
Diverse Strategies Used by Picornaviruses to Escape Host RNA Decay Pathways

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
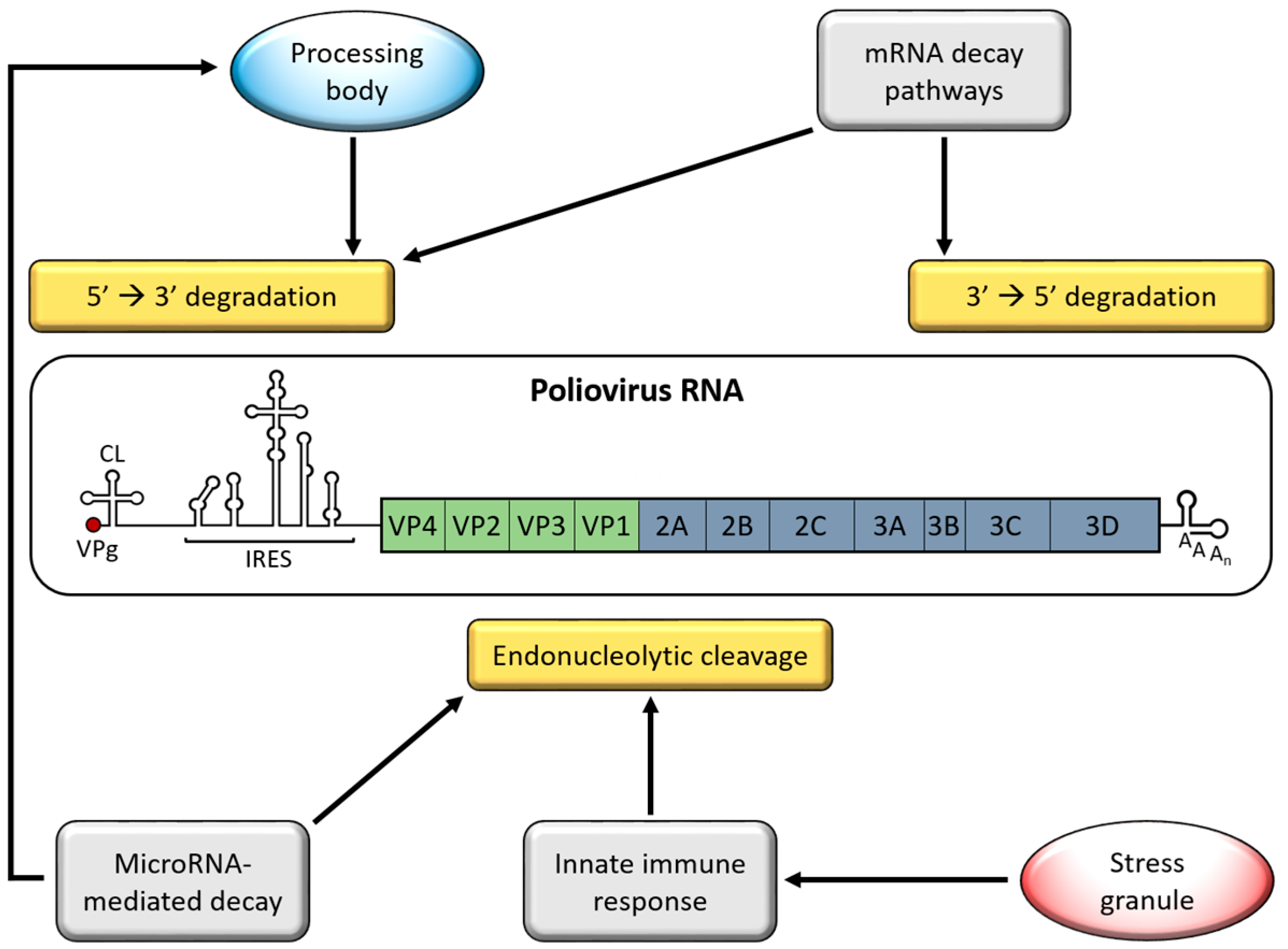
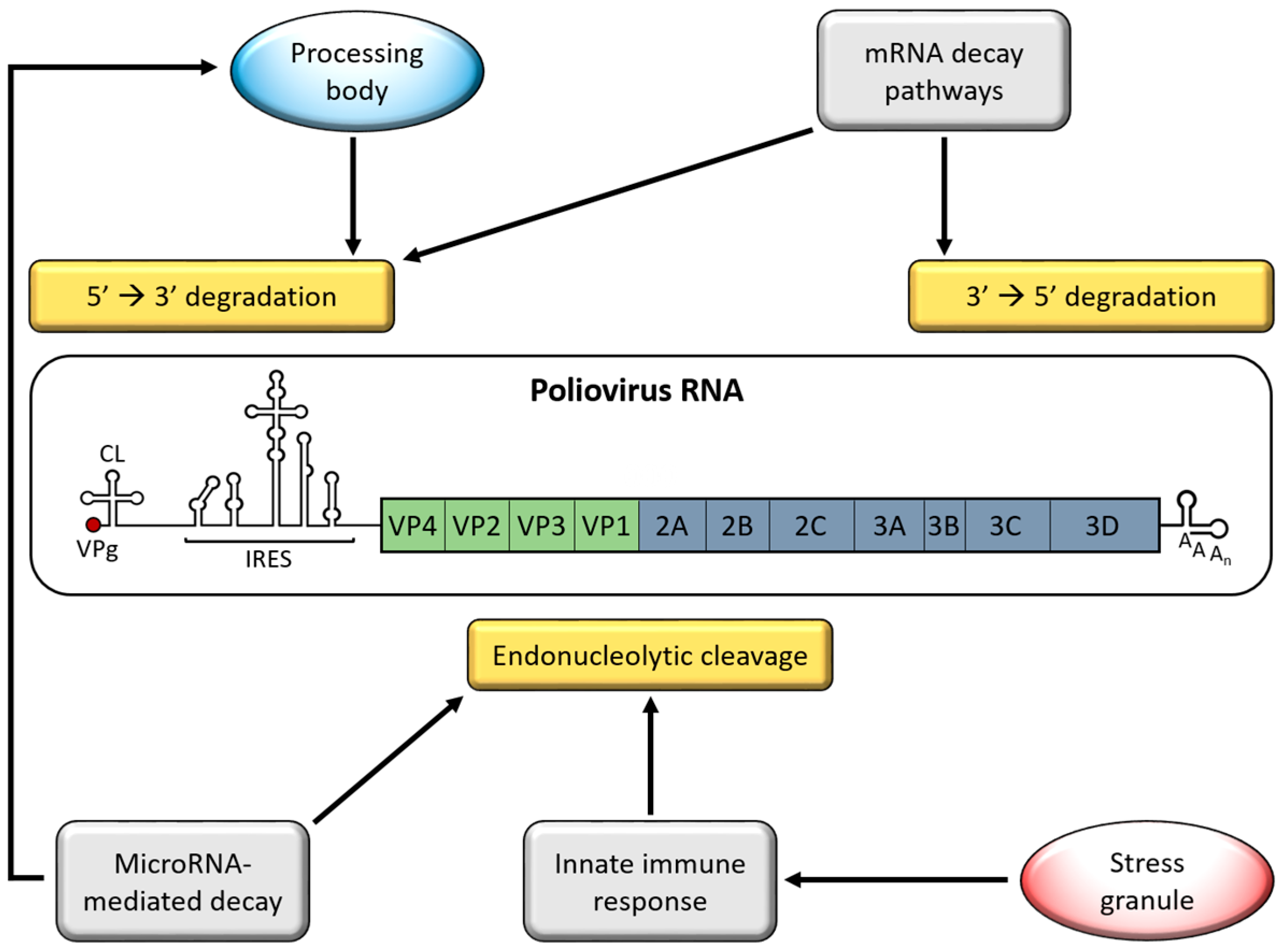
2. Genome Stabilizing Features for Picornaviruses
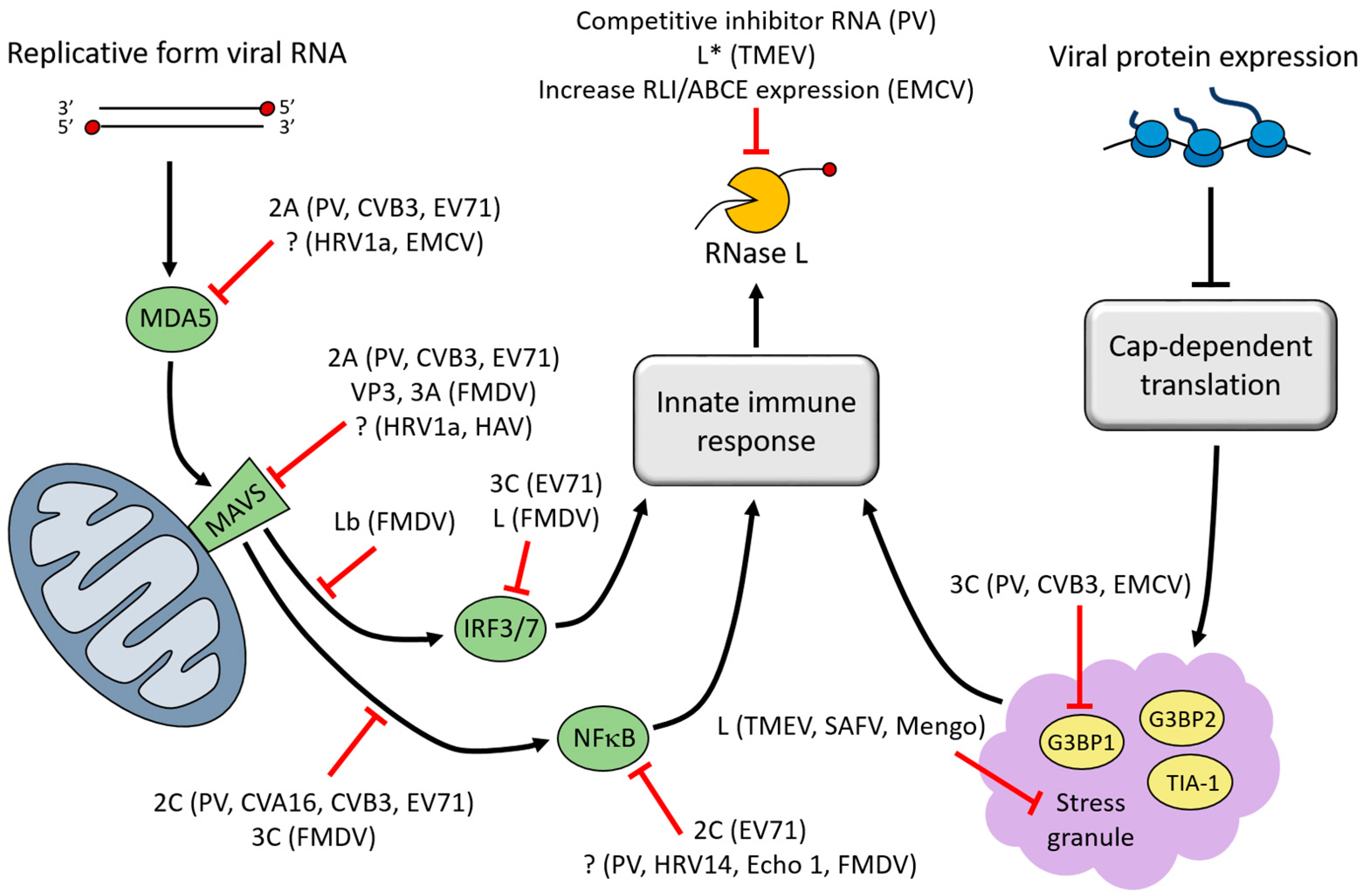
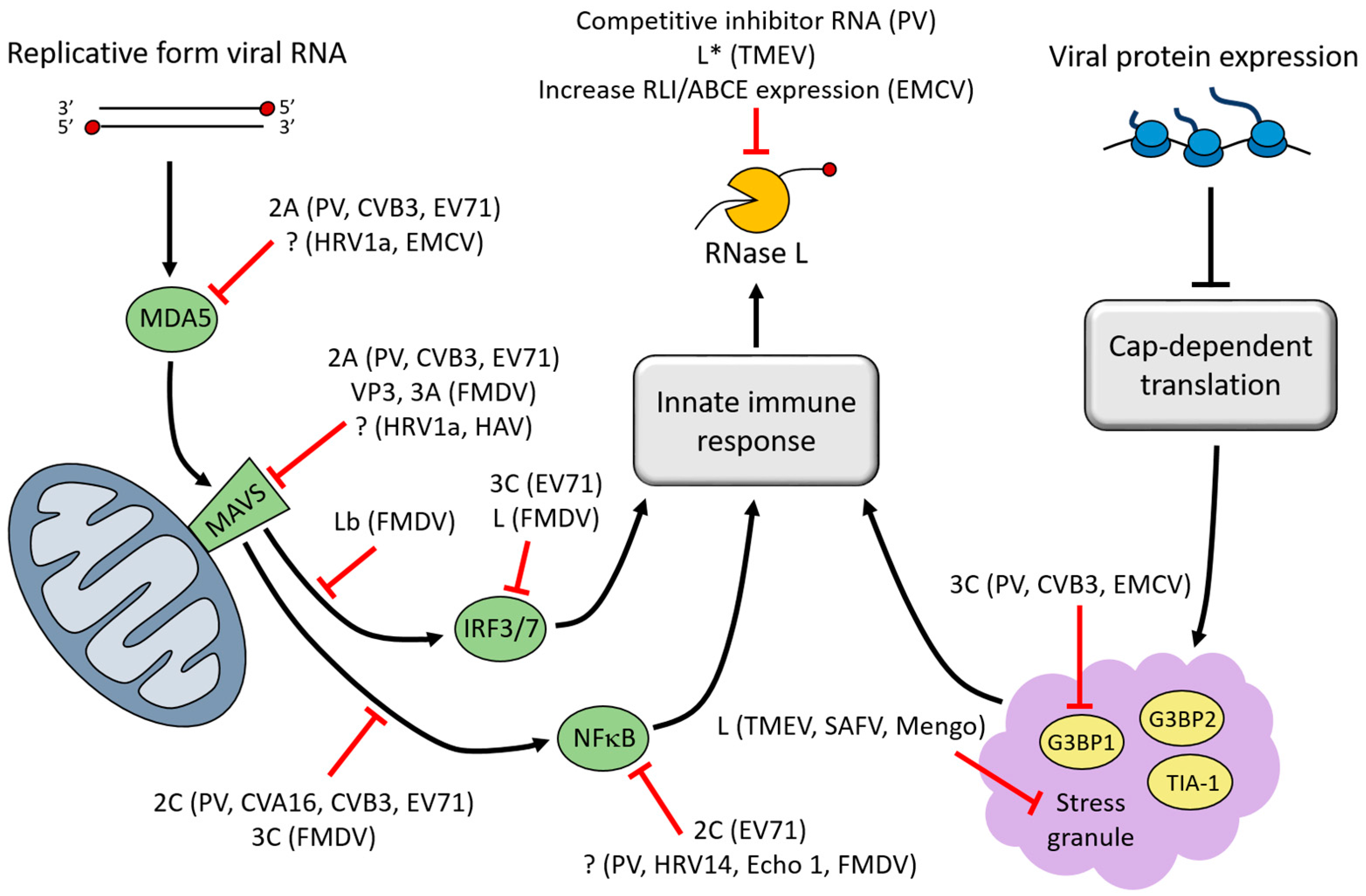
3. Interferon (IFN)-Induced Viral RNA (vRNA) Degradation
4. Stress Granules (SGs)
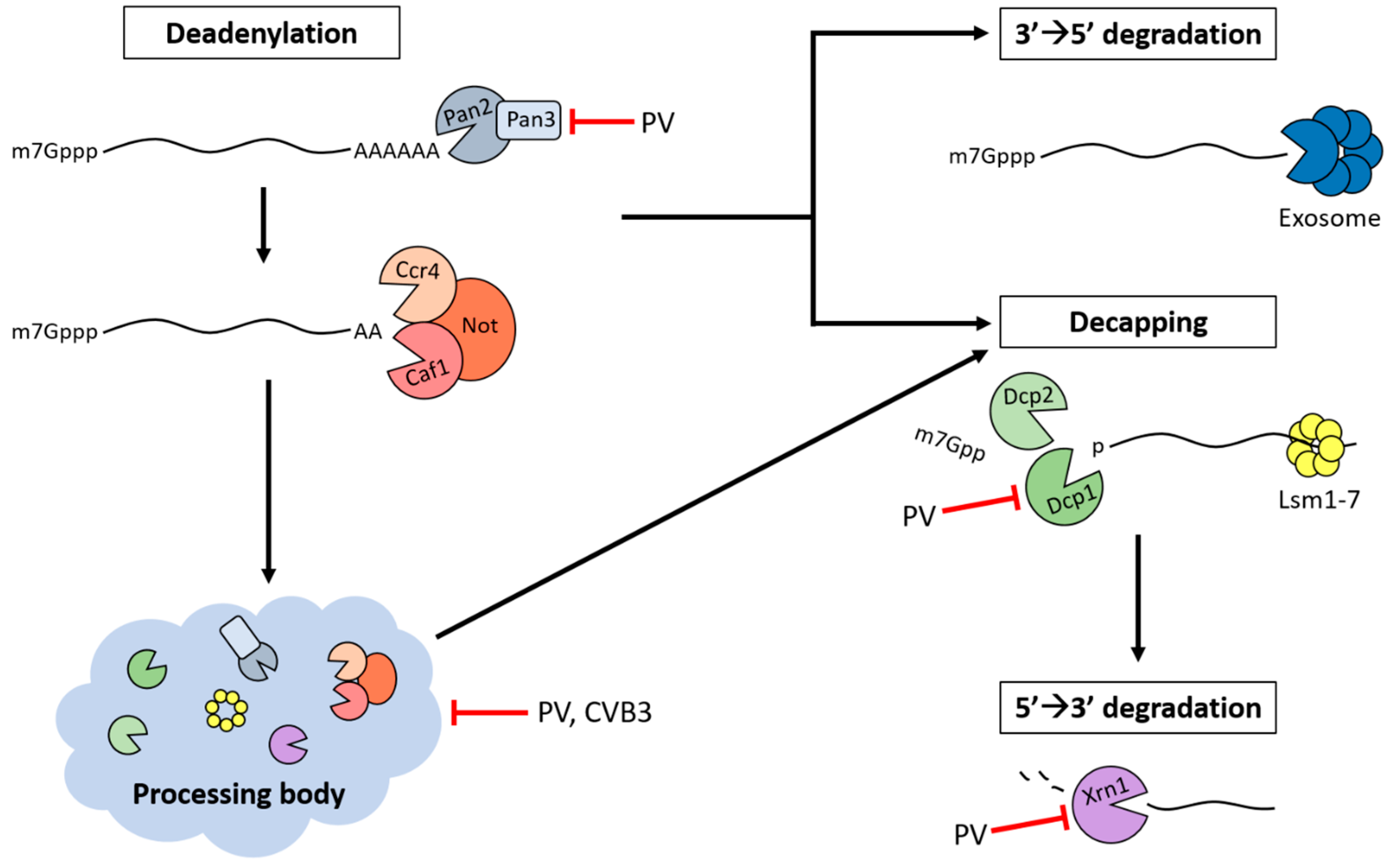
5. Processing Bodies (PBs)
6. Adenylate Uridylate-Rich Element (ARE)-Mediated mRNA Decay (AMD)
7. MicroRNA-Mediated Decay
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flather, D.; Semler, B.L. Picornaviruses and nuclear functions: Targeting a cellular compartment distinct from the replication site of a positive-strand RNA virus. Front. Microbiol. 2015, 6, 594. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Chen, T.C.; Weng, K.F.; Chang, S.C.; Chen, L.L.; Shih, S.R. Viral and host proteins involved in picornavirus life cycle. J. Biomed. Sci. 2009, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Racaniello, V.R. Picornaviridae: The viruses and their replication. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 453–489. [Google Scholar]
- Lee, Y.F.; Nomoto, A.; Detjen, B.M.; Wimmer, E. A protein covalently linked to poliovirus genome RNA. Proc. Natl. Acad. Sci. USA 1977, 74, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Flanegan, J.B.; Petterson, R.F.; Ambros, V.; Hewlett, N.J.; Baltimore, D. Covalent linkage of a protein to a defined nucleotide sequence at the 5’-terminus of virion and replicative intermediate RNAs of poliovirus. Proc. Natl. Acad. Sci. USA 1977, 74, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Virgen-Slane, R.; Rozovics, J.M.; Fitzgerald, K.D.; Ngo, T.; Chou, W.; van der Heden van Noort, G.J.; Filippov, D.V.; Gershon, P.D.; Semler, B.L. An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 14634–14639. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, A.; Lee, Y.F.; Wimmer, E. The 5′ end of poliovirus mRNA is not capped with m7G(5′)ppp(5′)Np. Proc. Natl. Acad. Sci. USA 1976, 73, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Munoz, R.; Darnell, J.E. Structural difference between the 5′ termini of viral and cellular mRNA in poliovirus-infected cells: Possible basis for the inhibition of host protein synthesis. J. Virol. 1976, 18, 719–726. [Google Scholar] [PubMed]
- Hewlett, M.J.; Rose, J.K.; Baltimore, D. 5′-terminal structure of poliovirus polyribosomal RNA is pUp. Proc. Natl. Acad. Sci. USA 1976, 73, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.D.; White, J.P.; Lloyd, R.E. Poliovirus-mediated disruption of cytoplasmic processing bodies. J. Virol. 2011, 85, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Flodström-Tullberg, M.; Hultcrantz, M.; Stotland, A.; Maday, A.; Tsai, D.; Fine, C.; Williams, B.; Silverman, R.; Sarvetnick, N. RNase L and double-stranded RNA-dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. J. Immunol. 2005, 174, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Romeo, G.; Nissim, A.; Marziali, G.; Albertini, R.; Affabris, E.; Battistini, A.; Fiorucci, G.; Orsatti, R.; Rossi, G.B.; et al. A full-length murine 2-5A synthetase cDNA transfected in NIH-3T3 cells impairs EMCV but not VSV replication. Virology 1990, 179, 228–233. [Google Scholar] [CrossRef]
- Townsend, H.L.; Jha, B.K.; Han, J.-Q.; Maluf, N.K.; Silverman, R.H.; Barton, D.J. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 2008, 14, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-Q.; Townsend, H.L.; Jha, B.K.; Paranjape, J.M.; Silverman, R.H.; Barton, D.J. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J. Virol. 2007, 81, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Townsend, H.L.; Jha, B.K.; Silverman, R.H.; Barton, D.J. A putative loop E motif and an H-H kissing loop interaction are conserved and functional features in a group C enterovirus RNA that inhibits ribonuclease L. RNA Biol. 2008, 5, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.A.; Jha, B.K.; Silverman, R.H.; Hesselberth, J.R.; Barton, D.J. Ribonuclease L and metal-ion–independent endoribonuclease cleavage sites in host and viral RNAs. Nucleic Acids Res. 2014, 42, 5202–5216. [Google Scholar] [CrossRef] [PubMed]
- Blyn, L.B.; Swiderek, K.M.; Richards, O.; Stahl, D.C.; Semler, B.L.; Ehrenfeld, E. Poly(rC) binding protein 2 binds to stem-loop IV of the poliovirus RNA 5′ noncoding region: Identification by automated liquid chromatography-tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 1996, 93, 11115–11120. [Google Scholar] [CrossRef] [PubMed]
- Parsley, T.B.; Towner, J.S.; Blyn, L.B.; Ehrenfeld, E.; Semler, B.L. Poly (rC) binding protein 2 forms a ternary complex with the 5′-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA 1997, 3, 1124–1134. [Google Scholar] [PubMed]
- Gamarnik, A.V.; Andino, R. Two functional complexes formed by KH domain containing proteins with the 5’ noncoding region of poliovirus RNA. RNA 1997, 3, 882–892. [Google Scholar] [PubMed]
- Murray, K.E.; Roberts, A.W.; Barton, D.J. Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA 2001, 7, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Kempf, B.J.; Barton, D.J. Poly(rC) binding proteins and the 5′ cloverleaf of uncapped poliovirus mRNA function during de novo assembly of polysomes. J. Virol. 2008, 82, 5835–5846. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Ihle, Y.; Seitz, S.; Gündel, U.; Wutzler, P.; Görlach, M. Poly(rC)-binding protein 2 interacts with the oligo(rC) tract of coxsackievirus B3. Biochem. Biophys. Res. Commun. 2008, 366, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.L.; Hsuan, J.J.; Totty, N.; Jackson, R.J. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev. 1999, 13, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Y.; Li, M.-L.; Huang, P.-N.; Chien, K.-Y.; Horng, J.-T.; Shih, S.-R. Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5′ untranslated region and participates in virus replication. J. Gen. Virol. 2008, 89, 2540–2549. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.L.; Nguyen, J.H.; Ehrenfeld, E.; Semler, B.L. Differential utilization of poly(rC) binding protein 2 in translation directed by picornavirus IRES elements. RNA 1999, 5, 1570–1585. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Y.; Brewer, G.; Li, M.-L. HuR and Ago2 bind the internal ribosome entry site of enterovirus 71 and promote virus translation and replication. PLoS ONE 2015, 10, e0140291. [Google Scholar] [CrossRef] [PubMed]
- Sokoloski, K.J.; Dickson, A.M.; Chaskey, E.L.; Garneau, N.L.; Wilusz, C.J.; Wilusz, J. Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 2010, 8, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Lenarcic, E.M.; Landry, D.M.; Greco, T.M.; Cristea, I.M.; Thompson, S.R. Thiouracil cross-linking mass spectrometry: A cell-based method to identify host factors involved in viral amplification. J. Virol. 2013, 87, 8697–8712. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; van Kuppeveld, F.J.M. Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev. 2014, 25, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yi, L.; Ke, C.; Zhang, Y.; Liu, R.; Chen, J.; Kung, H.-f.; He, M.-L. The interaction between human enteroviruses and type I IFN signaling pathway. Crit. Rev. Microbiol. 2015, 41, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Bisbal, C.; Silverman, R.H. Diverse functions of RNase L and implications in pathology. Biochimie 2007, 89, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Sorgeloos, F.; Jha, B.K.; Silverman, R.H.; Michiels, T. Evasion of antiviral innate immunity by Theiler’s virus L* protein through direct inhibition of RNase L. PLoS Pathog. 2013, 9, e1003474. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Kong, W.P.; Zhang, L.; Ward, P.L.; Roos, R.P. A picornaviral protein synthesized out of frame with the polyprotein plays a key role in a virus-induced immune-mediated demyelinating disease. Nat. Med. 1995, 1, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Martinand, C.; Salehzada, T.; Silhol, M.; Lebleu, B.; Bisbal, C. RNase L inhibitor (RLI) antisense constructions block partially the down regulation of the 2-5A/RNase L pathway in encephalomyocarditis-virus-(EMCV)-infected cells. Eur. J. Biochem. 1998, 254, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of MDA-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.-H.; Kim, S.J.; So, E.Y.; Meng, L.; Colonna, M.; Kim, B.S. Melanoma differentiation-associated gene 5 is critical for protection against Theiler’s virus-induced demyelinating disease. J. Virol. 2012, 86, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.-L.; Kao, L.-T.; Lin, S.-J.; Wang, R.Y.-L.; Shih, S.-R. MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS ONE 2013, 8, e63431. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Fujii, K.; Nagata, N.; Takeuchi, O.; Akira, S.; Oshiumi, H.; Matsumoto, M.; Seya, T.; Koike, S. The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J. Virol. 2012, 86, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Drahos, J.; Racaniello, V.R. Cleavage of IPS-1 in cells infected with human rhinovirus. J. Virol. 2009, 83, 11581–11587. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 2007, 104, 7253–7258. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, W.; Yang, F.; Liu, H.; Zhu, Z.; Lian, K.; Lei, C.; Li, S.; Liu, X.; Zheng, H.; et al. The VP3 structural protein of foot-and-mouth disease virus inhibits the IFN-β signaling pathway. FASEB J. 2016, 30, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lei, C.; Xu, Z.; Yang, F.; Liu, H.; Zhu, Z.; Li, S.; Liu, X.; Shu, H.; Zheng, H. Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-β signaling pathway. Sci. Rep. 2016, 6, 21888. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Li, P.; Sun, L.; Fan, J.; Zhang, Q.; Luo, R.; Liu, X.; Li, K.; Chen, H.; et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 2011, 85, 3758–3766. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Luo, R.; Ye, R.; Fang, Y.; Xie, L.; Chen, H.; Xiao, S. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem. Biophys. Res. Commun. 2010, 399, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zheng, Z.; Liu, Y.; Zhang, Z.; Liu, Q.; Meng, J.; Ke, X.; Hu, Q.; Wang, H. 2C proteins of enteroviruses suppress IKKβ phosphorylation by recruiting protein phosphatase 1. J. Virol. 2016, 90, 5141–5151. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, H.; Zhang, Z.; Meng, J.; Mao, D.; Bai, B.; Lu, B.; Mao, P.; Hu, Q.; Wang, H. Enterovirus 71 2C protein inhibits TNF-α–mediated activation of NF-κB by suppressing IκB kinase β phosphorylation. J. Immunol. 2011, 187, 2202–2212. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Li, K.; Zhong, H.; Fan, J.; Ouyang, C.; Zhang, H.; Duan, E.; Luo, R.; Zhang, Z.; et al. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 2012, 86, 9311–9322. [Google Scholar] [CrossRef] [PubMed]
- De Los Santos, T.; Diaz-San Segundo, F.; Grubman, M.J. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J. Virol. 2007, 81, 12803–12815. [Google Scholar] [CrossRef] [PubMed]
- Neznanov, N.; Chumakov, K.M.; Neznanova, L.; Almasan, A.; Banerjee, A.K.; Gudkov, A.V. Proteolytic cleavage of the p65-RelA subunit of NF-κB during poliovirus infection. J. Biol. Chem. 2005, 280, 24153–24158. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yin, P.; Yang, X.; Zhang, L.; Jin, Q.; Zhu, G. Enterovirus 71 2C protein inhibits NF-κB activation by binding to RelA(p65). Sci. Rep. 2015, 5, 14302. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding Proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Sukarieh, R.; Bordeleau, M.-E.; Kaufman, R.J.; Northcote, P.; Tanaka, J.; Gallouzi, I.; Pelletier, J. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2α phosphorylation. Mol. Biol. Cell 2006, 17, 4212–4219. [Google Scholar] [CrossRef] [PubMed]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.-J.; Anderson, P.; Kaufman, R.J. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 2005, 280, 16925–16933. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc.Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Kimball, S.R.; Horetsky, R.L.; Ron, D.; Jefferson, L.S.; Harding, H.P. Mammalian stress granules represent sites of accumulation of stalled translation initiation complexes. Am. J. Physiol. Cell Physiol. 2003, 284, C273–C284. [Google Scholar] [CrossRef] [PubMed]
- Hilliker, A.; Gao, Z.; Jankowsky, E.; Parker, R. The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol. Cell 2011, 43, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Cho, M.R.; Li, W.; Yacono, P.W.; Chen, S.; Gilks, N.; Golan, D.E.; Anderson, P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 2000, 151, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Mollet, S.; Cougot, N.; Wilczynska, A.; Dautry, F.; Kress, M.; Bertrand, E.; Weil, D. Translationally repressed mRNA transiently cycles through stress granules during stress. Mol. Biol. Cell 2008, 19, 4469–4479. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Peng, Y.; Murray, E.L.; Otsuka, Y.; Kedersha, N.; Schoenberg, D.R. Polysome-bound endonuclease PMR1 is targeted to stress granules via stress-specific binding to TIA-1. Mol. Cell. Biol. 2006, 26, 8803–8813. [Google Scholar] [CrossRef] [PubMed]
- Rothé, F.; Gueydan, C.; Bellefroid, E.; Huez, G.; Kruys, V. Identification of FUSE-binding proteins as interacting partners of TIA proteins. Biochem. Biophys. Res. Commun. 2006, 343, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Stoecklin, G.; Stubbs, T.; Kedersha, N.; Wax, S.; Rigby, W.F.; Blackwell, T.K.; Anderson, P. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004, 23, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Wilczynska, A.; Aigueperse, C.; Kress, M.; Dautry, F.; Weil, D. The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. J. Cell Sci. 2005, 118, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Gareau, C.; Fournier, M.-J.; Filion, C.; Coudert, L.; Martel, D.; Labelle, Y.; Mazroui, R. p21WAF1/CIP1 upregulation through the stress granule-associated protein CUGBP1 confers resistance to bortezomib-mediated apoptosis. PLoS ONE 2011, 6, e20254. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.-E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2007, 2, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, J.; Hansen, S.J.; Park, N.; Jamka, K.; Sarnow, P.; Gustin, K.E. Stable formation of compositionally unique stress granules in virus-infected cells. J. Virol. 2010, 84, 3654–3665. [Google Scholar] [CrossRef] [PubMed]
- Fung, G.; Ng, C.S.; Zhang, J.; Shi, J.; Wong, J.; Piesik, P.; Han, L.; Chu, F.; Jagdeo, J.; Jan, E.; et al. Production of a dominant-negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during CVB3 infection. PLoS ONE 2013, 8, e79546. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, Y.; Lin, L.; Si, X.; Wang, T.; Zhong, X.; Tong, L.; Luan, Y.; Chen, Y.; Li, X.; et al. Protease 2A induces stress granule formation during coxsackievirus B3 and enterovirus 71 infections. Virol. J. 2014, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.S.; Jogi, M.; Yoo, J.-S.; Onomoto, K.; Koike, S.; Iwasaki, T.; Yoneyama, M.; Kato, H.; Fujita, T. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 2013, 87, 9511–9522. [Google Scholar] [CrossRef] [PubMed]
- Borghese, F.; Michiels, T. The leader protein of cardioviruses inhibits stress granule assembly. J. Virol. 2011, 85, 9614–9622. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.D.; Semler, B.L. Poliovirus infection induces the co-localization of cellular protein SRp20 with TIA-1, a cytoplasmic stress granule protein. Virus Res. 2013, 176, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.; Tsai, W.-C.; Lloyd, R. Multiple poliovirus proteins repress cytoplasmic RNA granules. Viruses 2015, 7, 2922. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, H.; Takahashi, M.; Higuchi, M.; Makokha, G.N.; Oie, M.; Fujii, M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells 2013, 18, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Tourrière, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Jogi, M.; Yoo, J.-S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Reineke, L.C.; Lloyd, R.E. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J. Virol. 2015, 89, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Kedersha, N.; Langereis, M.A.; van Kuppeveld, F.J.; Lloyd, R.E. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio 2015, 6, e02486-14. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; García, M.A.; Gomez-Puertas, P.; Guerra, S.; Rullas, J.; Nakano, H.; Alcamí, J.; Esteban, M. TRAF family proteins link PKR with NF-κB activation. Mol. Cell. Biol. 2004, 24, 4502–4512. [Google Scholar] [CrossRef] [PubMed]
- Pindel, A.; Sadler, A. The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 2010, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Pichlmair, A.; Rehwinkel, J.; Rogers, N.C.; Scheuner, D.; Kato, H.; Takeuchi, O.; Akira, S.; Kaufman, R.J.; Reis e Sousa, C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 2010, 7, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Ingelfinger, D.; Arndt-Jovin, D.J.; Lührmann, R.; Achsel, T. The human LSm1–7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA 2002, 8, 1489–1501. [Google Scholar] [PubMed]
- Jain, S.; Parker, R. The discovery and analysis of P bodies. In Ten Years of Progress in GW/P Body Research; Chan, L.E.K., Fritzler, J.M., Eds.; Springer: New York, NY, USA, 2013; pp. 23–43. [Google Scholar]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Ezzeddine, N.; Chen, C.-Y.A.; Zhu, W.; He, X.; Shyu, A.-B. Deadenylation is prerequisite for P-body formation and mRNA decay in mammalian cells. J. Cell Biol. 2008, 182, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Cougot, N.; Babajko, S.; Séraphin, B. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 2004, 165, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Stalder, L.; Mühlemann, O. Processing bodies are not required for mammalian nonsense-mediated mRNA decay. RNA 2009, 15, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Behm-Ansmant, I.; Schweizer, D.; Izaurralde, E. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol. Cell. Biol. 2007, 27, 3970–3981. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Valencia-Sanchez, M.A.; Hannon, G.J.; Parker, R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 2005, 7, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.S.; Bhattacharyya, S.N.; Artus, C.G.; Zoller, T.; Cougot, N.; Basyuk, E.; Bertrand, E.; Filipowicz, W. Inhibition of Translational Initiation by Let-7 MicroRNA in Human Cells. Science 2005, 309, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Sheth, U.; Parker, R. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell 2006, 125, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Brengues, M.; Teixeira, D.; Parker, R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 2005, 310, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Andrei, M.A.; Ingelfinger, D.; Heintzmann, R.; Achsel, T.; Rivera-Pomar, R.; Lurhmann, R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA 2005, 11, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Beisang, D.; Bohjanen, P.R. Perspectives on the ARE as it turns 25 years old. Wiley Interdiscip. Rev. RNA 2012, 3, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Gherzi, R.; Ong, S.-E.; Chan, E.L.; Raijmakers, R.; Pruijn, G.J.M.; Stoecklin, G.; Moroni, C.; Mann, M.; Karin, M. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 2001, 107, 451–464. [Google Scholar] [CrossRef]
- Mukherjee, D.; Gao, M.; O’Connor, J.P.; Raijmakers, R.; Pruijn, G.; Lutz, C.S.; Wilusz, J. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J. 2002, 21, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wilusz, C.J.; Peltz, S.W.; Wilusz, J. A novel mRNA-decapping activity in HeLa cytoplasmic extracts is regulated by AU-rich elements. EMBO J. 2001, 20, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Stoecklin, G.; Mayo, T.; Anderson, P. ARE-mRNA degradation requires the 5′–3′ decay pathway. EMBO Rep. 2006, 7, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Gherzi, R.; Lee, K.-Y.; Briata, P.; Wegmüller, D.; Moroni, C.; Karin, M.; Chen, C.-Y. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol. Cell 2004, 14, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J.; Wagner, E. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 2005, 19, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G. An A + U-rich element RNA-binding factor regulates c-myc mRNA stability in vitro. Mol. Cell. Biol. 1991, 11, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.; Saccani, S.; Sarkar, S.; Lewis, A.; Pestka, S. Increased interleukin-10 mRNA stability in melanoma cells is associated with decreased levels of A + U-rich element binding factor AUF1. J. Interferon Cytokine Res. 2003, 23, 553–564. [Google Scholar] [PubMed]
- Chen, T.-M.; Hsu, C.-H.; Tsai, S.-J.; Sun, H.S. AUF1 p42 isoform selectively controls both steady-state and PGE2-induced FGF9 mRNA decay. Nucleic Acids Res. 2010, 38, 8061–8071. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wang, W.; Wilson, G.M.; Yang, X.; Brewer, G.; Holbrook, N.J.; Gorospe, M. Down-regulation of cyclin D1 expression by prostaglandin A2 is mediated by enhanced cyclin D1 mRNA turnover. Mol. Cell. Biol. 2000, 20, 7903–7913. [Google Scholar] [CrossRef] [PubMed]
- Sela-Brown, A.; Silver, J.; Brewer, G.; Naveh-Many, T. Identification of AUF1 as a parathyroid hormone mRNA 3′-untranslated region-binding protein that determines parathyroid hormone mRNA stability. J. Biol. Chem. 2000, 275, 7424–7429. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Brown, J.A.; Gong, C.; Fan, H.; Brewer, G.; Gnarra, J.R. Association of the von Hippel–Lindau protein with AUF1 and posttranscriptional regulation of VEGFA mRNA. Mol. Cancer Res. 2012, 10, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Hu, Y.; Brewer, G. Competitive binding of AUF1 and TIAR to MYC mRNA controls its translation. Nat. Struct. Mol. Biol. 2007, 14, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.J.; DeMaria, C.T.; Sun, Y.; Wilson, G.M.; Brewer, G. Structure and genomic organization of the human AUF1 gene: Alternative pre-mRNA splicing generates four protein isoforms. Genomics 1998, 48, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, B.; Lu, J.-Y.; Schneider, R.J. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. J. Biol. Chem. 2003, 278, 20700–20707. [Google Scholar] [CrossRef] [PubMed]
- Zucconi, B.E.; Ballin, J.D.; Brewer, B.Y.; Ross, C.R.; Huang, J.; Toth, E.A.; Wilson, G.M. Alternatively expressed domains of AU-rich element RNA-binding protein 1 (AUF1) regulate RNA-binding affinity, RNA-induced protein oligomerization, and the local conformation of bound RNA ligands. J. Biol. Chem. 2010, 285, 39127–39139. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Kuriyama, R.; Kayama, F.; Kato, S. A nuclear matrix-associated factor, SAF-B, interacts with specific isoforms of AUF1/hnRNP D. Arch. Biochem. Biophys. 2000, 380, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; De, S.; Srikantan, S.; Abdelmohsen, K.; Grammatikakis, I.; Kim, J.; Kim, K.M.; Noh, J.H.; White, E.J.; Martindale, J.L.; et al. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat. Commun. 2014, 5, 5248. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Pananá, E.M.; Peng, R.; Brewer, G.; Tan, J.; Ling, P.D. Regulation of the Epstein-Barr virus C promoter by AUF1 and the cyclic AMP/protein kinase A signaling pathway. J. Virol. 2000, 74, 8166–8175. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Pimienta, G.; Steitz, J.A. AUF1/hnRNP D is a novel protein partner of the EBER1 noncoding RNA of Epstein-Barr virus. RNA 2012, 18, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Lund, N.; Milev, M.P.; Wong, R.; Sanmuganantham, T.; Woolaway, K.; Chabot, B.; Abou Elela, S.; Mouland, A.J.; Cochrane, A. Differential effects of hnRNP D/AUF1 isoforms on HIV-1 gene expression. Nucleic Acids Res. 2012, 40, 3663–3675. [Google Scholar] [CrossRef] [PubMed]
- Paek, K.Y.; Kim, C.S.; Park, S.M.; Kim, J.H.; Jang, S.K. RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J. Virol. 2008, 82, 12082–12093. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, S.; Schmidt, T.; Geissler, R.; Lilie, H.; Chabierski, S.; Ulbert, S.; Liebert, U.G.; Golbik, R.P.; Behrens, S.-E. AUF1 p45 promotes West Nile virus replication by an RNA chaperone activity that supports cyclization of the viral genome. J. Virol. 2014, 88, 11586–11599. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, S.; Schmidt, T.; Schierhorn, A.; Lilie, H.; Szczepankiewicz, G.; Bergs, S.; Liebert, U.G.; Golbik, R.P.; Behrens, S.E. Arginine methylation enhances the RNA chaperone activity of the West Nile virus host factor AUF1 p45. RNA 2016, 22, 1574–1591. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, A.L.; Rozovics, J.M.; Semler, B.L. Cellular mRNA decay protein AUF1 negatively regulates enterovirus and human rhinovirus infections. J. Virol. 2013, 87, 10423–10434. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, A.L.; Semler, B.L. Differential restriction patterns of mRNA decay factor AUF1 during picornavirus infections. J. Gen. Virol. 2014, 95, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Si, X.; Angeles, A.; Zhang, J.; Shi, J.; Fung, G.; Jagdeo, J.; Wang, T.; Zhong, Z.; Jan, E.; et al. Cytoplasmic redistribution and cleavage of AUF1 during coxsackievirus infection enhance the stability of its viral genome. FASEB J. 2013, 27, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Y.; Li, M.-L.; Brewer, G. mRNA decay factor AUF1 binds the internal ribosomal entry site of enterovirus 71 and inhibits virus replication. PLoS ONE 2014, 9, e103827. [Google Scholar] [CrossRef] [PubMed]
- Rozovics, J.M.; Chase, A.J.; Cathcart, A.L.; Chou, W.; Gershon, P.D.; Palusa, S.; Wilusz, J.; Semler, B.L. Picornavirus modification of a host mRNA decay protein. mBio 2012, 3, e00431-12. [Google Scholar] [CrossRef] [PubMed]
- Spurrell, J.C.L.; Wiehler, S.; Zaheer, R.S.; Sanders, S.P.; Proud, D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L85–L95. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Y.; Shih, S.-R.; Pan, M.; Li, C.; Lue, C.-F.; Stollar, V.; Li, M.-L. hnRNP A1 interacts with the 5′ untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J. Virol. 2009, 83, 6106–6114. [Google Scholar] [CrossRef] [PubMed]
- Levengood, J.D.; Tolbert, M.; Li, M.-L.; Tolbert, B.S. High-affinity interaction of hnRNP A1 with conserved RNA structural elements is required for translation and replication of enterovirus 71. RNA Biol. 2013, 10, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Cammas, A.; Pileur, F.; Bonnal, S.; Lewis, S.M.; Lévêque, N.; Holcik, M.; Vagner, S. Cytoplasmic relocalization of heterogeneous nuclear ribonucleoprotein A1 controls translation initiation of specific mRNAs. Mol. Biol. Cell 2007, 18, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Turck, C.W.; Nikolic, J.M.; Black, D.L. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev. 1997, 11, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Davis-Smyth, T.; Duncan, R.C.; Zheng, T.; Michelotti, G.; Levens, D. The far upstream element-binding proteins comprise an ancient family of single-strand DNA-binding transactivators. J. Biol. Chem. 1996, 271, 31679–31687. [Google Scholar] [CrossRef] [PubMed]
- Trabucchi, M.; Briata, P.; Garcia-Mayoral, M.; Haase, A.D.; Filipowicz, W.; Ramos, A.; Gherzi, R.; Rosenfeld, M.G. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 2009, 459, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Y.; Li, M.-L.; Shih, S.-R. Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res. 2009, 37, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Kung, Y.-A.; Weng, K.-F.; Lin, J.-Y.; Horng, J.-T.; Shih, S.-R. Enterovirus 71 infection cleaves a negative regulator for viral internal ribosomal entry site-driven translation. J. Virol. 2013, 87, 3828–3838. [Google Scholar] [CrossRef] [PubMed]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.T.; Graber, T.E.; Jordan, L.E.; Cloutier, M.; Lewis, S.M.; Goulet, I.; Cote, J.; Holcik, M. hnRNP A1 regulates UV-induced NF-[kappa]B signalling through destabilization of cIAP1 mRNA. Cell Death Differ. 2008, 16, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Geissler, R.; Simkin, A.; Floss, D.; Patel, R.; Fogarty, E.A.; Scheller, J.; Grimson, A. A widespread sequence-specific mRNA decay pathway mediated by hnRNPs A1 and A2/B1. Genes Dev. 2016, 30, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.; Pileur, F.; Orsini, C.; Parker, F.; Pujol, F.; Prats, A.-C.; Vagner, S. Heterogeneous nuclear ribonucleoprotein A1 is a novel internal ribosome entry site trans-acting factor that modulates alternative initiation of translation of the fibroblast growth factor 2 mRNA. J. Biol. Chem. 2005, 280, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Kunze, M.M.; Benz, F.; Brauß, T.F.; Lampe, S.; Weigand, J.E.; Braun, J.; Richter, F.M.; Wittig, I.; Brüne, B.; Schmid, T. sST2 translation is regulated by FGF2 via an hnRNP A1-mediated IRES-dependent mechanism. BBA Gene Regul. Mech. 2016, 1859, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, H.-o.; Tomari, Y. The functions of microRNAs: MRNA decay and translational repression. Trends Cell Biol. 2015, 25, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; Kim, V.N. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Azlan, A.; Dzaki, N.; Azzam, G. Argonaute: The executor of small RNA function. J. Genet. Genom. 2016, 43, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.-J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.V.; Ciaudo, C.; Marchais, A.; Li, Y.; Jay, F.; Ding, S.W.; Voinnet, O. Antiviral RNA interference in mammalian cells. Science 2013, 342, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Sun, J.; Wang, B.; Wu, M.; Zhang, J.; Duan, Z.; Wang, H.; Hu, N.; Hu, Y. Novel microRNA-like viral small regulatory RNAs arising during human hepatitis A virus infection. FASEB J. 2014, 28, 4381–4393. [Google Scholar] [CrossRef] [PubMed]
- Weng, K.-F.; Hung, C.-T.; Hsieh, P.-T.; Li, M.-L.; Chen, G.-W.; Kung, Y.-A.; Huang, P.-N.; Kuo, R.-L.; Chen, L.-L.; Lin, J.-Y.; et al. A cytoplasmic RNA virus generates functional viral small RNAs and regulates viral IRES activity in mammalian cells. Nucleic Acids Res. 2014, 42, 12789–12805. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullmer, W.; Semler, B.L. Diverse Strategies Used by Picornaviruses to Escape Host RNA Decay Pathways. Viruses 2016, 8, 335. https://doi.org/10.3390/v8120335
Ullmer W, Semler BL. Diverse Strategies Used by Picornaviruses to Escape Host RNA Decay Pathways. Viruses. 2016; 8(12):335. https://doi.org/10.3390/v8120335
Chicago/Turabian StyleUllmer, Wendy, and Bert L. Semler. 2016. "Diverse Strategies Used by Picornaviruses to Escape Host RNA Decay Pathways" Viruses 8, no. 12: 335. https://doi.org/10.3390/v8120335
APA StyleUllmer, W., & Semler, B. L. (2016). Diverse Strategies Used by Picornaviruses to Escape Host RNA Decay Pathways. Viruses, 8(12), 335. https://doi.org/10.3390/v8120335