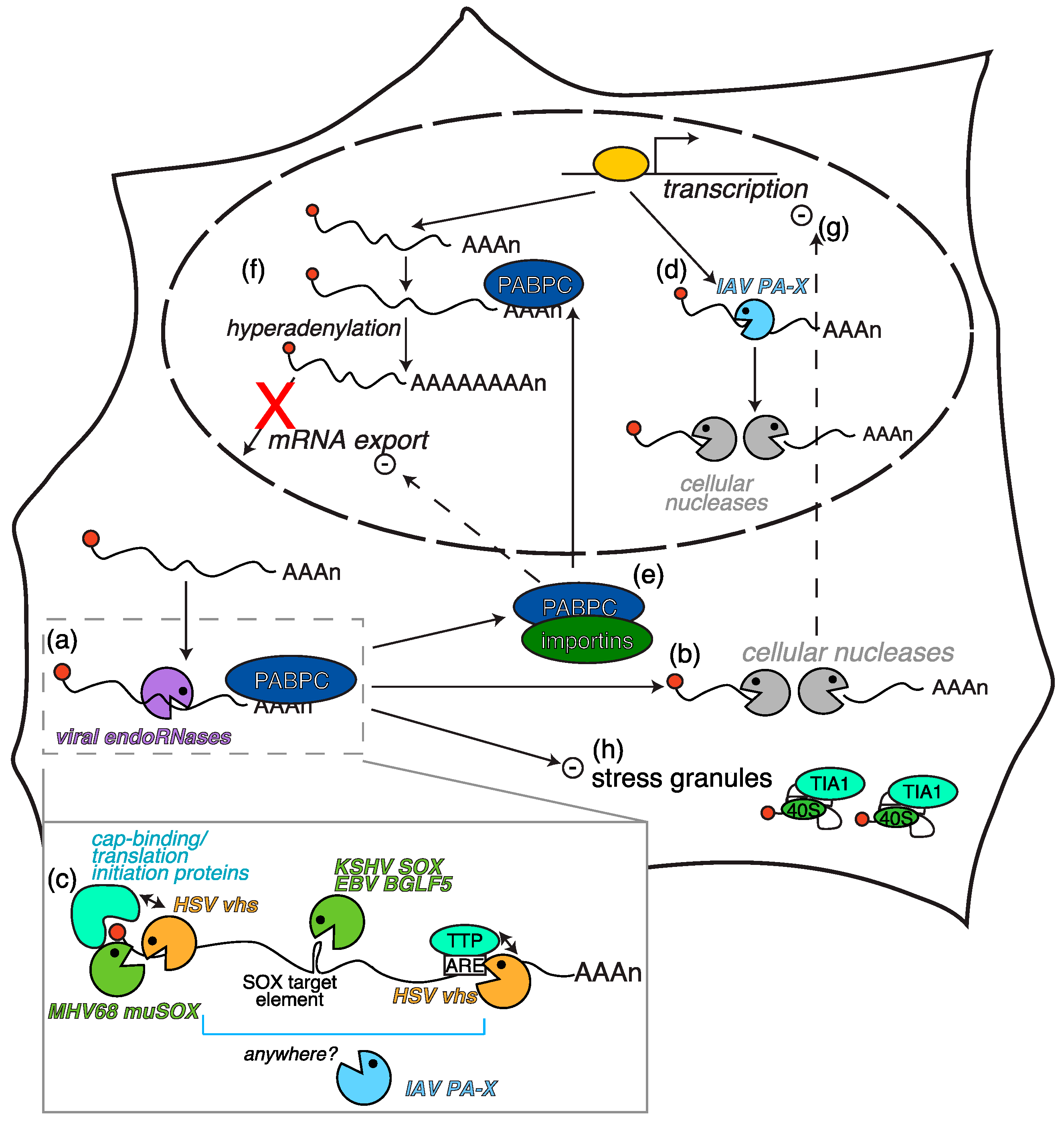
2.1. Herpesvirus-Triggered RNA Degradation: Two Proteins, One Mechanism
2.2. Secondary Consequences of RNA Degradation by Vhs and SOX
2.3. Other Host Shutoff Mechanisms in γ-Herpesviruses?
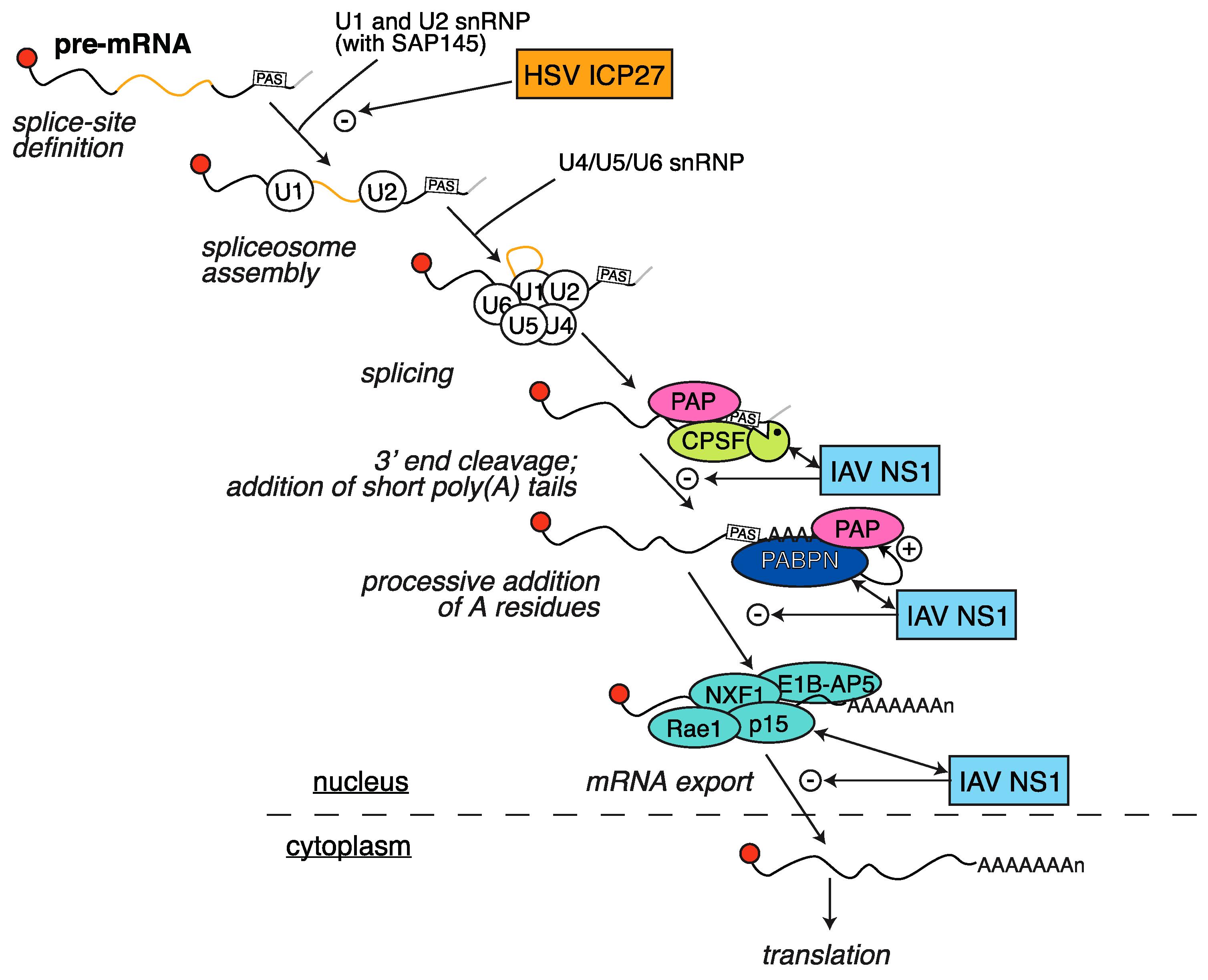
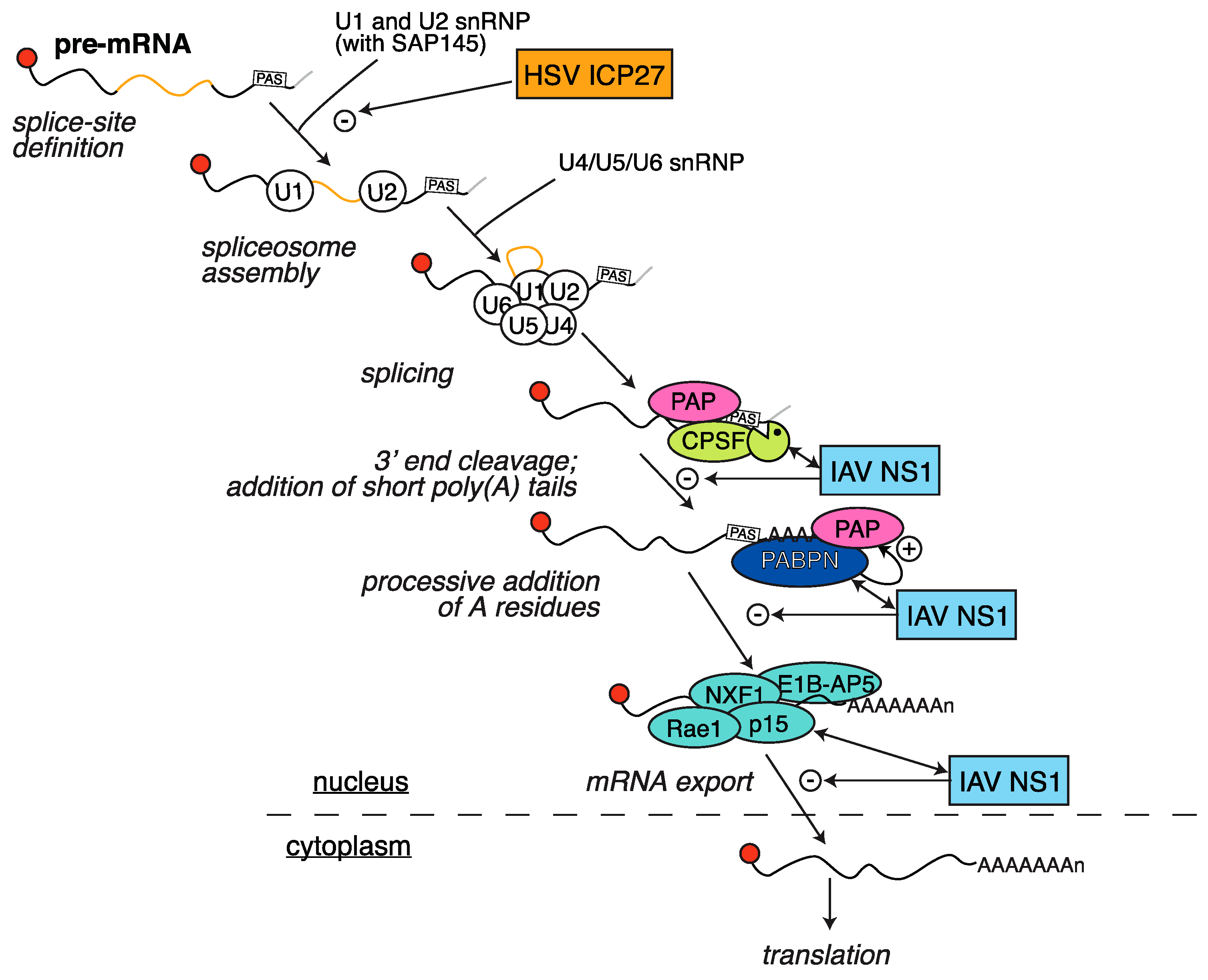
2.4. HSV ICP27 Inhibition of RNA Splicing
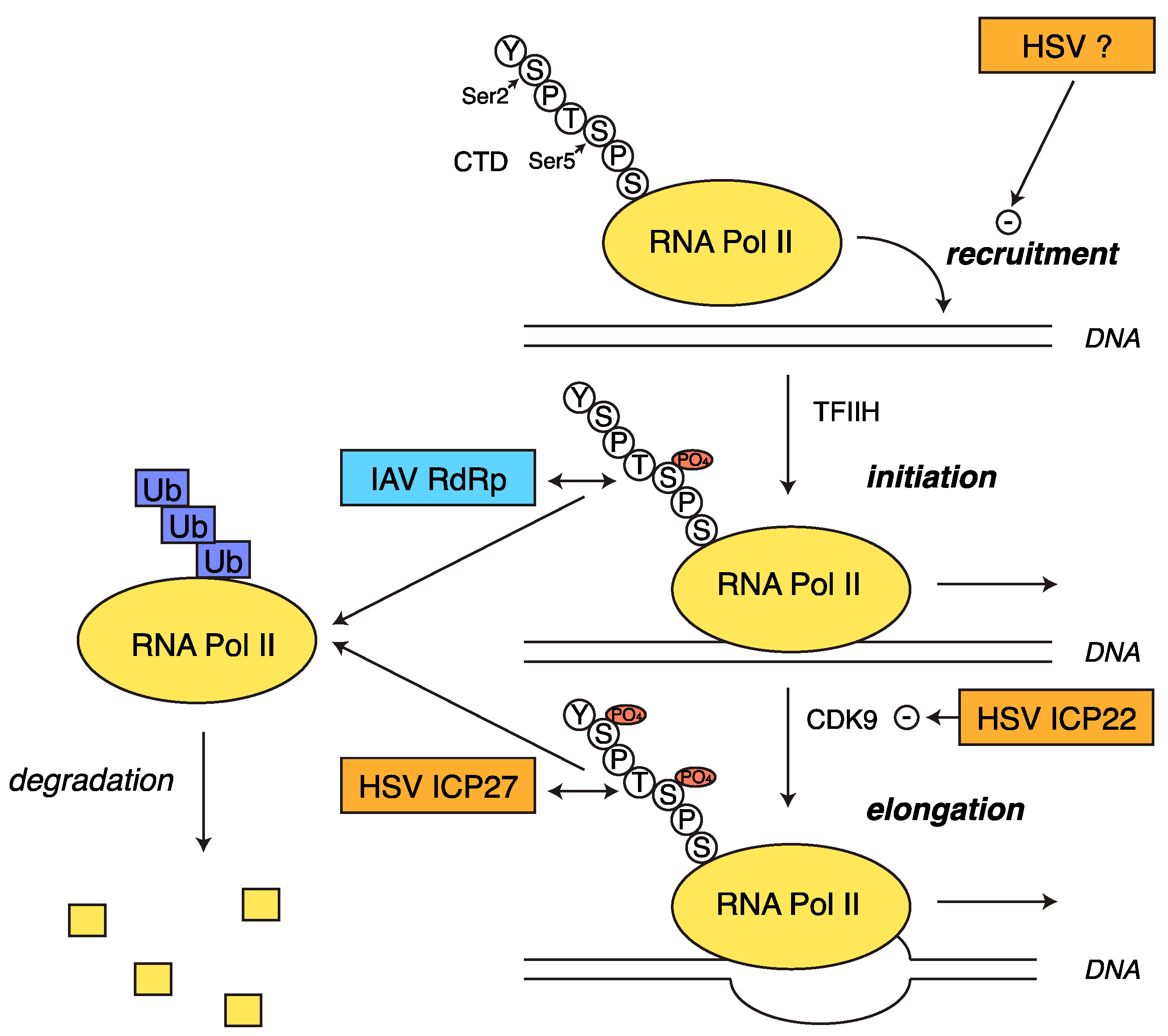
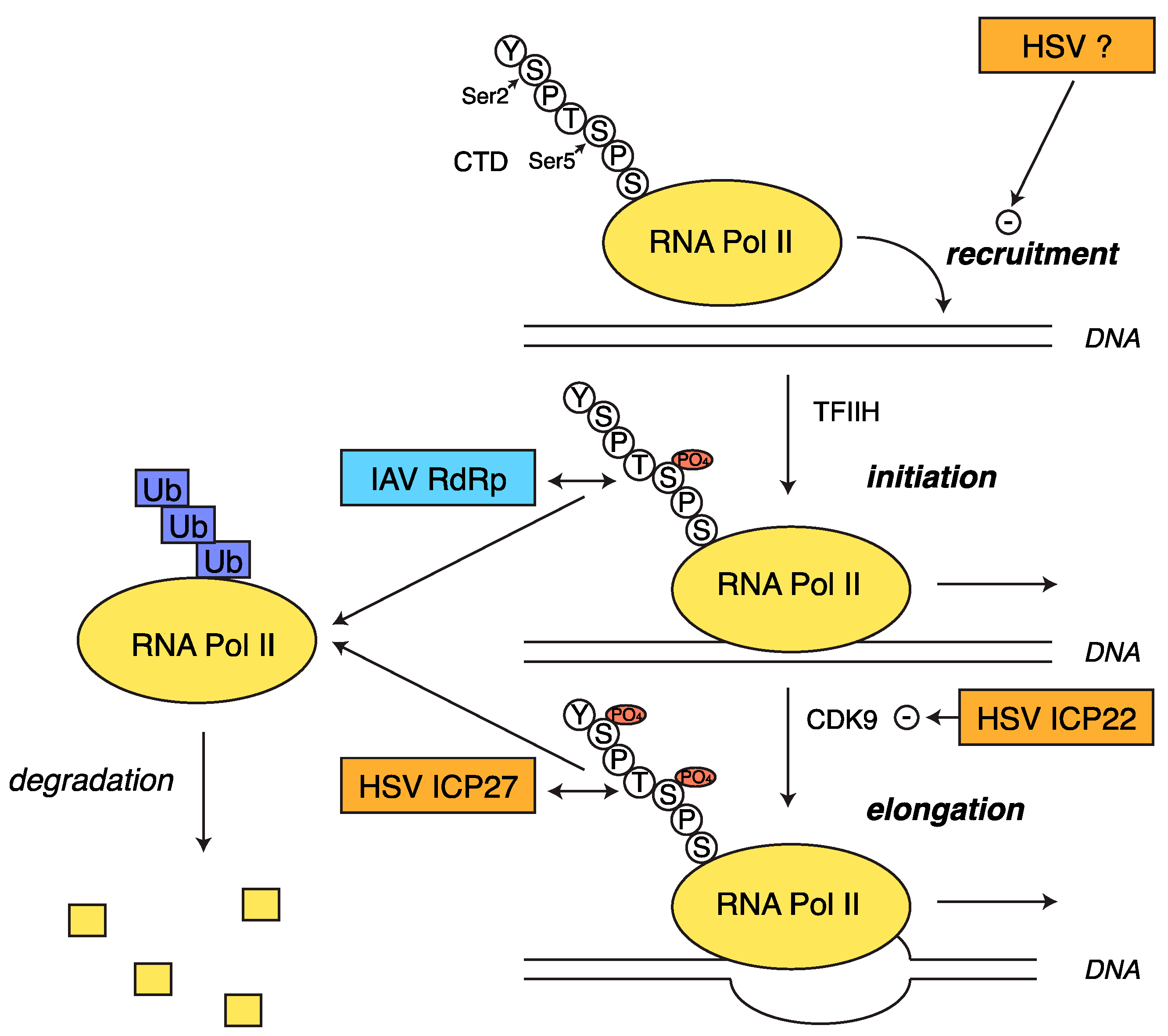
2.5. Transcriptional Shutoff and Termination Defects during HSV-1 Infection
2.6. Herpesviruses: All Hands on Deck to Regulate Host Gene Expression?
3. Host Shutoff in Influenza A Virus
3.1. Inhibition of Polyadenylation and Export of Host mRNAs by NS1
3.2. RNA Pol II Degradation by the Viral RdRp
3.3. Widespread RNA Degradation by PA-X
3.4. Host Shutoff in IAV: One Virus, Multiple Mechanisms, or Different Strains, Different Mechanisms?
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dauber, B.; Saffran, H.A.; Smiley, J.R. The herpes simplex virus 1 virion host shutoff protein enhances translation of viral late mRNAs by preventing mRNA overload. J. Virol. 2014, 88, 9624–9632. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective degradation of host RNA polymerase II transcripts by influenza A virus PA-X host shutoff protein. PLoS Pathog. 2016, 12, e1005427. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef] [PubMed]
- De Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G.; Weber, F. The interferon response circuit: Induction and suppression by pathogenic viruses. Virology 2006, 344, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Tigges, M.A.; Leng, S.; Johnson, D.C.; Burke, R.L. Human herpes simplex virus (HSV)-specific CD8+ CTL clones recognize HSV-2-infected fibroblasts after treatment with IFN-gamma or when virion host shutoff functions are disabled. J. Immunol. 1996, 156, 3901–3910. [Google Scholar] [PubMed]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.H.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [PubMed]
- Read, G.S.; Frenkel, N. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J. Virol. 1983, 46, 498–512. [Google Scholar] [PubMed]
- Kwong, A.D.; Frenkel, N. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 1926–1930. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef]
- Covarrubias, S.; Richner, J.M.; Clyde, K.; Lee, Y.J.; Glaunsinger, B.A. Host shutoff is a conserved phenotype of gammaherpesvirus infection and is orchestrated exclusively from the cytoplasm. J. Virol. 2009, 83, 9554–9566. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Callanan, L.D.; Pesnicak, L.; Krogmann, T.; Cohen, J.I. Varicella-zoster virus (VZV) ORF17 protein induces RNA cleavage and is critical for replication of VZV at 37 degrees C but not 33 degrees C. J. Virol. 2002, 76, 11012–11023. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.W.; Chang, Y.Y.; Wong, M.L.; Lin, J.W.; Chang, T.J. Functional analysis of virion host shutoff protein of pseudorabies virus. Virology 2004, 324, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Elgadi, M.M.; Hayes, C.E.; Smiley, J.R. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 1999, 73, 7153–7164. [Google Scholar] [PubMed]
- Everly, D.N.; Feng, P.; Mian, I.S.; Read, G.S. mRNA degradation by the virion host shutoff (vhs) protein of herpes simplex virus: Genetic and biochemical evidence that vhs is a nuclease. J. Virol. 2002, 76, 8560–8571. [Google Scholar] [CrossRef] [PubMed]
- Schek, N.; Bachenheimer, S.L. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J. Virol. 1985, 55, 601–610. [Google Scholar] [PubMed]
- Strom, T.; Frenkel, N. Effects of herpes simplex virus on mRNA stability. J. Virol. 1987, 61, 2198–2207. [Google Scholar] [PubMed]
- Fenwick, M.L.; McMenamin, M.M. Early virion-associated suppression of cellular protein synthesis by herpes simplex virus is accompanied by inactivation of mRNA. J. Gen. Virol. 1984, 65, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Oroskar, A.A.; Read, G.S. A mutant of herpes simplex virus type 1 exhibits increased stability of immediate-early (alpha) mRNAs. J. Virol. 1987, 61, 604–606. [Google Scholar] [PubMed]
- Oroskar, A.A.; Read, G.S. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J. Virol. 1989, 63, 1897–1906. [Google Scholar] [PubMed]
- Korom, M.; Wylie, K.M.; Morrison, L.A. Selective ablation of virion host shutoff protein RNase activity attenuates herpes simplex virus 2 in mice. J. Virol. 2008, 82, 3642–3653. [Google Scholar] [CrossRef] [PubMed]
- Strelow, L.I.; Leib, D.A. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J. Virol. 1995, 69, 6779–6786. [Google Scholar] [PubMed]
- Smith, T.J.; Ackland-Berglund, C.E.; Leib, D.A. Herpes simplex virus virion host shutoff (vhs) activity alters periocular disease in mice. J. Virol. 2000, 74, 3598–3604. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Morrison, L.A.; Leib, D.A. Pathogenesis of herpes simplex virus type 2 virion host shutoff (vhs) mutants. J. Virol. 2002, 76, 2054–2061. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murphy, J.A.; Duerst, R.J.; Smith, T.J.; Morrison, L.A. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J. Virol. 2003, 77, 9337–9345. [Google Scholar] [CrossRef] [PubMed]
- Pasieka, T.J.; Lu, B.; Crosby, S.D.; Wylie, K.M.; Morrison, L.A.; Alexander, D.E.; Menachery, V.D.; Leib, D.A. Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J. Virol. 2008, 82, 5527–5535. [Google Scholar] [CrossRef] [PubMed]
- Duerst, R.J.; Morrison, L.A. Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness. Virology 2004, 322, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Buisson, M.; Géoui, T.; Flot, D.; Tarbouriech, N.; Ressing, M.E.; Wiertz, E.J.; Burmeister, W.P. A bridge crosses the active-site canyon of the Epstein-Barr virus nuclease with DNase and RNase activities. J. Mol. Biol. 2009, 391, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Bagnéris, C.; Briggs, L.C.; Savva, R.; Ebrahimi, B.; Barrett, T.E. Crystal structure of a KSHV-SOX-DNA complex: Insights into the molecular mechanisms underlying DNase activity and host shutoff. Nucleic Acids Res. 2011, 39, 5744–5756. [Google Scholar] [CrossRef] [PubMed]
- Dahlroth, S.-L.; Gurmu, D.; Schmitzberger, F.; Engman, H.; Haas, J.; Erlandsen, H.; Nordlund, P. Crystal structure of the shutoff and exonuclease protein from the oncogenic Kaposi’s sarcoma-associated herpesvirus. FEBS J. 2009, 276, 6636–6645. [Google Scholar] [CrossRef] [PubMed]
- Bujnicki, J.M.; Rychlewski, L. The herpesvirus alkaline exonuclease belongs to the restriction endonuclease PD-(D/E)XK superfamily: Insight from molecular modeling and phylogenetic analysis. Virus Genes 2001, 22, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Chavez, L.; Ganem, D. The exonuclease and host shutoff functions of the SOX protein of Kaposi’s sarcoma-associated herpesvirus are genetically separable. J. Virol. 2005, 79, 7396–7401. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, S.; Gaglia, M.M.; Kumar, G.R.; Wong, W.; Jackson, A.O.; Glaunsinger, B.A. Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the mammalian exonuclease Xrn1. PLoS Pathog. 2011, 7, e1002339. [Google Scholar] [CrossRef] [PubMed]
- Richner, J.M.; Clyde, K.; Pezda, A.C.; Cheng, B.Y.H.; Wang, T.; Kumar, G.R.; Covarrubias, S.; Coscoy, L.; Glaunsinger, B. Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog. 2011, 7, e1002150. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.H.J.; Ressing, M.E.; Rowe, M. The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 2008, 82, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Burmeister, W.P.; Boer, I.G.J.; van Leeuwen, D.; Buisson, M.; Gorbalenya, A.E.; Wiertz, E.J.H.J.; Ressing, M.E. The “bridge” in the Epstein-Barr virus alkaline exonuclease protein BGLF5 contributes to shutoff activity during productive infection. J. Virol. 2012, 86, 9175–9187. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, E.; Clyde, K.; Yeasmin, R.; Krug, L.T.; Burlingame, A.; Coscoy, L.; Glaunsinger, B. Gammaherpesviral gene expression and virion composition are broadly controlled by accelerated mRNA degradation. PLoS Pathog. 2014, 10, e1003882. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, M.; Griffin, B.D.; Berkhoff, E.G.; van Leeuwen, D.; Boer, I.G.J.; Buisson, M.; Hartgers, F.C.; Burmeister, W.P.; Wiertz, E.J.; Ressing, M.E. EBV lytic-phase protein BGLF5 contributes to TLR9 downregulation during productive infection. J. Immunol. 2011, 186, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, M.; Gram, A.M.; Boer, I.G.J.; Geerdink, R.J.; Lindenbergh, M.F.S.; Lebbink, R.J.; Wiertz, E.J.; Ressing, M.E. Silencing the shutoff protein of Epstein-Barr virus in productively infected B cells points to (innate) targets for immune evasion. J. Gen. Virol. 2015, 96, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Ressing, M.E.; Horst, D.; Griffin, B.D.; Tellam, J.; Zuo, J.; Khanna, R.; Rowe, M.; Wiertz, E.J.H.J. Epstein-Barr virus evasion of CD8+ and CD4+ T cell immunity via concerted actions of multiple gene products. Semin. Cancer Biol. 2008, 18, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Covarrubias, S.; Wong, W.; Glaunsinger, B.A. A common strategy for host RNA degradation by divergent viruses. J. Virol. 2012, 86, 9527–9530. [Google Scholar] [CrossRef] [PubMed]
- Shiflett, L.A.; Read, G.S. mRNA decay during herpes simplex virus (HSV) infections: Mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (vhs) protein. J. Virol. 2013, 87, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Esclatine, A.; Zhang, W.; Roizman, B. The stress-inducible immediate-early responsive gene IEX-1 is activated in cells infected with herpes simplex virus 1, but several viral mechanisms, including 3′ degradation of its RNA, preclude expression of the gene. J. Virol. 2003, 77, 6178–6187. [Google Scholar] [CrossRef] [PubMed]
- Esclatine, A.; Taddeo, B.; Roizman, B. The UL41 protein of herpes simplex virus mediates selective stabilization or degradation of cellular mRNAs. Proc. Natl. Acad. Sci. USA 2004, 101, 18165–18170. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Taddeo, B.; Roizman, B. Tristetraprolin recruits the herpes simplex virion host shutoff RNase to AU-rich elements in stress response mRNAs to enable their cleavage. J. Virol. 2015, 89, 5643–5650. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Everly, D.N.; Read, G.S. mRNA decay during herpesvirus infections: Interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 2001, 75, 10272–10280. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Everly, D.N.; Read, G.S. mRNA decay during herpes simplex virus (HSV) infections: Protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J. Virol. 2005, 79, 9651–9664. [Google Scholar] [CrossRef] [PubMed]
- Page, H.G.; Read, G.S. The virion host shutoff endonuclease (UL41) of herpes simplex virus interacts with the cellular cap-binding complex eIF4F. J. Virol. 2010, 84, 6886–6890. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Saffran, H.A.; Smiley, J.R. The vhs1 mutant form of herpes simplex virus virion host shutoff protein retains significant internal ribosome entry site-directed RNA cleavage activity. J. Virol. 2001, 75, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Sarma, N.; Agarwal, D.; Shiflett, L.A.; Read, G.S. Small interfering RNAs that deplete the cellular translation factor eIF4H impede mRNA degradation by the virion host shutoff protein of herpes simplex virus. J. Virol. 2008, 82, 6600–6609. [Google Scholar] [CrossRef] [PubMed]
- Von Roretz, C.; Marco, S.D.; Mazroui, R.; Gallouzi, I.E. Turnover of AU-rich-containing mRNAs during stress: A matter of survival. Wiley Interdiscip. Rev. RNA 2011, 2, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Schott, J.; Stoecklin, G. Networks controlling mRNA decay in the immune system. Wiley Interdiscip. Rev. RNA 2010, 1, 432–456. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Taddeo, B.; Roizman, B. The nuclear-cytoplasmic shuttling of virion host shutoff RNase is enabled by pUL47 and an Embedded nuclear export signal and defines the sites of degradation of AU-rich and stable cellular mRNAs. J. Virol. 2013, 87, 13569–13578. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Rycroft, C.H.; Glaunsinger, B.A. Transcriptome-wide cleavage site mapping on cellular mRNAs reveals features underlying sequence-specific cleavage by the viral ribonuclease SOX. PLoS Pathog. 2015, 11, e1005305. [Google Scholar] [CrossRef] [PubMed]
- Clyde, K.; Glaunsinger, B.A. Deep sequencing reveals direct targets of gammaherpesvirus-induced mRNA decay and suggests that multiple mechanisms govern cellular transcript escape. PLoS ONE 2011, 6, e19655. [Google Scholar] [CrossRef] [PubMed]
- Smibert, C.A.; Popova, B.; Xiao, P.; Capone, J.P.; Smiley, J.R. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J. Virol. 1994, 68, 2339–2346. [Google Scholar] [PubMed]
- Lam, Q.; Smibert, C.A.; Koop, K.E.; Lavery, C.; Capone, J.P.; Weinheimer, S.P.; Smiley, J.R. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 1996, 15, 2575–2581. [Google Scholar] [PubMed]
- Knez, J.; Bilan, P.T.; Capone, J.P. A single amino acid substitution in herpes simplex virus type 1 VP16 inhibits binding to the virion host shutoff protein and is incompatible with virus growth. J. Virol. 2003, 77, 2892–2902. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Sciortino, M.T.; Zhang, W.; Roizman, B. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. USA 2007, 104, 12163–12168. [Google Scholar] [CrossRef] [PubMed]
- Sciortino, M.T.; Taddeo, B.; Giuffrè-Cuculletto, M.; Medici, M.A.; Mastino, A.; Roizman, B. Replication-competent herpes simplex virus 1 isolates selected from cells transfected with a bacterial artificial chromosome DNA lacking only the UL49 gene vary with respect to the defect in the UL41 gene encoding host shutoff RNase. J. Virol. 2007, 81, 10924–10932. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Zhang, W.; Roizman, B. The herpes simplex virus host shutoff RNase degrades cellular and viral mRNAs made before infection but not viral mRNA made after infection. J. Virol. 2013, 87, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Taddeo, B.; Zhang, W.; Roizman, B. Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc. Natl. Acad. Sci. USA 2013, 110, E1669–E1675. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Mehl-Lautscham, A.M.; Bannert, H.; Delecluse, H.J. The Epstein-Barr virus protein kinase BGLF4 and the exonuclease BGLF5 have opposite effects on the regulation of viral protein production. J. Virol. 2009, 83, 10877–10891. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Bannert, H.; Lips, H.; Müller-Lantzsch, N.; Delecluse, H.J. The Epstein-Barr virus alkaline exonuclease BGLF5 serves pleiotropic functions in virus replication. J. Virol. 2009, 83, 4952–4962. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. KSHV and the pathogenesis of Kaposi sarcoma: Listening to human biology and medicine. J. Clin. Investig. 2010, 120, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Casper, C. Human herpesvirus 8-associated neoplasms: The roles of viral replication and antiviral treatment. Curr. Opin. Infect. Dis. 2011, 24, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Highly selective escape from KSHV-mediated host mRNA shutoff and its implications for viral pathogenesis. J. Exp. Med. 2004, 200, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Asou, H.; Said, J.W.; Yang, R.; Munker, R.; Park, D.J.; Kamada, N.; Koeffler, H.P. Mechanisms of growth control of Kaposi’s sarcoma–associated herpes virus–associated primary effusion lymphoma cells. Blood 1998, 91, 2475–2481. [Google Scholar] [PubMed]
- Leger-Ravet, M.B.; Peuchmaur, M.; Devergne, O.; Audouin, J.; Raphael, M.; Damme, J.V.; Galanaud, P.; Diebold, J.; Emilie, D. Interleukin-6 gene expression in Castleman’s disease. Blood 1991, 78, 2923–2930. [Google Scholar] [PubMed]
- Hutin, S.; Lee, Y.; Glaunsinger, B.A. An RNA element in human interleukin 6 confers escape from degradation by the gammaherpesvirus SOX protein. J. Virol. 2013, 87, 4672–4682. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Hutin, S.; Marigold, O.; Li, K.H.; Burlingame, A.; Glaunsinger, B.A. A ribonucleoprotein complex protects the interleukin-6 mRNA from degradation by distinct herpesviral endonucleases. PLoS Pathog. 2015, 11, e1004899. [Google Scholar] [CrossRef] [PubMed]
- Saffran, H.A.; Pare, J.M.; Corcoran, J.A.; Weller, S.K.; Smiley, J.R. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 2007, 8, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Duguay, B.A.; Saffran, H.A.; Ponomarev, A.; Duley, S.A.; Eaton, H.E.; Smiley, J.R. Elimination of mitochondrial DNA is not required for herpes simplex virus 1 replication. J. Virol. 2014, 88, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Shao, L.; Bronstein, J.C.; Weber, P.C.; Weller, S.K. The product of a 1.9-kb mRNA which overlaps the HSV-1 alkaline nuclease gene (UL12) cannot relieve the growth defects of a null mutant. Virology 1996, 215, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.B.; Antoku, S.; Weller, S.K. The UL12.5 gene product of herpes simplex virus type 1 exhibits nuclease and strand exchange activities but does not localize to the nucleus. J. Virol. 2004, 78, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.A.; Saffran, H.A.; Duguay, B.A.; Smiley, J.R. Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. J. Virol. 2009, 83, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Duguay, B.A.; Smiley, J.R. Mitochondrial nucleases ENDOG and EXOG participate in mitochondrial DNA depletion initiated by herpes simplex virus 1 UL12.5. J. Virol. 2013, 87, 11787–11797. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Goldstein, J.N.; Weller, S.K. The product of the UL12.5 gene of herpes simplex virus type 1 is not essential for lytic viral growth and is not specifically associated with capsids. Virology 2002, 298, 248–257. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Glaunsinger, B.A. Aberrant herpesvirus-induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol. 2009, 7, e1000107. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.R.; Glaunsinger, B.A. Nuclear import of cytoplasmic poly(A) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol. Cell. Biol. 2010, 30, 4996–5008. [Google Scholar] [CrossRef] [PubMed]
- Afonina, E.; Stauber, R.; Pavlakis, G.N. The human poly(A)-binding protein 1 shuttles between the nucleus and the cytoplasm. J. Biol. Chem. 1998, 273, 13015–13021. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Bhattacharjee, R.B.; Bag, J. Expression of poly(A)-binding protein is upregulated during recovery from heat shock in HeLa cells. FEBS J. 2009, 276, 552–570. [Google Scholar] [CrossRef] [PubMed]
- Salaun, C.; MacDonald, A.I.; Larralde, O.; Howard, L.; Lochtie, K.; Burgess, H.M.; Brook, M.; Malik, P.; Gray, N.K.; Graham, S.V. Poly(A)-binding protein 1 partially relocalizes to the nucleus during herpes simplex virus type 1 infection in an ICP27-independent manner and does not inhibit virus replication. J. Virol. 2010, 84, 8539–8548. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.R.; Shum, L.; Glaunsinger, B.A. Importin α-mediated nuclear import of cytoplasmic poly(A) binding protein occurs as a direct consequence of cytoplasmic mRNA depletion. Mol. Cell. Biol. 2011, 31, 3113–3125. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin α. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Emara, M.M.; Johnston, B.P.; Anderson, P.; Hatchette, T.F.; McCormick, C. Influenza A virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog. 2014, 10, e1004217. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, E.; Gilbertson, S.; Alla, R.; Glaunsinger, B. Viral nucleases induce an mRNA degradation-transcription feedback loop in mammalian cells. Cell Host Microbe 2015, 18, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Pelletier, J.; Smiley, J.R. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J. Virol. 2011, 85, 5363–5373. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Hay, T.J.M.; Dauber, B.; Smiley, J.R.; Banfield, B.W. The herpes simplex virus 2 virion-associated ribonuclease vhs interferes with stress granule formation. J. Virol. 2014, 88, 12727–12739. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Beckham, C.J.; Parker, R. P bodies, stress granules, and viral life cycles. Cell Host Microbe 2008, 3, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Pangka, K.R.; Banfield, B.W. Herpes simplex virus 2 infection impacts stress granule accumulation. J. Virol. 2012, 86, 8119–8130. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Y.; Kedersha, N.; Anderson, P.; Emara, M.; Swiderek, K.M.; Moreno, G.T.; Brinton, M.A. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 2002, 76, 11989–12000. [Google Scholar] [CrossRef] [PubMed]
- Chandriani, S.; Ganem, D. Host transcript accumulation during lytic KSHV infection reveals several classes of host responses. PLoS ONE 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; Hsia, D.A.; Izumiya, Y.; Wu, C.Y.; Hsieh, W.P.; Lin, S.F.; Campbell, M.; Lam, K.S.; Luciw, P.A.; et al. Histone demethylase JMJD2A regulates Kaposi’s sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J. Virol. 2011, 85, 3283–3293. [Google Scholar] [CrossRef] [PubMed]
- Sandri-Goldin, R.M. The many roles of the regulatory protein ICP27 during herpes simplex virus infection. Front. Biosci. J. Virtual Libr. 2008, 13, 5241–5256. [Google Scholar] [CrossRef]
- Sandri-Goldin, R.M. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol. 2011, 6, 1261–1277. [Google Scholar] [CrossRef] [PubMed]
- Hardwicke, M.A.; Sandri-Goldin, R.M. The herpes simplex virus regulatory protein ICP27 contributes to the decrease in cellular mRNA levels during infection. J. Virol. 1994, 68, 4797–4810. [Google Scholar] [PubMed]
- Hardy, W.R.; Sandri-Goldin, R.M. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J. Virol. 1994, 68, 7790–7799. [Google Scholar] [PubMed]
- Luo, M.; Reed, R. Splicing is required for rapid and efficient mRNA export in metazoans. Proc. Natl. Acad. Sci. USA 1999, 96, 14937–14942. [Google Scholar] [CrossRef] [PubMed]
- Nojima, T.; Oshiro-Ideue, T.; Nakanoya, H.; Kawamura, H.; Morimoto, T.; Kawaguchi, Y.; Kataoka, N.; Hagiwara, M. Herpesvirus protein ICP27 switches PML isoform by altering mRNA splicing. Nucleic Acids Res. 2009, 37, 6515–6527. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Lalli, J.; Sedlackova-Slavikova, L.; Rice, S.A. Functional comparison of herpes simplex virus 1 (HSV-1) and HSV-2 ICP27 homologs reveals a role for ICP27 in virion release. J. Virol. 2015, 89, 2892–2905. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, A.; Kreivi, J.-P. Splicing inhibition at the level of spliceosome assembly in the presence of herpes simplex virus protein ICP27. Virology 2002, 294, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Wadd, S.E.; Lamond, A.I.; Silverstein, S.J.; Clements, J.B. Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome-associated protein 145 and inhibits splicing prior to the first catalytic step. J. Virol. 2001, 75, 4376–4385. [Google Scholar] [CrossRef] [PubMed]
- Sciabica, K.S.; Dai, Q.J.; Sandri-Goldin, R.M. ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBO J. 2003, 22, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Champion-Arnaud, P.; Reed, R. The prespliceosome components SAP 49 and SAP 145 interact in a complex implicated in tethering U2 snRNP to the branch site. Genes Dev. 1994, 8, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Graveley, B.R. Sorting out the complexity of SR protein functions. RNA 2000, 6, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Roscigno, R.F.; Garcia-Blanco, M.A. SR proteins escort the U4/U6.U5 tri-snRNP to the spliceosome. RNA 1995, 1, 692–706. [Google Scholar] [PubMed]
- Prasad, J.; Colwill, K.; Pawson, T.; Manley, J.L. The protein kinase Clk/Sty directly modulates SR protein activity: Both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol. 1999, 19, 6991–7000. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, A.J.; Erhard, F.; L’Hernault, A.; Bonfert, T.; Schilhabel, M.; Crump, C.; Rosenstiel, P.; Efstathiou, S.; Zimmer, R.; Friedel, C.C.; et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.M.; Newton, A.A. The effects of herpes simplex virus type 1 on cellular DNA-dependent RNA polymerase activities. J. Gen. Virol. 1976, 33, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.K.; Roizman, B. Ribonucleic acid synthesis in cells infected with herpes simplex virus I. Patterns of ribonucleic acid synthesis in productively infected cells. J. Virol. 1969, 4, 36–46. [Google Scholar] [PubMed]
- Abrisch, R.G.; Eidem, T.M.; Yakovchuk, P.; Kugel, J.F.; Goodrich, J.A. Infection by herpes simplex virus 1 causes near-complete loss of RNA polymerase II occupancy on the host cell genome. J. Virol. 2016, 90, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Heidemann, M.; Hintermair, C.; Voß, K.; Eick, D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta 2013, 1829, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.A.; Long, M.C.; Lam, V.; Spencer, C.A. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J. Virol. 1994, 68, 988–1001. [Google Scholar] [PubMed]
- Fraser, K.A.; Rice, S.A. Herpes simplex virus type 1 infection leads to loss of serine-2 phosphorylation on the carboxyl-terminal domain of RNA polymerase II. J. Virol. 2005, 79, 11323–11334. [Google Scholar] [CrossRef] [PubMed]
- Fraser, K.A.; Rice, S.A. Herpes simplex virus immediate-early protein ICP22 triggers loss of serine 2-phosphorylated RNA polymerase II. J. Virol. 2007, 81, 5091–5101. [Google Scholar] [CrossRef] [PubMed]
- Dai-Ju, J.Q.; Li, L.; Johnson, L.A.; Sandri-Goldin, R.M. ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. J. Virol. 2006, 80, 3567–3581. [Google Scholar] [CrossRef] [PubMed]
- Durand, L.O.; Advani, S.J.; Poon, A.P.W.; Roizman, B. The carboxyl-terminal domain of RNA polymerase II is phosphorylated by a complex containing CDK9 and infected-cell protein 22 of herpes simplex virus 1. J. Virol. 2005, 79, 6757–6762. [Google Scholar] [CrossRef] [PubMed]
- Zaborowska, J.; Baumli, S.; Laitem, C.; O’Reilly, D.; Thomas, P.H.; O’Hare, P.; Murphy, S. Herpes simplex virus 1 (HSV-1) ICP22 protein directly interacts with cyclin-dependent kinase (CDK)9 to inhibit RNA polymerase II transcription elongation. PLOS ONE 2014, 9, e107654. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, D.C.; Horng, T.; Medzhitov, R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 2009, 138, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Eick, D.; Bensaude, O. CTD serine-2 plays a critical role in splicing and termination factor recruitment to RNA polymerase II in vivo. Nucleic Acids Res. 2013, 41, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Vasin, A.V.; Temkina, O.A.; Egorov, V.V.; Klotchenko, S.A.; Plotnikova, M.A.; Kiselev, O.I. Molecular mechanisms enhancing the proteome of influenza A viruses: An overview of recently discovered proteins. Virus Res. 2014, 185, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Inglis, S.C. Inhibition of host protein synthesis and degradation of cellular mRNAs during infection by influenza and herpes simplex virus. Mol. Cell. Biol. 1982, 2, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Egorov, A.; Brandt, S.; Sereinig, S.; Romanova, J.; Ferko, B.; Katinger, D.; Grassauer, A.; Alexandrova, G.; Katinger, H.; Muster, T. Transfectant influenza A viruses with long deletions in the NS1 protein grow efficiently in Vero cells. J. Virol. 1998, 72, 6437–6441. [Google Scholar] [PubMed]
- García-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Krug, R.M. The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J. Virol. 1994, 68, 2425–2432. [Google Scholar] [PubMed]
- Fortes, P.; Beloso, A.; Ortín, J. Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO J. 1994, 13, 704–712. [Google Scholar] [PubMed]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′ end formation of cellular pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Choi, E.A.; Shi, Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2011, 2, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Mata, M.A.; Zhang, L.; Fontoura, B.M.A. Nuclear imprisonment: Viral strategies to arrest host mRNA nuclear export. Viruses 2013, 5, 1824–1849. [Google Scholar] [CrossRef] [PubMed]
- Satterly, N.; Tsai, P.L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M.A. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Stutz, F.; Izaurralde, E. The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol. 2003, 13, 319–327. [Google Scholar] [CrossRef]
- Zhang, L.; Das, P.; Schmolke, M.; Manicassamy, B.; Wang, Y.; Deng, X.; Cai, L.; Tu, B.P.; Forst, C.V.; Roth, M.G.; et al. Inhibition of pyrimidine synthesis reverses viral virulence factor-mediated block of mRNA nuclear export. J. Cell Biol. 2012, 196, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.; Pritlove, D.C.; Fodor, E.; Brownlee, G.G. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J. Virol. 1999, 73, 3473–3476. [Google Scholar] [PubMed]
- York, A.; Fodor, E. Biogenesis, assembly, and export of viral messenger ribonucleoproteins in the influenza A virus infected cell. RNA Biol. 2013, 10, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Kainov, D.E.; Müller, K.H.; Theisen, L.L.; Anastasina, M.; Kaloinen, M.; Muller, C.P. Differential effects of NS1 proteins of human pandemic H1N1/2009, avian highly pathogenic H5N1, and low pathogenic H5N2 influenza A viruses on cellular pre-mRNA polyadenylation and mRNA translation. J. Biol. Chem. 2011, 286, 7239–7247. [Google Scholar] [CrossRef] [PubMed]
- Twu, K.Y.; Kuo, R.L.; Marklund, J.; Krug, R.M. The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J. Virol. 2007, 81, 8112–8121. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Steel, J.; Medina, R.A.; Manicassamy, B.; Ye, J.; Hickman, D.; Hai, R.; Schmolke, M.; Lowen, A.C.; Perez, D.R.; et al. Inefficient control of host gene expression by the 2009 pandemic H1N1 influenza A virus NS1 protein. J. Virol. 2010, 84, 6909–6922. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; García-Sastre, A.; Martínez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Domingues, P.; Rajsbaum, R.; Miorin, L.; Schmolke, M.; Hale, B.G.; García-Sastre, A. A single amino acid substitution in the novel H7N9 influenza A virus NS1 protein increases CPSF30 binding and virulence. J. Virol. 2014, 88, 12146–12151. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Ma, L.C.; Xiao, R.; Radvansky, B.; Aramini, J.; Zhao, L.; Marklund, J.; Kuo, R.L.; Twu, K.Y.; Arnold, E.; et al. Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. USA 2008, 105, 13093–13098. [Google Scholar] [CrossRef] [PubMed]
- Spesock, A.; Malur, M.; Hossain, M.J.; Chen, L.M.; Njaa, B.L.; Davis, C.T.; Lipatov, A.S.; York, I.A.; Krug, R.M.; Donis, R.O. The virulence of 1997 H5N1 influenza viruses in the mouse model is increased by correcting a defect in their NS1 proteins. J. Virol. 2011, 85, 7048–7058. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.Y.; Vreede, F.T.; Smith, M.; Engelhardt, O.G.; Fodor, E. Influenza virus inhibits RNA polymerase II elongation. Virology 2006, 351, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, O.G.; Smith, M.; Fodor, E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 2005, 79, 5812–5818. [Google Scholar] [CrossRef] [PubMed]
- Spooner, L.L.R.; Barry, R.D. Participation of DNA-dependent RNA polymerase II in replication of influenza viruses. Nature 1977, 268, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Plotch, S.J.; Bouloy, M.; Ulmanen, I.; Krug, R.M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23, 847–858. [Google Scholar] [CrossRef]
- Rodriguez, A.; Pérez-González, A.; Nieto, A. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J. Virol. 2007, 81, 5315–5324. [Google Scholar] [CrossRef] [PubMed]
- Vreede, F.T.; Chan, A.Y.; Sharps, J.; Fodor, E. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 2010, 396, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Vreede, F.T.; Fodor, E. The role of the influenza virus RNA polymerase in host shut-off. Virulence 2010, 1, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Llompart, C.M.; Nieto, A.; Rodriguez-Frandsen, A. Specific residues of PB2 and PA influenza virus polymerase subunits confer the ability for RNA polymerase II degradation and virus pathogenicity in mice. J. Virol. 2014, 88, 3455–3463. [Google Scholar] [CrossRef] [PubMed]
- Beloso, A.; Martínez, C.; Valcárcel, J.; Santarén, J.F.; Ortín, J. Degradation of cellular mRNA during influenza virus infection: Its possible role in protein synthesis shutoff. J. Gen. Virol. 1992, 73, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.-A.; Wills, N.M.; Xiao, Y.-L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Jagger, B.W.; Wise, H.M.; Nelson, C.C.; Parsawar, K.; Wills, N.M.; Napthine, S.; Taubenberger, J.K.; Digard, P.; Atkins, J.F. Ribosomal frameshifting used in influenza A virus expression occurs within the sequence UCC_UUU_CGU and is in the +1 direction. Open Biol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Bouvier, D.; Crépin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W.H. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Bartlam, M.; Lou, Z.; Chen, S.; Zhou, J.; He, X.; Lv, Z.; Ge, R.; Li, X.; Deng, T.; et al. Crystal structure of an avian influenza polymerase PAN reveals an endonuclease active site. Nature 2009, 458, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Mapping of a region of the PA-X protein of influenza A virus that is important for its shutoff activity. J. Virol. 2015, 89, 8661–8665. [Google Scholar] [CrossRef] [PubMed]
- Desmet, E.A.; Bussey, K.A.; Stone, R.; Takimoto, T. Identification of the N-terminal domain of the influenza virus PA responsible for the suppression of host protein synthesis. J. Virol. 2013, 87, 3108–3118. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, H.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; Seng, L.G.; et al. The 20 amino acids at the C-terminus of PA-X are associated with increased influenza A virus replication and pathogenicity. J. Gen. Virol. 2015, 96, 2036–2049. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A virus protein PA-X contributes to viral growth and suppression of the host antiviral and immune responses. J. Virol. 2015, 89, 6442–6452. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, Y.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; et al. The contribution of PA-X to the virulence of pandemic 2009 H1N1 and highly pathogenic H5N1 avian influenza viruses. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Mo, Y.; Wang, X.; Gu, M.; Hu, Z.; Zhong, L.; Wu, Q.; Hao, X.; Hu, S.; Liu, W.; et al. PA-X decreases the pathogenicity of highly pathogenic H5N1 influenza A virus in avian species by inhibiting virus replication and host response. J. Virol. 2015, 89, 4126–4142. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xu, G.; Sun, Y.; Qi, L.; Wang, J.; Kong, W.; Sun, H.; Pu, J.; Chang, K.C.; Liu, J. PA-X is a virulence factor in avian H9N2 influenza virus. J. Gen. Virol. 2015, 96, 2587–2594. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, X.; Sun, Y.; Liu, Q.; Sun, H.; Xiong, X.; Jiang, M.; He, Q.; Wang, Y.; Pu, J.; Guo, X.; Yang, H.; Liu, J. Truncation of C-terminal 20 amino acids in PA-X contributes to adaptation of swine influenza virus in pigs. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Bavagnoli, L.; Cucuzza, S.; Campanini, G.; Rovida, F.; Paolucci, S.; Baldanti, F.; Maga, G. The novel influenza A virus protein PA-X and its naturally deleted variant show different enzymatic properties in comparison to the viral endonuclease PA. Nucleic Acids Res. 2015, 43, 9405–9417. [Google Scholar] [CrossRef] [PubMed]
- De la Luna, S.; Fortes, P.; Beloso, A.; Ortín, J. Influenza virus NS1 protein enhances the rate of translation initiation of viral mRNAs. J. Virol. 1995, 69, 2427–2433. [Google Scholar] [PubMed]
- Khaperskyy, D.A.; Hatchette, T.F.; McCormick, C. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. 2012, 26, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Jagger, B.W.; Wise, H.M.; Digard, P.; Holmes, E.C.; Taubenberger, J.K. Evolutionary conservation of the PA-X open reading frame in segment 3 of influenza A Virus. J. Virol. 2012, 86, 12411–12413. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; McCormick, C. Timing is everything: Coordinated control of host shutoff by influenza A virus NS1 and PA-X proteins. J. Virol. 2015, 89, 6528–6531. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level of Regulation | Viral Protein | Molecular Activity | Result | Interacting Cellular Proteins |
|---|---|---|---|---|
| Transcription | Unknown—maybe ICP22 and ICP27 | Unknown—maybe dephosphorylation of RNA Pol II? | Inhibition of transcription initiation; inhibition of transcription termination | Unknown—maybe RNA Pol II |
| RNA processing | ICP27 | Inhibits spliceosome formation | Inhibition of splicing; decreased mRNA levels in cytoplasm | SAP145, SRp20, SRPK1 |
| RNA stability | vhs | RNA endonuclease | Degradation of host and viral mRNAs; nuclear relocalization of PABPC | eIF4H, eIF4AI and eIF4AII, TTP |
| Level of Regulation | Viral Protein | Molecular Activity | Result | Interacting Cellular Proteins |
|---|---|---|---|---|
| Transcription | MHV68 muSOX (through feedback mechanism) | Unknown | Inhibition of transcription initiation | Xrn1 (functional interaction) |
| RNA stability | KSHV SOX, MHV68 muSOX, EBV BGLF5 | RNA endonuclease | Degradation of host and viral mRNAs; nuclear relocalization of PABPC | Unknown |
| Level of Regulation | Viral Protein | Molecular Activity | Result | Interacting Cellular Proteins |
|---|---|---|---|---|
| Transcription | RdRP (PA, PB1, PB2) | Ubiquitination and degradation of large subunit of RNA Pol II | Transcription inhibition | RNA Polymerase II |
| RNA processing | NS1 | Inhibition of mRNA 3′ end cleavage by CPSF and PABPN stimulation of poly(A) addition; interaction with nuclear mRNA export machinery | Inhibition of poly(A) tail addition and nuclear export of mRNA | CPSF30 |
| PABPN | ||||
| Nuclear export proteins (NXF1, p15, Rae1, E1B-AP5, Nup98) | ||||
| RNA stability | PA-X | RNA endonuclease | Degradation of host mRNA and ncRNAs; nuclear relocalization of PABPC | Unknown |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivas, H.G.; Schmaling, S.K.; Gaglia, M.M. Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes. Viruses 2016, 8, 102. https://doi.org/10.3390/v8040102
Rivas HG, Schmaling SK, Gaglia MM. Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes. Viruses. 2016; 8(4):102. https://doi.org/10.3390/v8040102
Chicago/Turabian StyleRivas, Hembly G., Summer K. Schmaling, and Marta M. Gaglia. 2016. "Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes" Viruses 8, no. 4: 102. https://doi.org/10.3390/v8040102
APA StyleRivas, H. G., Schmaling, S. K., & Gaglia, M. M. (2016). Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes. Viruses, 8(4), 102. https://doi.org/10.3390/v8040102