
4EBP-Dependent Signaling Supports West Nile Virus Growth and Protein Expression


Abstract
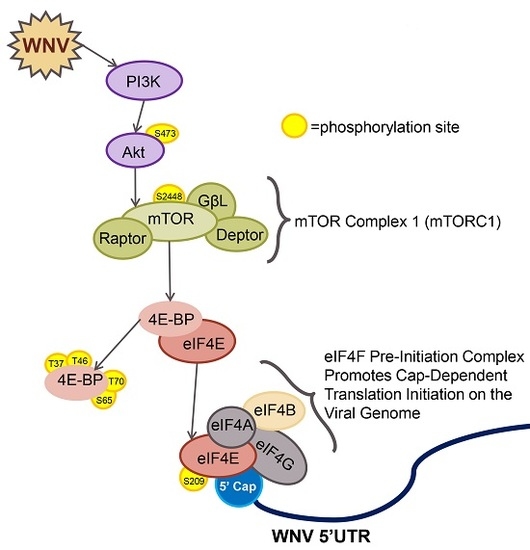
:
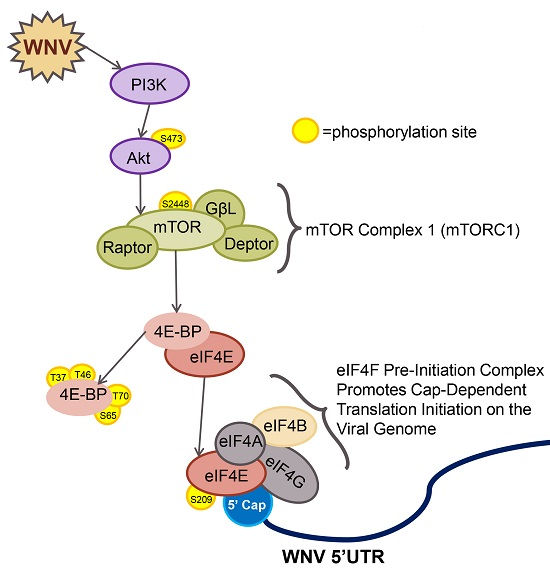{kind=link}
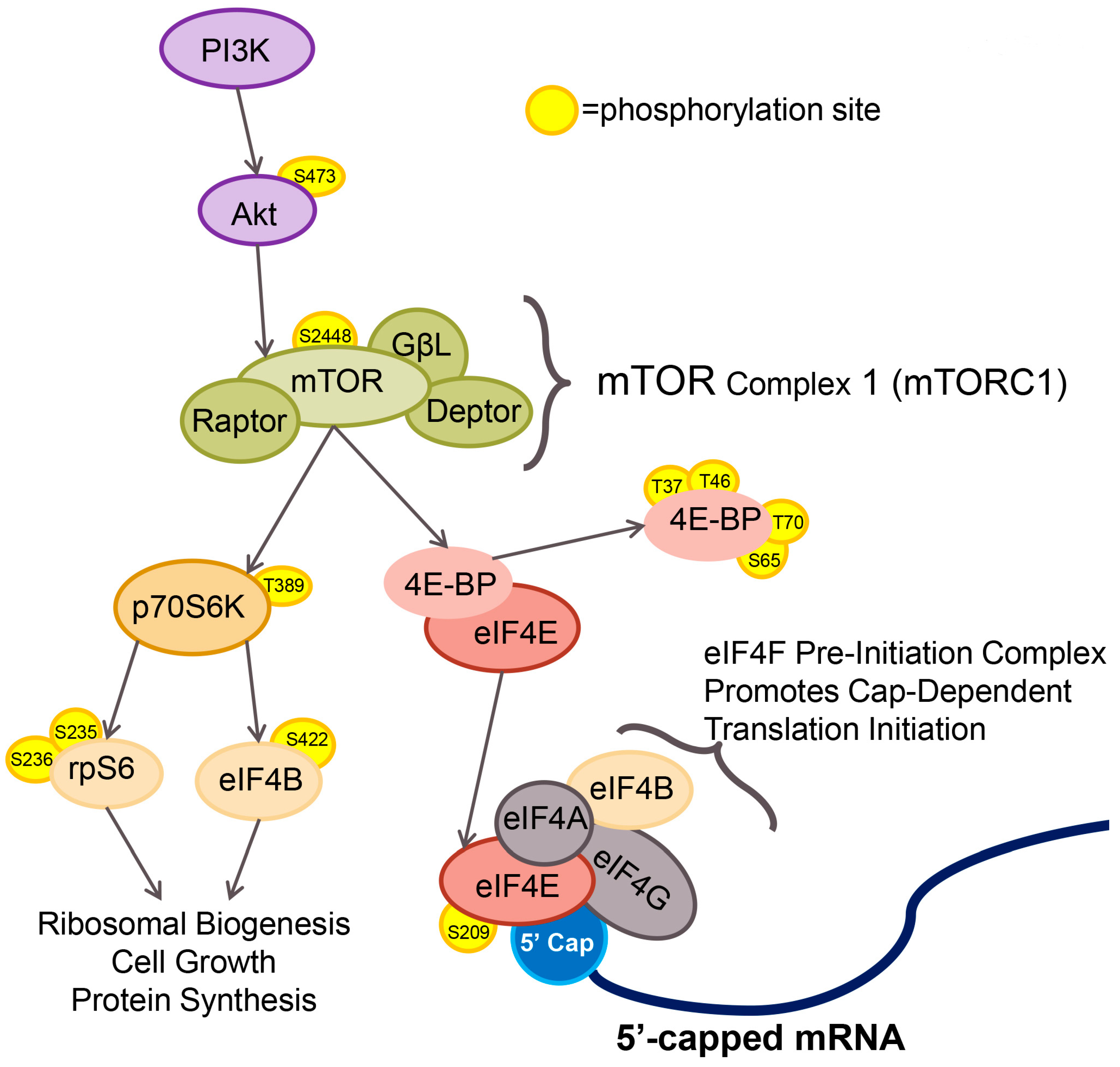{kind=link}
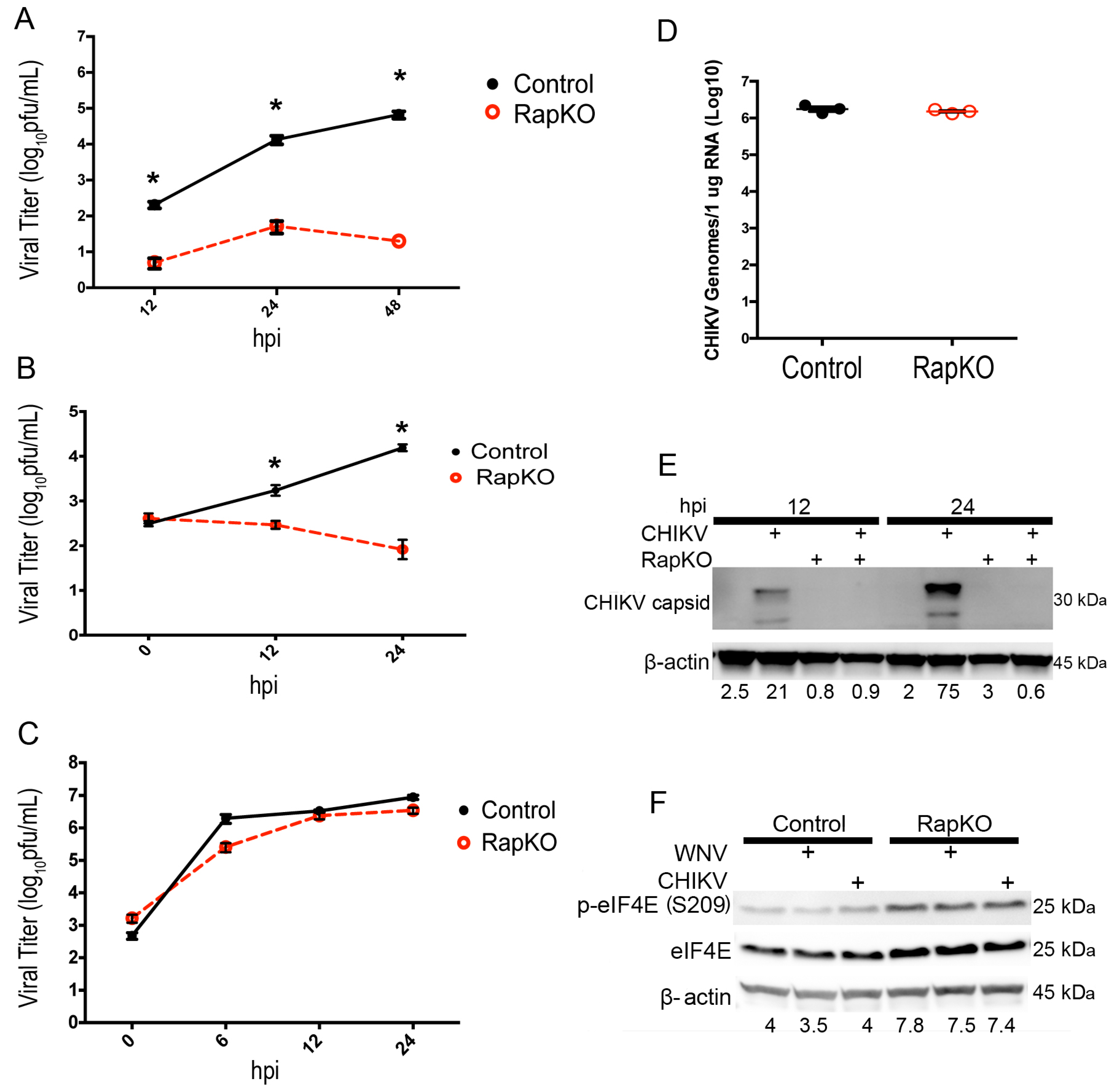{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Virus Propagation and Titration
2.2. Cell Lines
2.3. Inducible Raptor Murine Embryonic Fibroblasts
2.4. Western Blots
2.5. RNA Isolation and qRT-PCR
2.6. MTT Assay
2.7. Biochemical Inhibitor Studies
2.8. Statistical Analysis
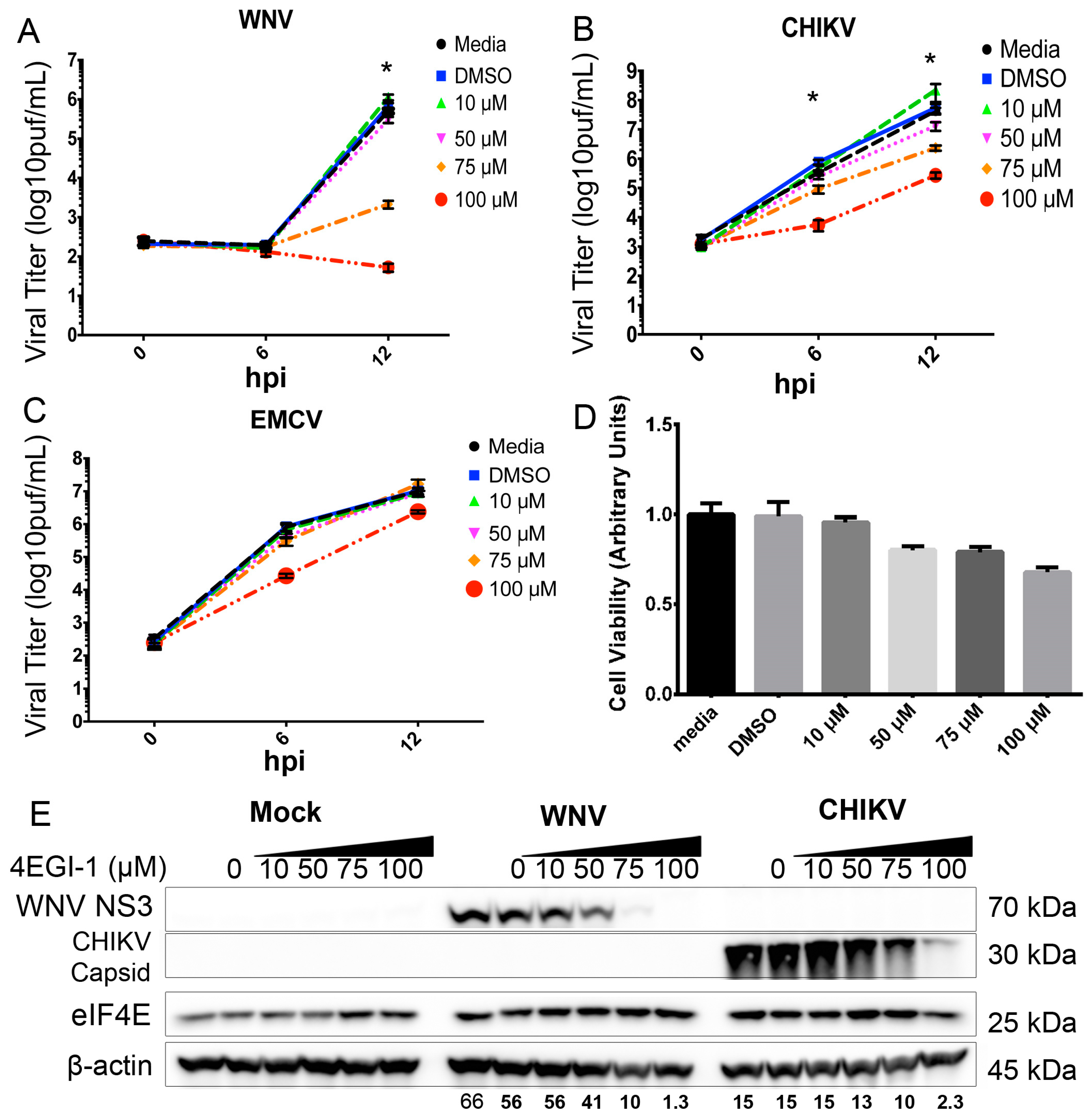
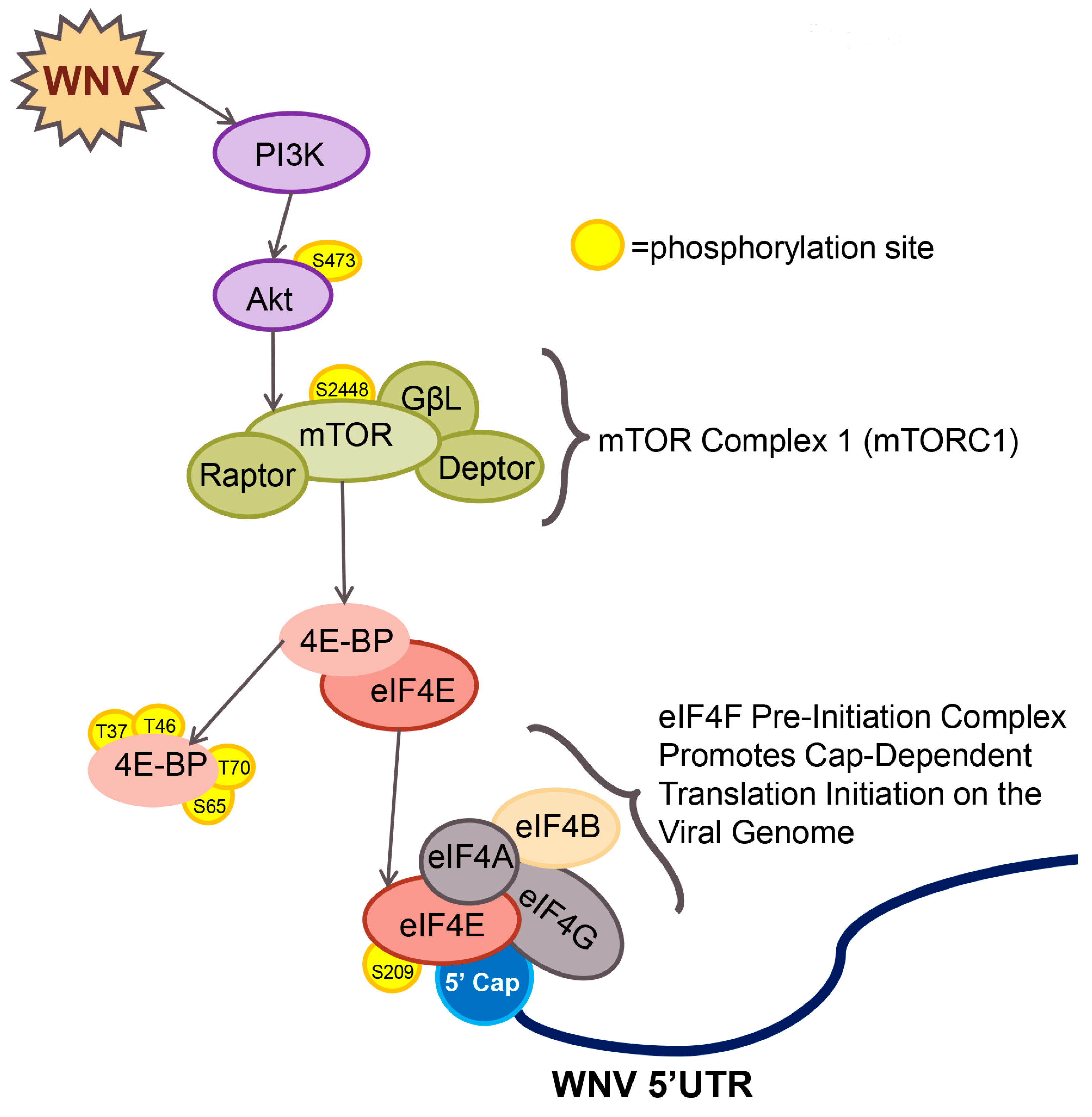
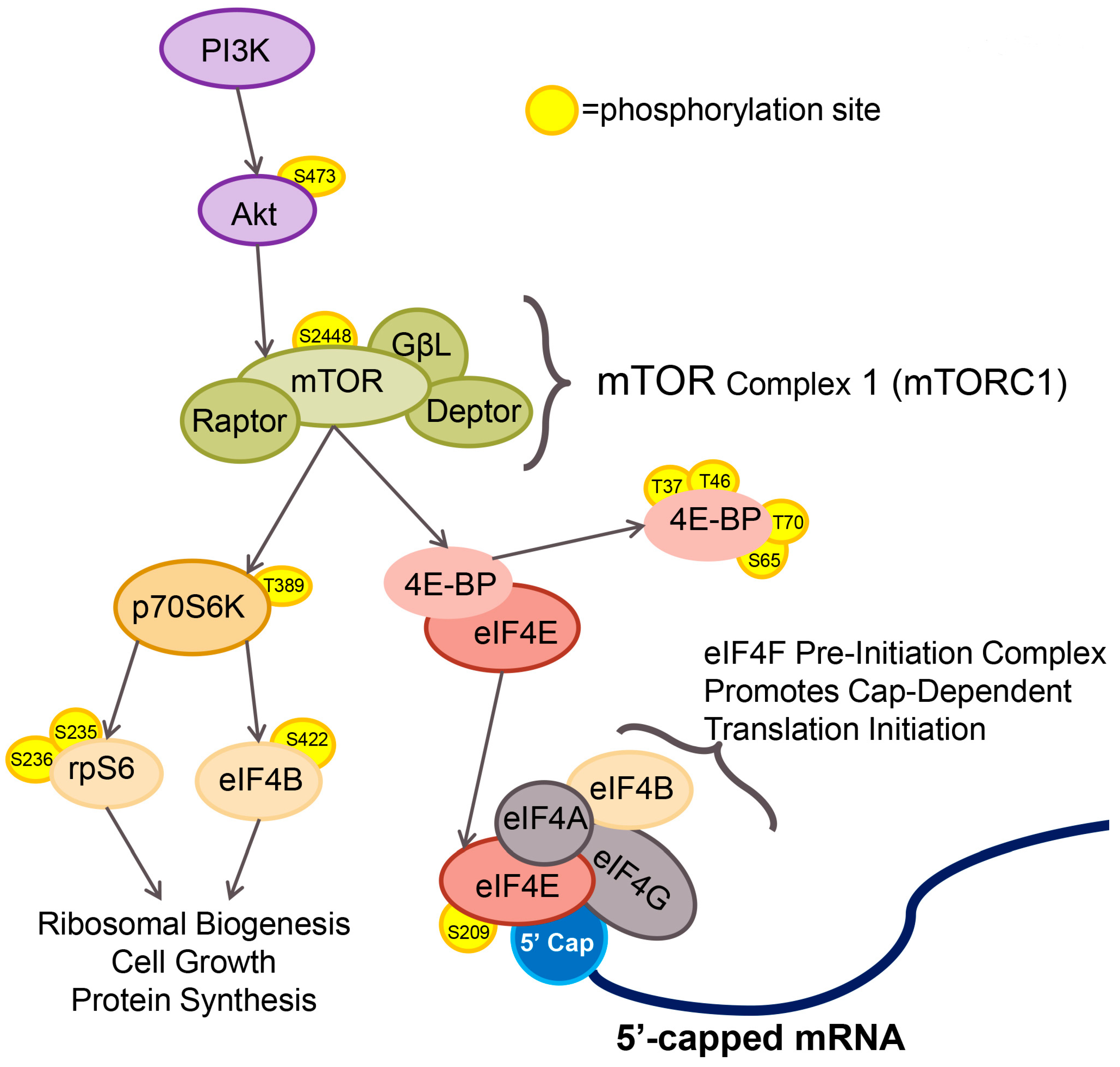
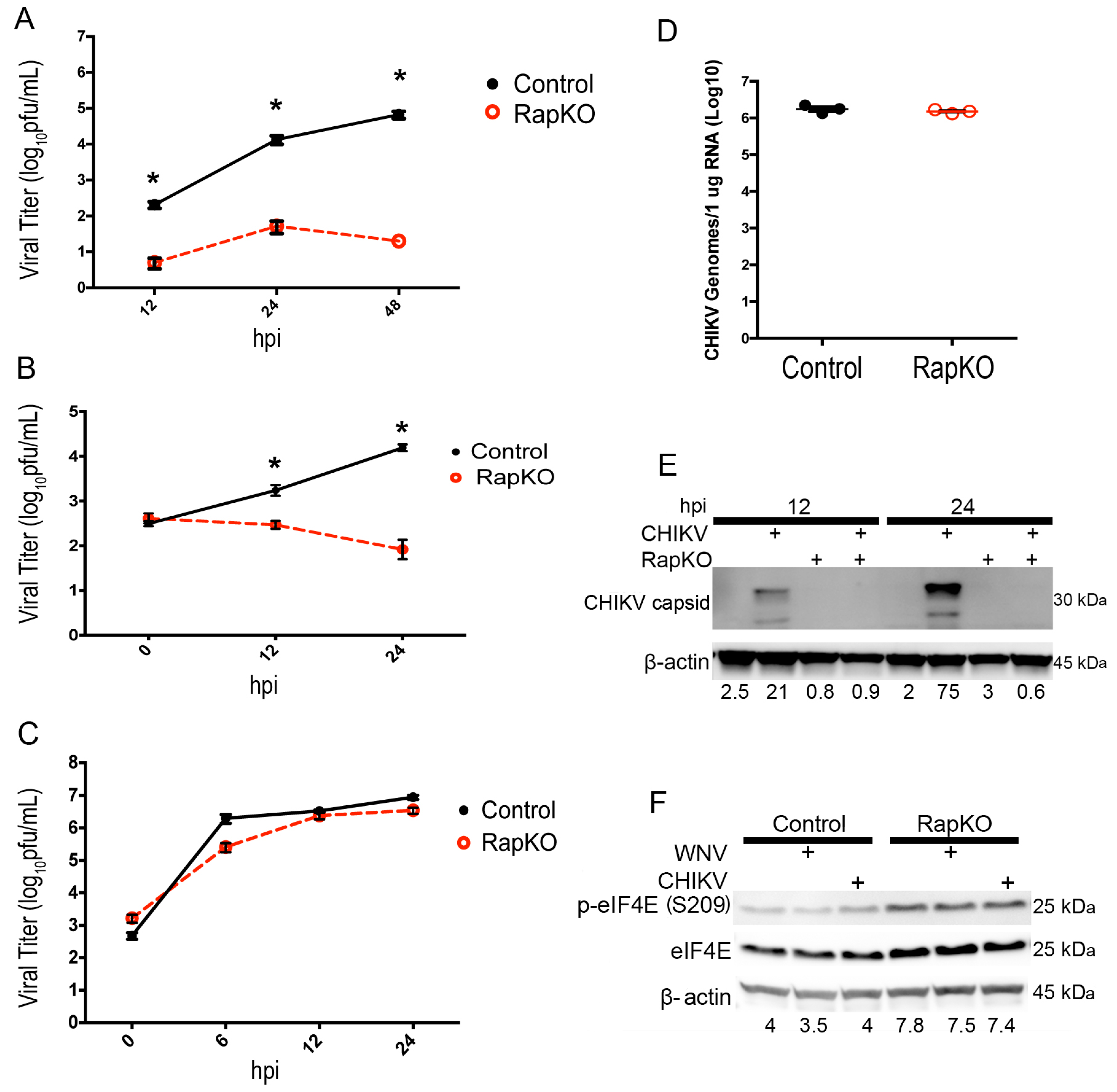
3. Results
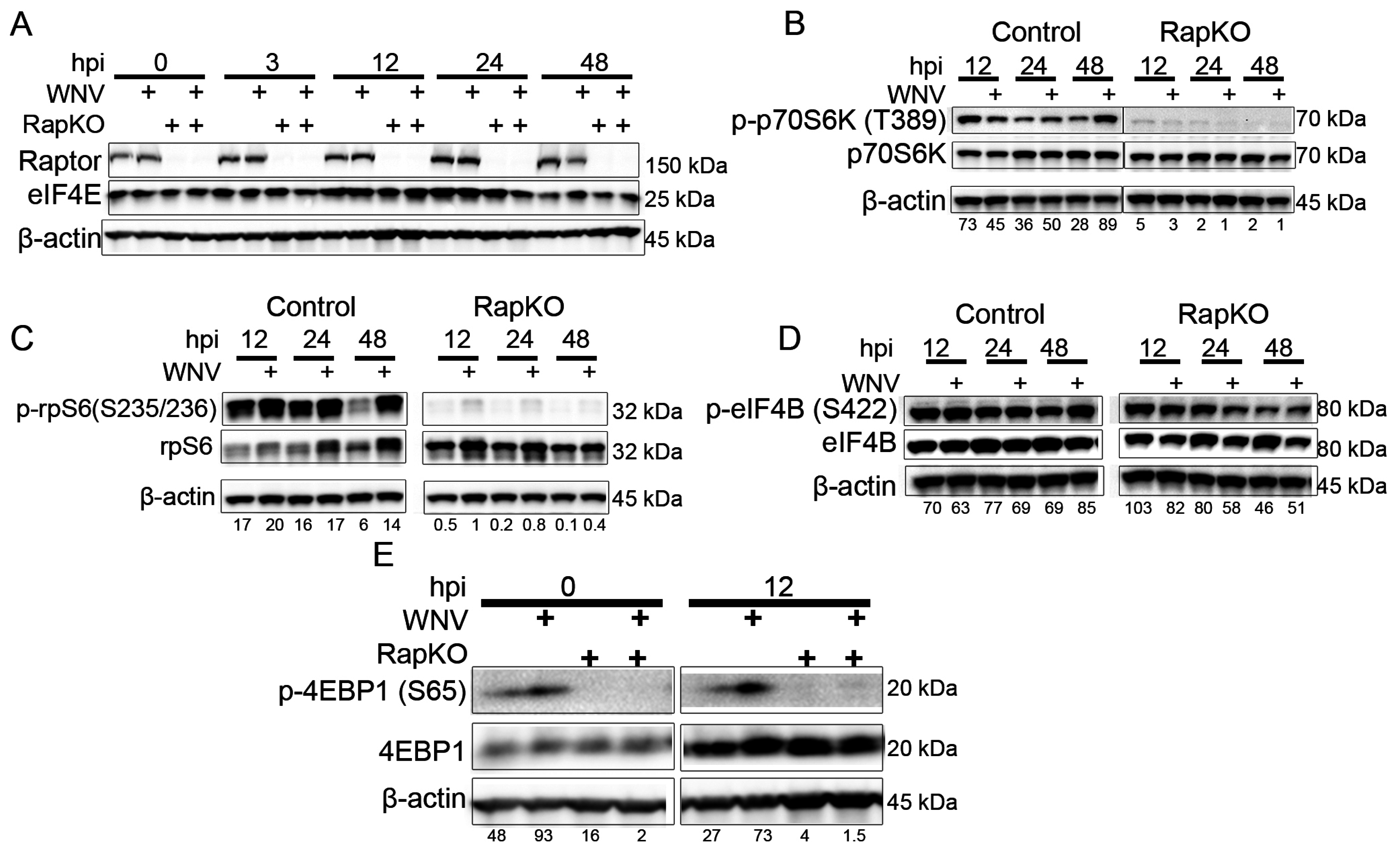
3.1. Raptor Deletion Reduces the Growth of 5′-Capped Viruses, but Not an IRES-Translated Virus
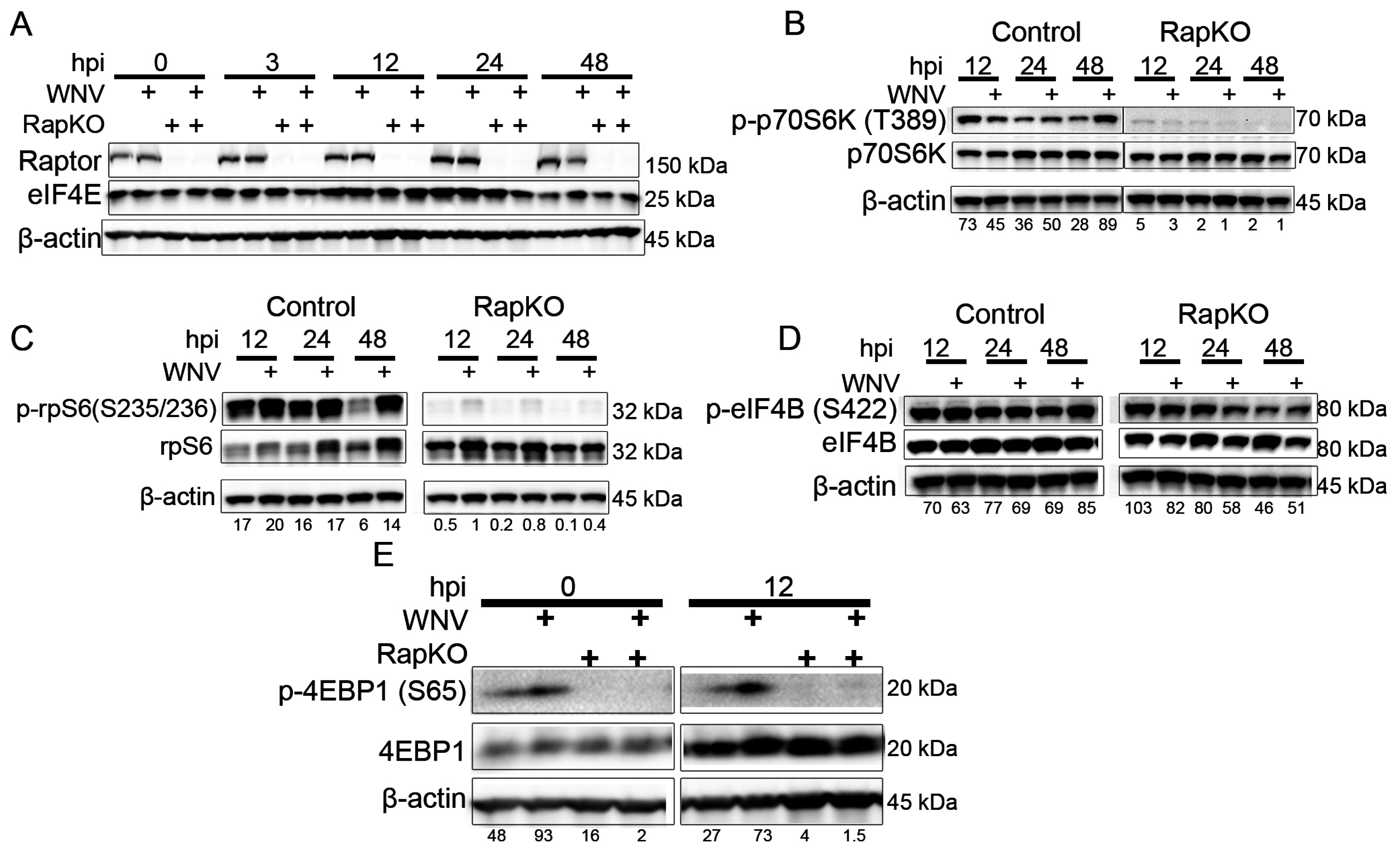
3.2. Impact of Raptor Knockout on mTORC1 Effectors
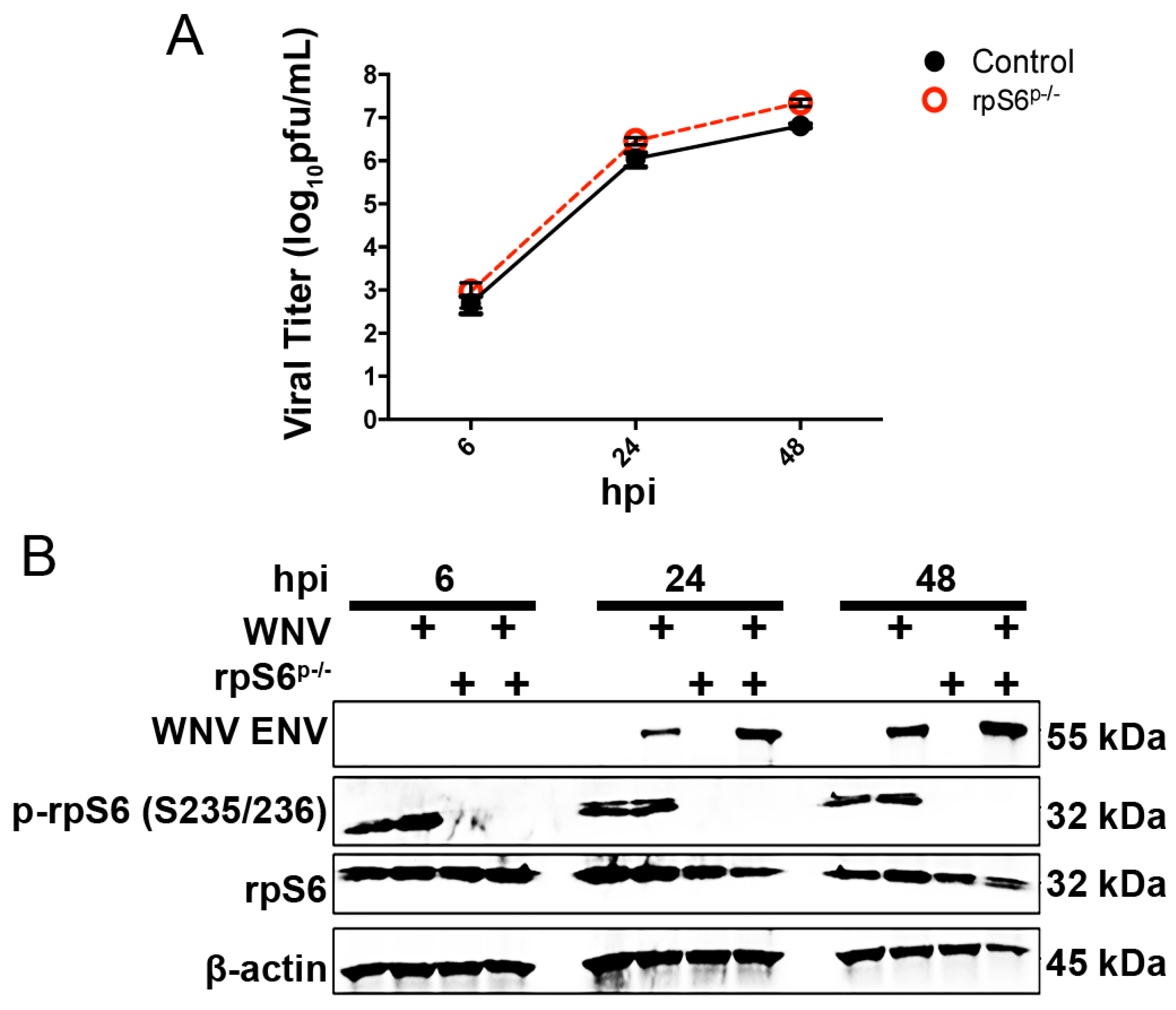
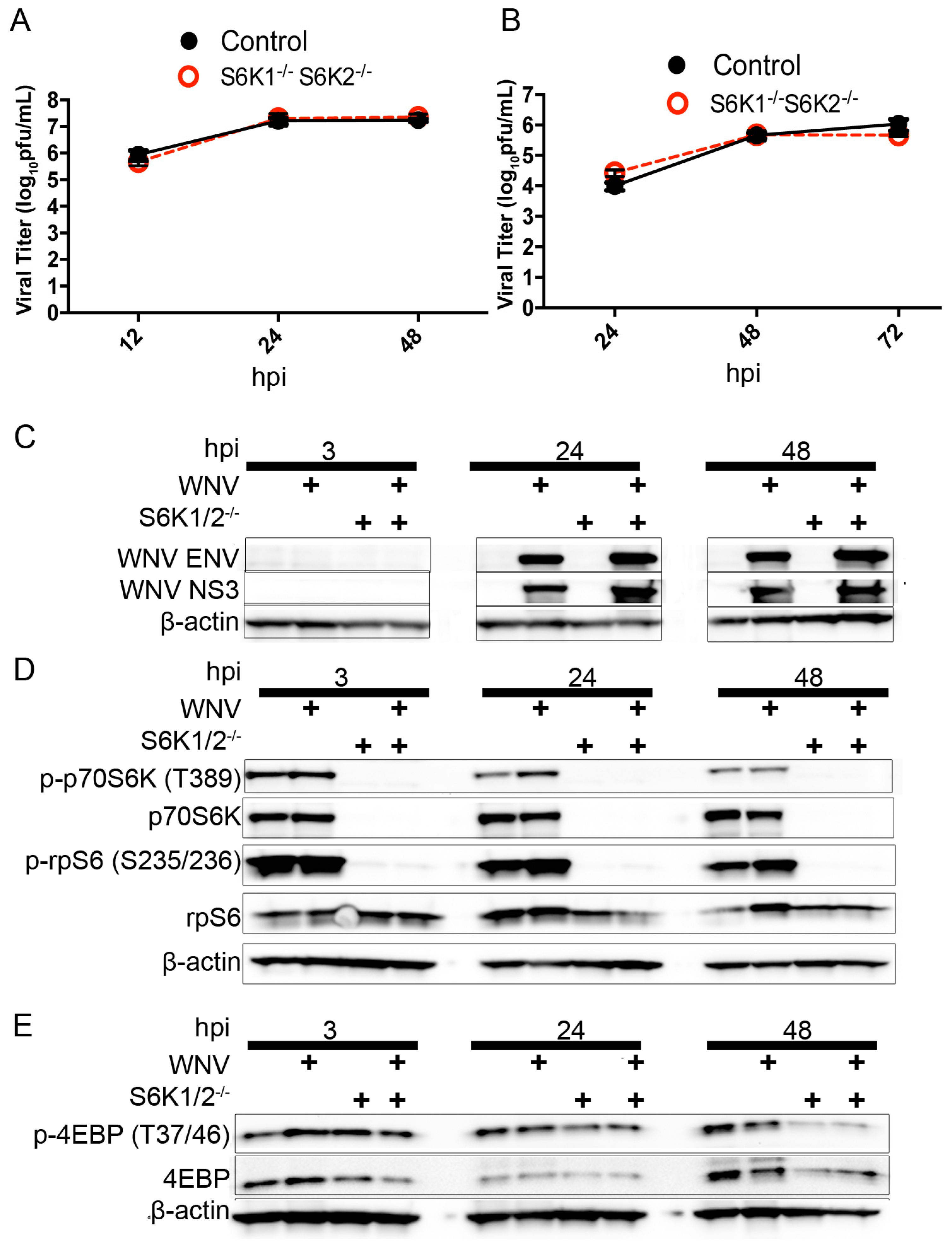
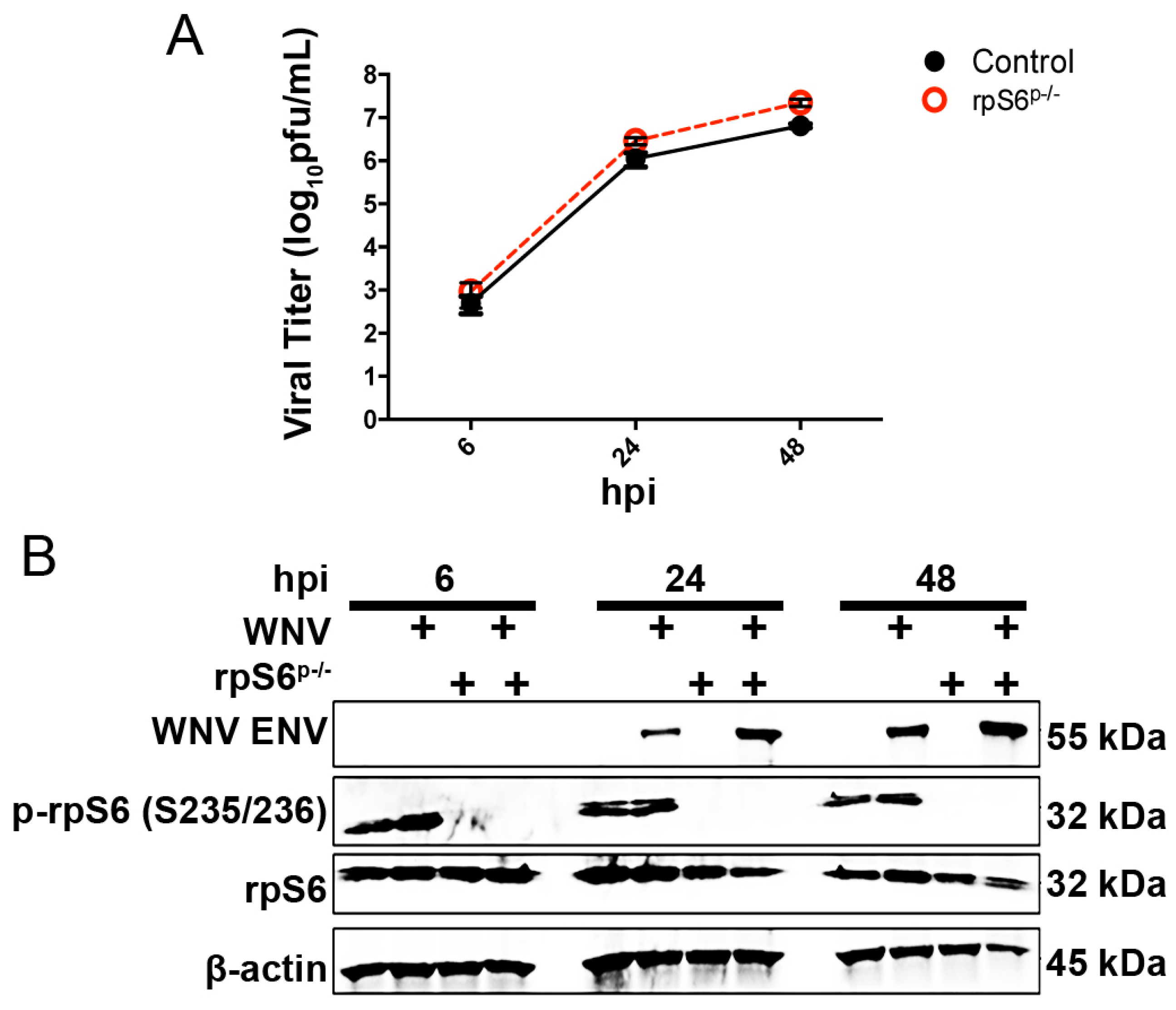
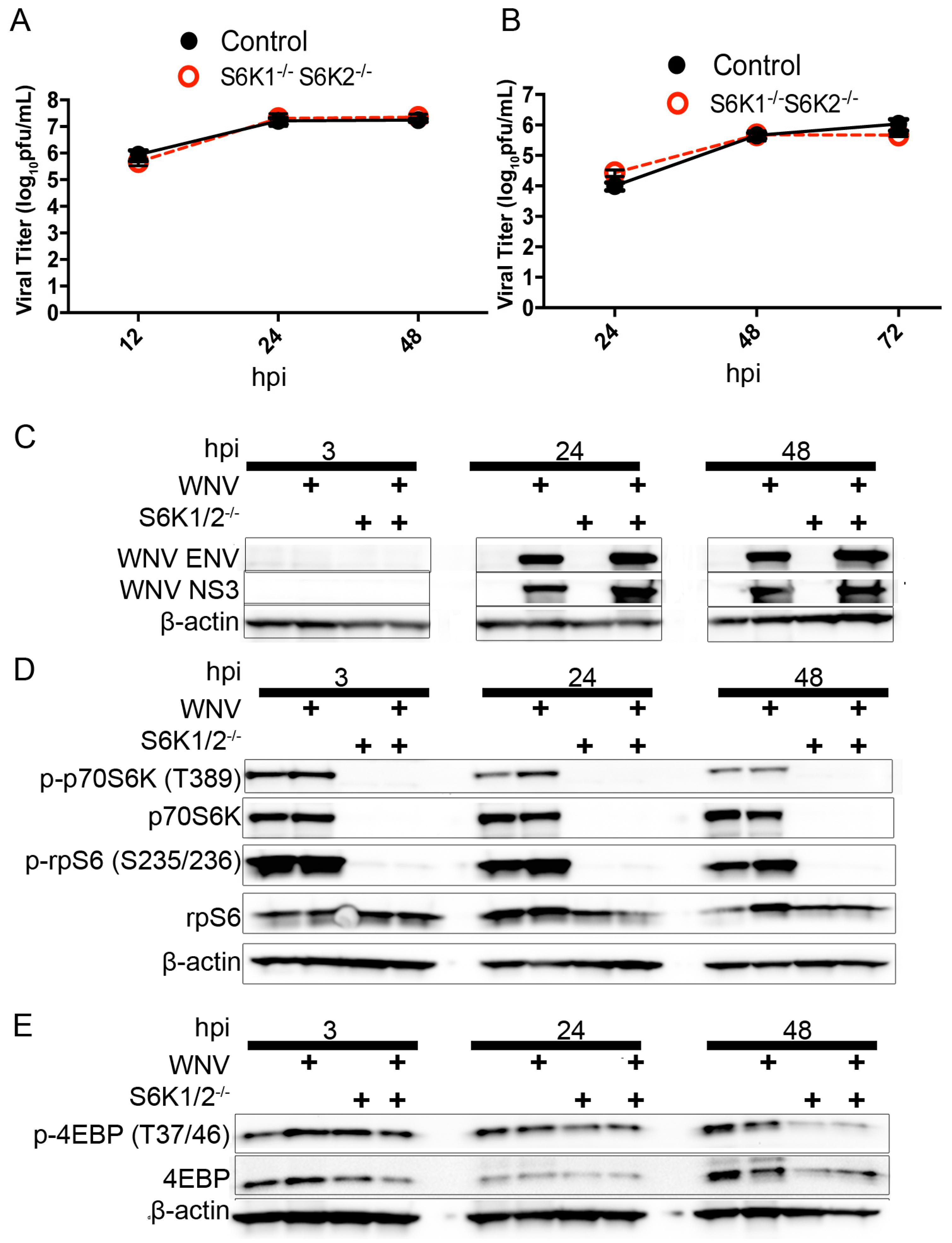
3.3. Loss of S6K/rpS6 Activity Does Not Impact WNV Growth
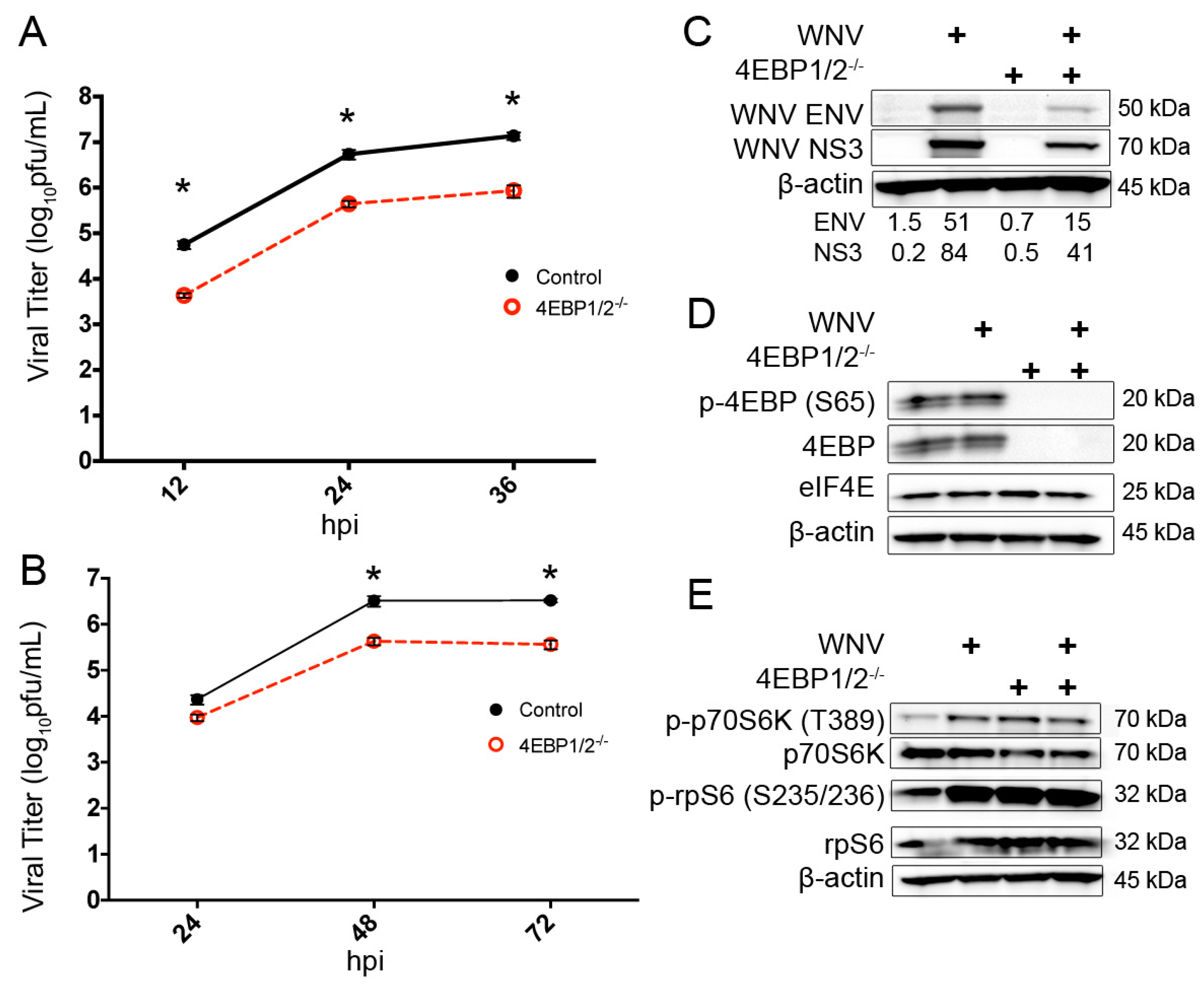
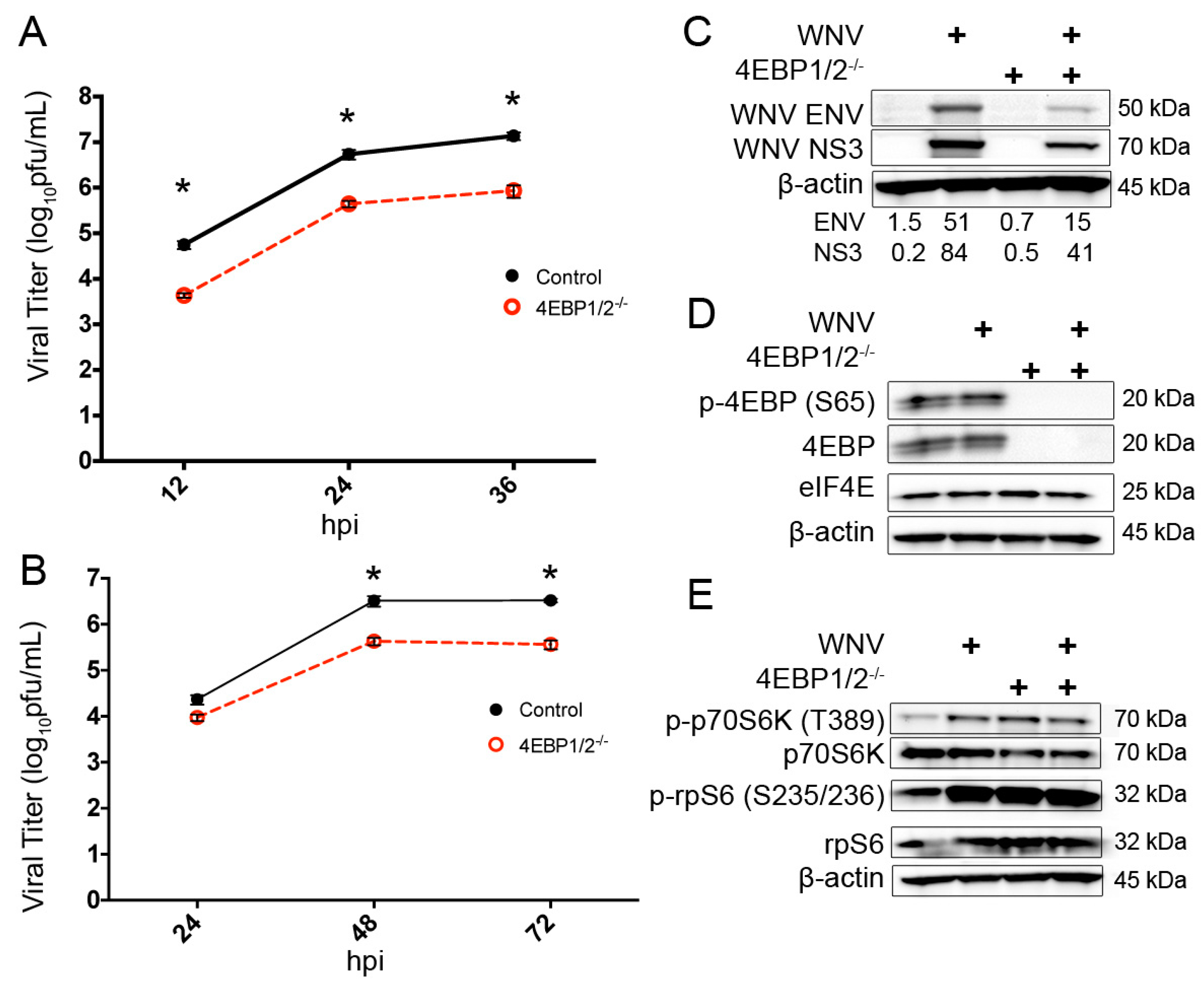
3.4. Knockout of 4EBP Expression Leads to Decreased WNV Growth and Decreased Viral Protein Production
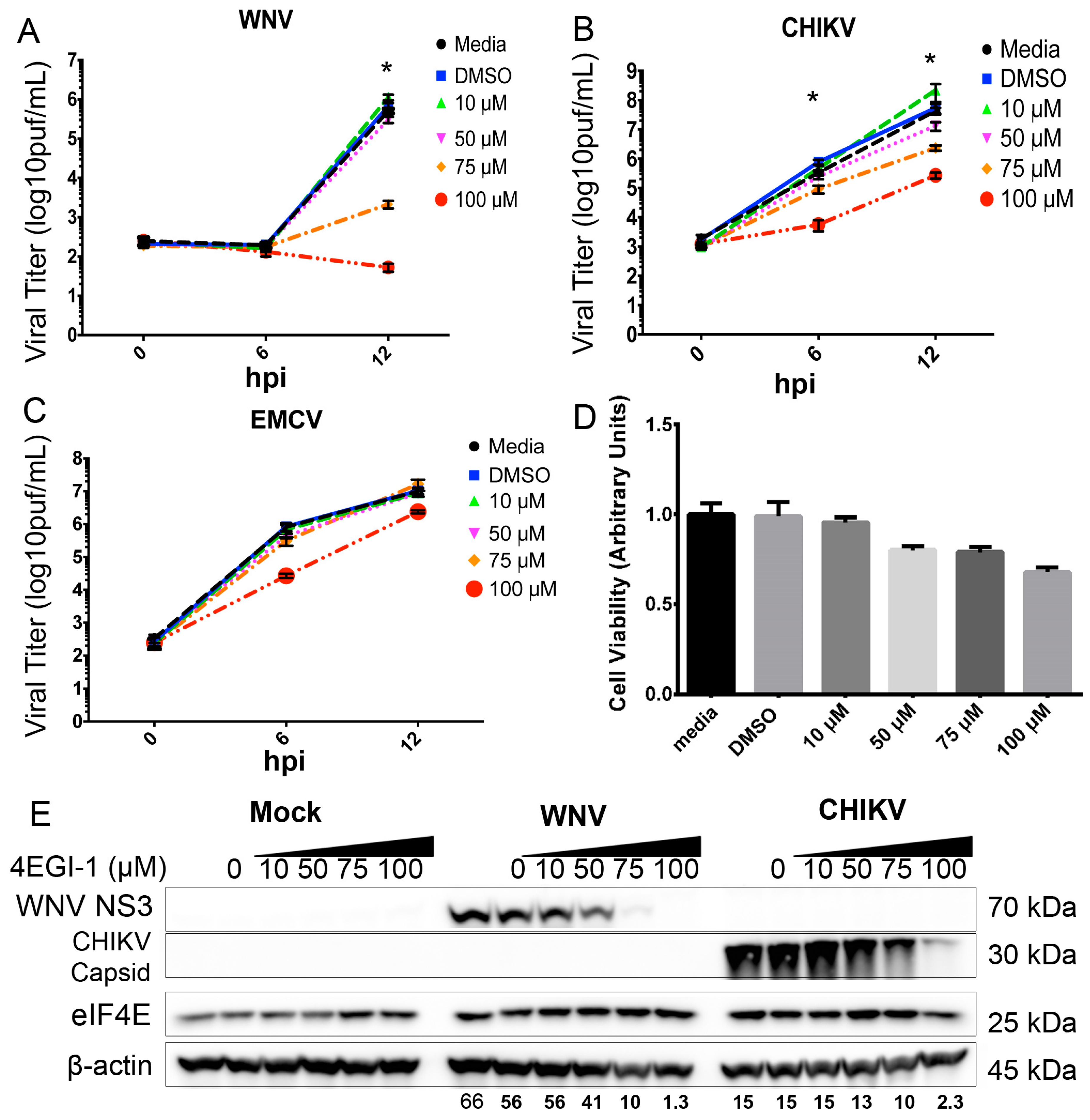
3.5. eIF4F Complex Formation Supports WNV Virus Growth and Protein Expression
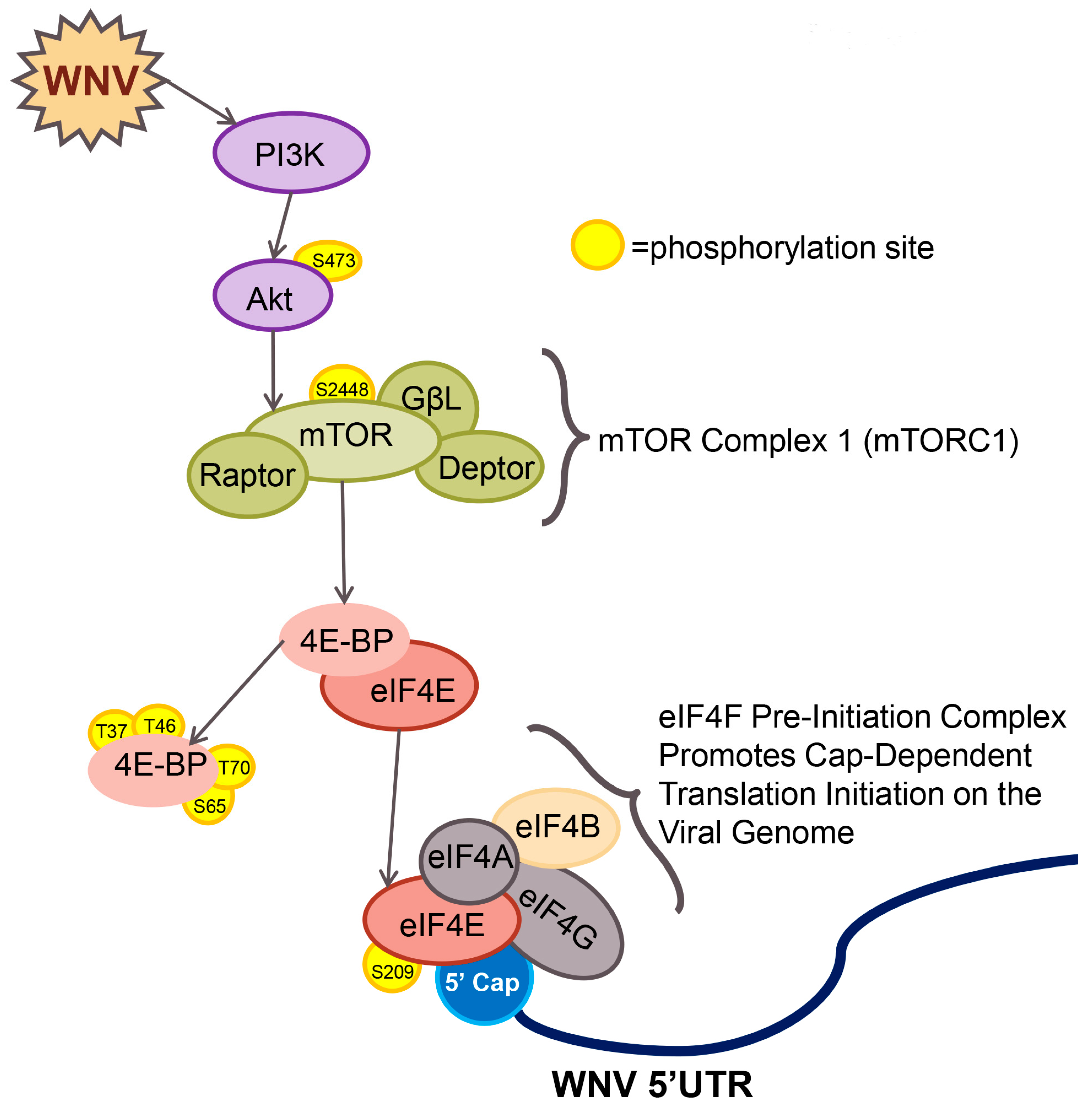
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Egloff, M.-P.; Benarroch, D.; Selisko, B.; Romette, J.-L.; Canard, B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 2002, 21, 2757–2768. [Google Scholar] [CrossRef] [PubMed]
- Wengler, G. The NS 3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. Virology 1993, 197, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Bartelma, G.; Padmanabhan, R. Expression, purification, and characterization of the RNA 5′-triphosphatase activity of dengue virus type 2 nonstructural protein 3. Virology 2002, 299, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale, M., Jr.; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2′-O methylation of the viral mRNA cap by West Nile virus evades IFIT1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef] [PubMed]
- Bowen, R.A.; Nemeth, N.M. Experimental infections with West Nile virus. Curr. Opin. Infect. Dis. 2007, 20, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Barrett, A.D.T. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2004, 2, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Edgil, D.; Polacek, C.; Harris, E. Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J. Virol. 2006, 80, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Shives, K.D.; Beatman, E.L.; Chamanian, M.; O’Brien, C.; Hobson-Peters, J.; Beckham, J.D. West Nile virus-induced activation of mammalian Target of Rapamycin (mTOR) Complex 1 supports viral growth and viral protein expression. J. Virol. 2014, 88, 9458–9471. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.G.; Thomas, G. The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett. 2006, 580, 2821–2829. [Google Scholar] [CrossRef] [PubMed]
- Montero, H.; García-Román, R.; Mora, S.I. eIF4E as a Control Target for Viruses. Viruses 2015, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Schalm, S.S.; Blenis, J. Identification of a conserved motif required for mTOR signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef]
- Dennis, M.D.; Kimball, S.R.; Jefferson, L.S. Mechanistic target of rapamycin complex 1 (mTORC1)-mediated phosphorylation is governed by competition between substrates for interaction with raptor. J. Biol. Chem. 2013, 288, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.Y.; Yoon, S.-O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed]
- Dufner, A.; Thomas, G. Ribosomal S6 Kinase Signaling and the Control of Translation. Exp. Cell Res. 1999, 253, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Shima, H.; Pende, M.; Chen, Y.; Fumagalli, S.; Thomas, G.; Kozma, S.C. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998, 17, 6649–6659. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, C.; Fernandez, A.; Lamb, N.J.; Thomas, G. Nuclear localization of p85s6k: Functional requirement for entry into S phase. EMBO J. 1994, 13, 1557–1565. [Google Scholar] [PubMed]
- Reinhard, C.; Thomas, G.; Kozma, S.C. A single gene encodes two isoforms of the p70 S6 kinase: Activation upon mitogenic stimulation. Proc. Natl. Acad. Sci. USA 1992, 89, 4052–4056. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.D.; Jefferson, L.S.; Kimball, S.R. Role of p70S6K1-mediated phosphorylation of eIF4B and PDCD4 proteins in the regulation of protein synthesis. J. Biol. Chem. 2012, 287, 42890–42899. [Google Scholar] [CrossRef] [PubMed]
- Parsyan, A.; Svitkin, Y.; Shahbazian, D.; Gkogkas, C.; Lasko, P.; Merrick, W.C.; Sonenberg, N. mRNA helicases: The tacticians of translational control. Nat. Rev. Mol. Cell Biol. 2011, 12, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar] [PubMed]
- Livingstone, M.; Bidinosti, M. Rapamycin-insensitive mTORC1 activity controls eIF4E:4E-BP1 binding. F1000Research 2012, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Sukarieh, R.; Sonenberg, N.; Pelletier, J. The eIF4E-binding proteins are modifiers of cytoplasmic eIF4E relocalization during the heat shock response. Am. J. Physiol. Cell Physiol. 2009, 296, C1207–C1217. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Livingstone, M.; Sukarieh, R.; Petroulakis, E.; Gingras, A.C.; Crosby, K.; Smith, B.; Polakiewicz, R.D.; Pelletier, J.; Ferraiuolo, M.A.; et al. Control of eIF4E cellular localization by eIF4E-binding proteins, 4E-BPs. RNA 2008, 14, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D. Manipulation of the host translation initiation complex eIF4F by DNA viruses. Biochem. Soc. Trans. 2010, 38, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Aumayr, M.; Fedosyuk, S.; Ruzicska, K.; Sousa-Blin, C.; Kontaxis, G.; Skern, T. NMR analysis of the interaction of picornaviral proteinases Lb and 2A with their substrate eukaryotic initiation factor 4GII. Protein Sci. 2015, 24, 1979–1996. [Google Scholar] [CrossRef] [PubMed]
- Brault, A.C.; Huang, C.Y.; Langevin, S.A.; Kinney, R.M.; Bowen, R.A.; Ramey, W.N.; Panella, N.A.; Holmes, E.C.; Powers, A.M.; Miller, B.R. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat. Genet. 2007, 39, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Burrack, K.A.S.; Hawman, D.W.; Jupille, H.J.; Oko, L.; Minor, M.; Shives, K.D.; Gunn, B.M.; Long, K.M.; Morrison, T.E. Attenuating mutations in nsP1 reveal tissue specific mechanisms for control of Ross River virus infection. J. Virol. 2014, 88, 3719–3732. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Palmenberg, A.C. Encephalomyocarditis viruses with short poly(C) tracts are more virulent than their mengovirus counterparts. J. Virol. 1995, 69, 2697–2699. [Google Scholar] [PubMed]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, N.; Zinzalla, V.; Hall, M.N. Inducible raptor and rictor knockout mouse embryonic fibroblasts. Methods Mol. Biol. 2012, 821, 267–278. [Google Scholar] [PubMed]
- Hawman, D.W.; Stoermer, K.A.; Montgomery, S.A.; Pal, P.; Oko, L.; Diamond, M.S.; Morrison, T.E. Chronic joint disease caused by persistent Chikungunya virus infection is controlled by the adaptive immune response. J. Virol. 2013, 87, 13878–13888. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative Phosphoproteomics Reveal mTORC1 Activates de Novo Pyrimidine Synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.-E.; Stapleford, K.; Guivel-Benhassine, F.; Vignuzzi, M.; Schwartz, O.; Albert, M.L. Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins via the Activation of the MnK/eIF4E Pathway. PLoS Pathog. 2015, 11, e1005091. [Google Scholar] [CrossRef] [PubMed]
- Thaa, B.; Biasiotto, R.; Eng, K.; Neuvonen, M.; Götte, B.; Rheinemann, L.; Mutso, M.; Utt, A.; Varghese, F.; Balistreri, G. Differential Phosphatidylinositol-3-Kinase-Akt-mTOR Activation by Semliki Forest and Chikungunya Viruses Is Dependent on nsP3 and Connected to Replication Complex Internalization. J. Virol. 2015, 89, 11420–11437. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yue, P.; Chan, C.-B.; Ye, K.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Fu, H.; Khuri, F.R.; Sun, S.Y. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol. Cell. Biol. 2007, 27, 7405–7413. [Google Scholar] [CrossRef] [PubMed]
- Colina, R.; Costa-Mattioli, M.; Dowling, R.J.O.; Jaramillo, M.; Tai, L.H.; Breitbach, C.J.; Martineau, Y.; Larsson, O.; Rong, L.; Svitkin, Y.V.; et al. Translational control of the innate immune response through IRF-7. Nature 2008, 452, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Pende, M.; Um, S.H.; Mieulet, V.; Sticker, M.; Goss, V.L.; Mestan, J.; Mueller, M.; Fumagalli, S.; Kozma, S.C.; Thomas, G. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell. Biol. 2004, 24, 3112–3124. [Google Scholar] [CrossRef] [PubMed]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; García-Moreno, M.; Sanz, M.A.; Carrasco, L. Translation of viral mRNAs that do not require eIF4E is blocked by the inhibitor 4EGI-1. Virology 2013, 444, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Svitkin, Y.; Belsham, G.J.; Pause, A.; Sonenberg, N. Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc. Natl. Acad. Sci. USA 1996, 93, 5578–5583. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.G.; Fingar, D.C. Mammalian target of rapamycin (mTOR): Conducting the cellular signaling symphony. J. Biol. Chem. 2010, 285, 14071–14077. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, C.R.; Ruggero, D. The cancerous translation apparatus. Curr. Opin. Genet. Dev. 2011, 21, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 11988–11993. [Google Scholar] [CrossRef] [PubMed]
- Grosso, S.; Pesce, E.; Brina, D.; Beugnet, A.; Loreni, F.; Biffo, S. Sensitivity of global translation to mTOR inhibition in REN cells depends on the equilibrium between eIF4E and 4E-BP1. PLoS ONE 2011, 6, e29136. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Shang, Y.C.; Zhang, L.; Wang, S.; Maiese, K. Mammalian target of rapamycin: Hitting the bull’s-eye for neurological disorders. Oxid. Med. Cell. Longev. 2010, 3, 374–391. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Ruggero, D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin. Cancer Res. 2010, 16, 4914–4920. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, K.C.; Tartell, M.A.; Herrmann, C.; Hackett, B.A.; Taschuk, F.; Panda, D.; Menghani, S.V.; Sabin, L.R.; Cherry, S. Virus-induced translational arrest through 4EBP1/2-dependent decay of 5′-TOP mRNAs restricts viral infection. Proc. Natl. Acad. Sci. USA 2015, 112, E2920–E2929. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. mTOR: On target for novel therapeutic strategies in the nervous system. Trends Mol. Med. 2013, 19, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Aktas, B.H.; Wang, Y.; He, X.; Sahoo, R.; Zhang, N.; Denoyelle, S.; Kabha, E.; Yang, H.; Freedman, R.; et al. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 2012, 3, 869–881. [Google Scholar] [CrossRef] [PubMed]
- De la Parra, C.; Borrero-Garcia, L.D.; Cruz-Collazo, A.; Schneider, R.J.; Dharmawardhane, S. Equol, an isoflavone metabolite, regulates cancer cell viability and protein synthesis initiation via c-Myc and eIF4G. J. Biol. Chem. 2015, 290, 6047–6057. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 2010, 17, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.-J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Martineau, Y.; Azar, R.; Müller, D.; Lasfargues, C.; El Khawand, S.; Anesia, R.; Pelletier, J.; Bousquet, C.; Pyronnet, S. Pancreatic tumours escape from translational control through 4E-BP1 loss. Oncogene 2014, 33, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Martineau, Y.; Azar, R.; Bousquet, C.; Pyronnet, S. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene 2013, 32, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Shatsky, I.N.; Dmitriev, S.E.; Andreev, D.E.; Terenin, I.M. Transcriptome-wide studies uncover the diversity of modes of mRNA recruitment to eukaryotic ribosomes. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 164–177. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shives, K.D.; Massey, A.R.; May, N.A.; Morrison, T.E.; Beckham, J.D. 4EBP-Dependent Signaling Supports West Nile Virus Growth and Protein Expression. Viruses 2016, 8, 287. https://doi.org/10.3390/v8100287
Shives KD, Massey AR, May NA, Morrison TE, Beckham JD. 4EBP-Dependent Signaling Supports West Nile Virus Growth and Protein Expression. Viruses. 2016; 8(10):287. https://doi.org/10.3390/v8100287
Chicago/Turabian StyleShives, Katherine D., Aaron R. Massey, Nicholas A. May, Thomas E. Morrison, and J. David Beckham. 2016. "4EBP-Dependent Signaling Supports West Nile Virus Growth and Protein Expression" Viruses 8, no. 10: 287. https://doi.org/10.3390/v8100287
APA StyleShives, K. D., Massey, A. R., May, N. A., Morrison, T. E., & Beckham, J. D. (2016). 4EBP-Dependent Signaling Supports West Nile Virus Growth and Protein Expression. Viruses, 8(10), 287. https://doi.org/10.3390/v8100287