
Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase—A Potential Strategy for Suppression of the Wnt Pathway

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
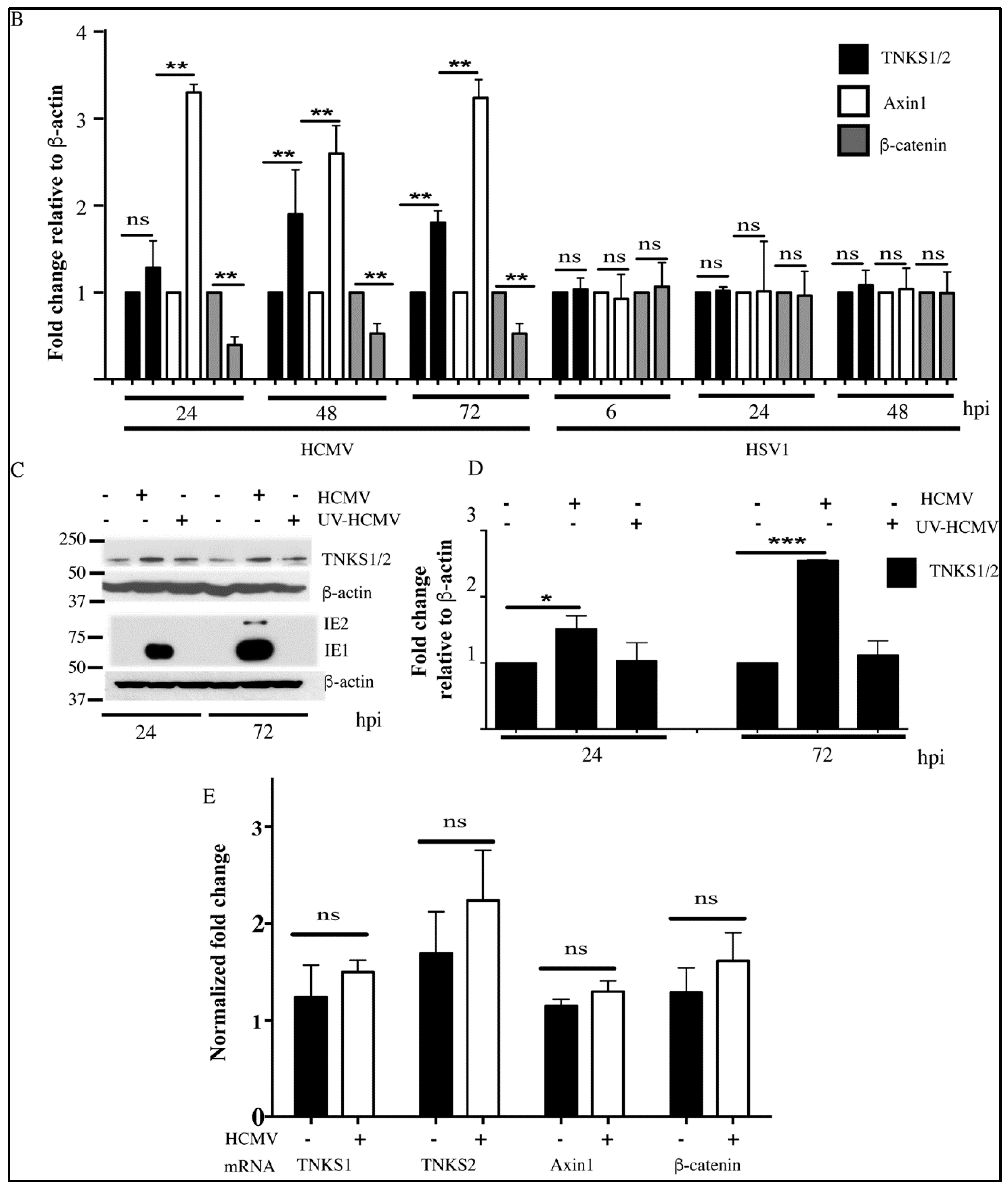
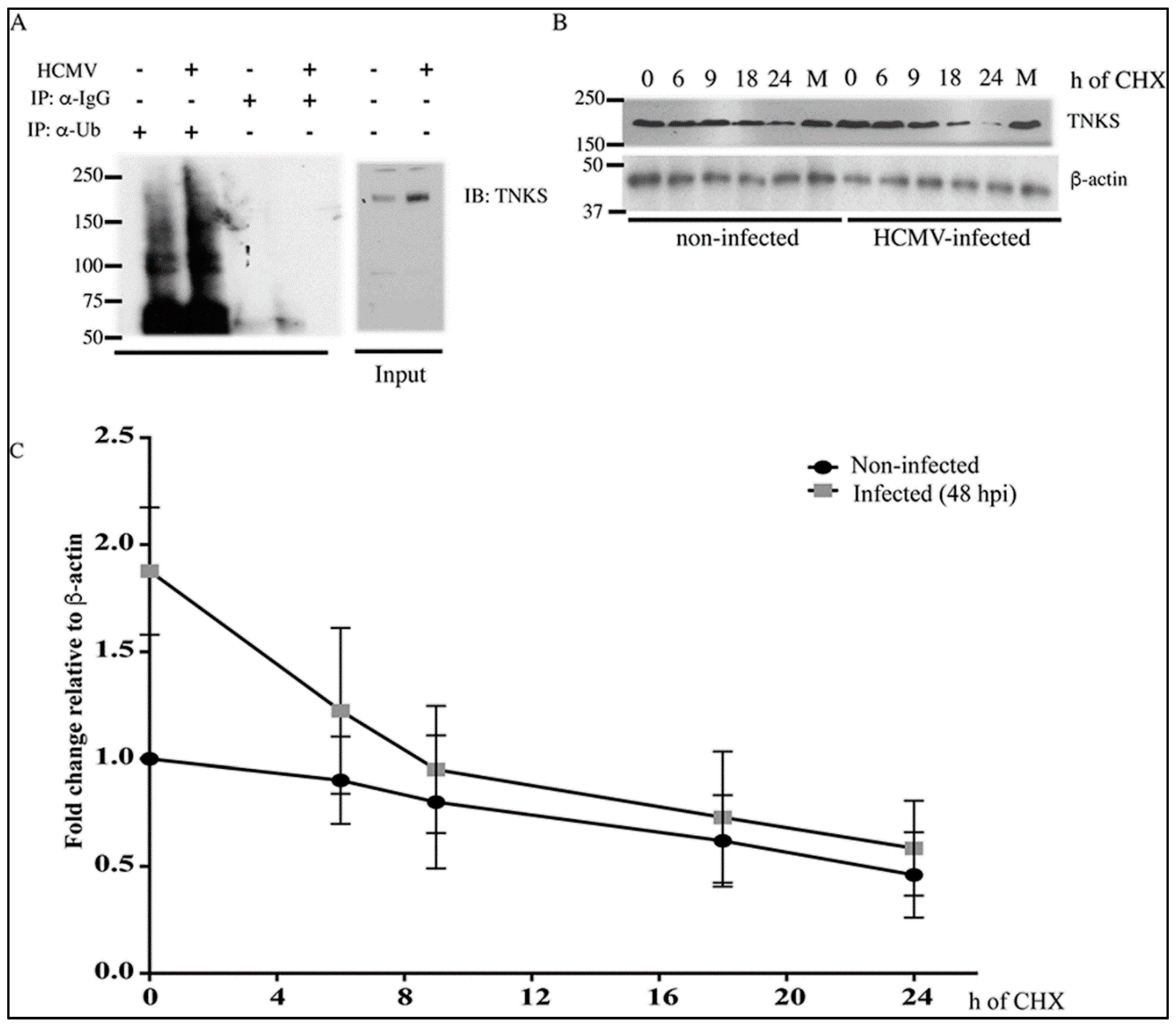
2.1. Human Cytomegalovirus (HCMV) Infection Stabilizes TNKS
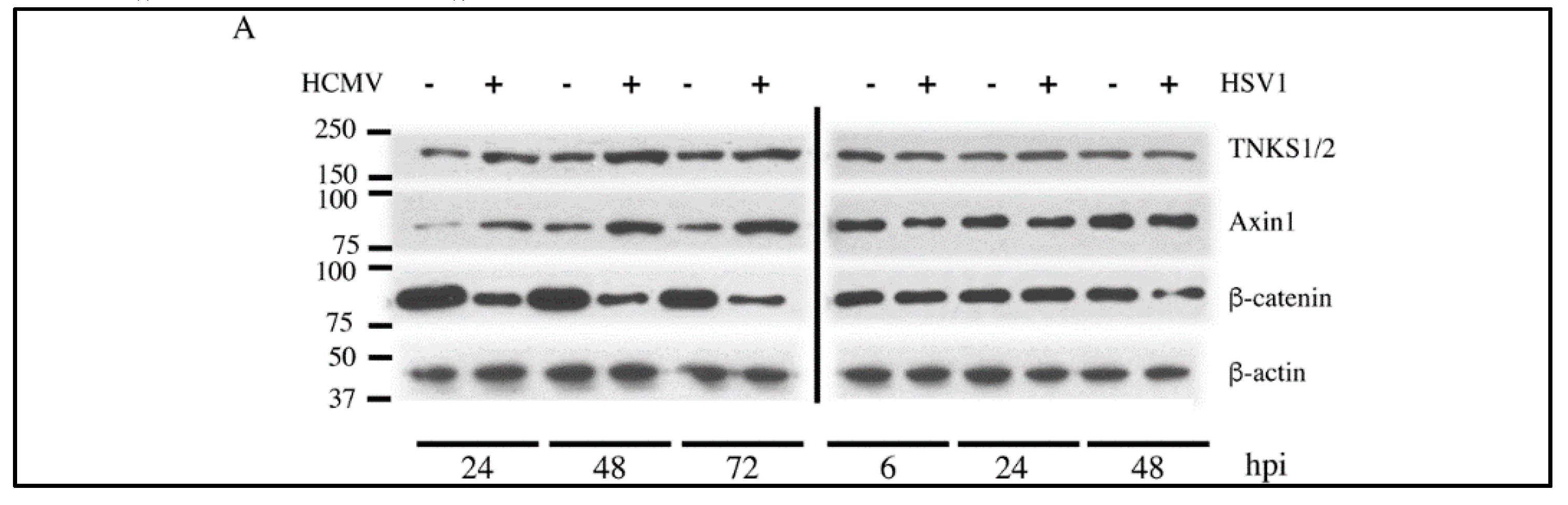
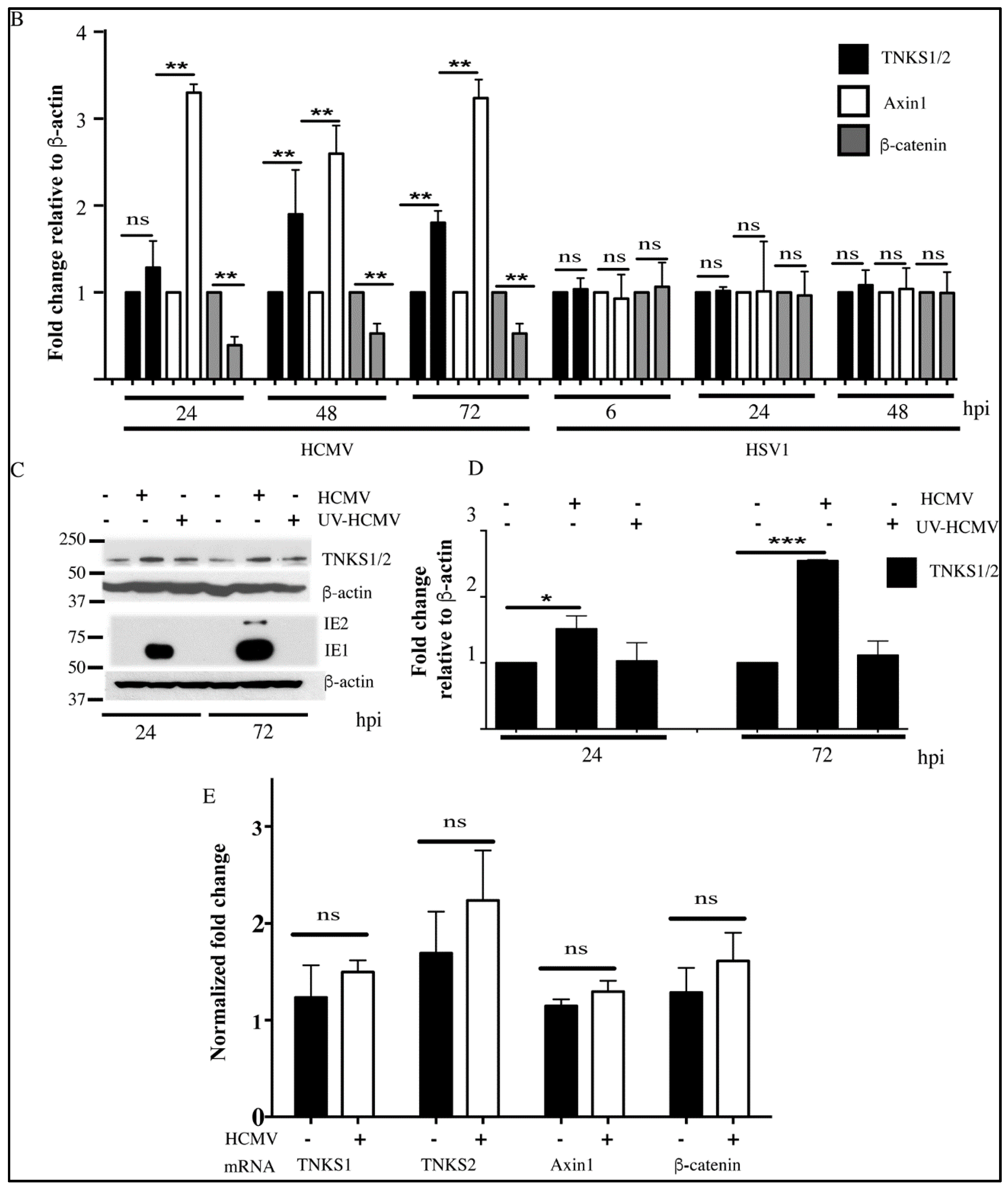
2.2. Effect of HCMV Infection on TNK Accumulation is Post-Translational

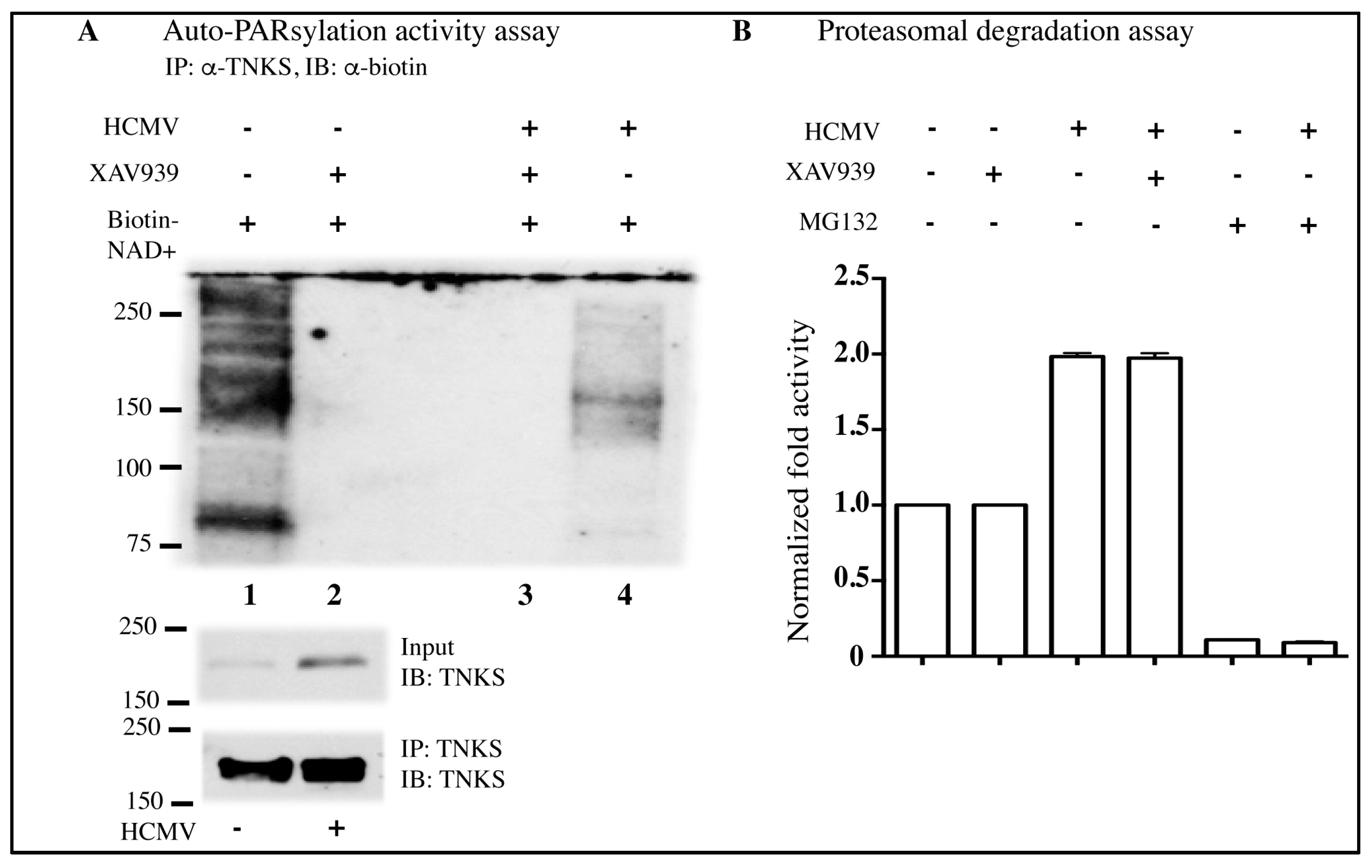
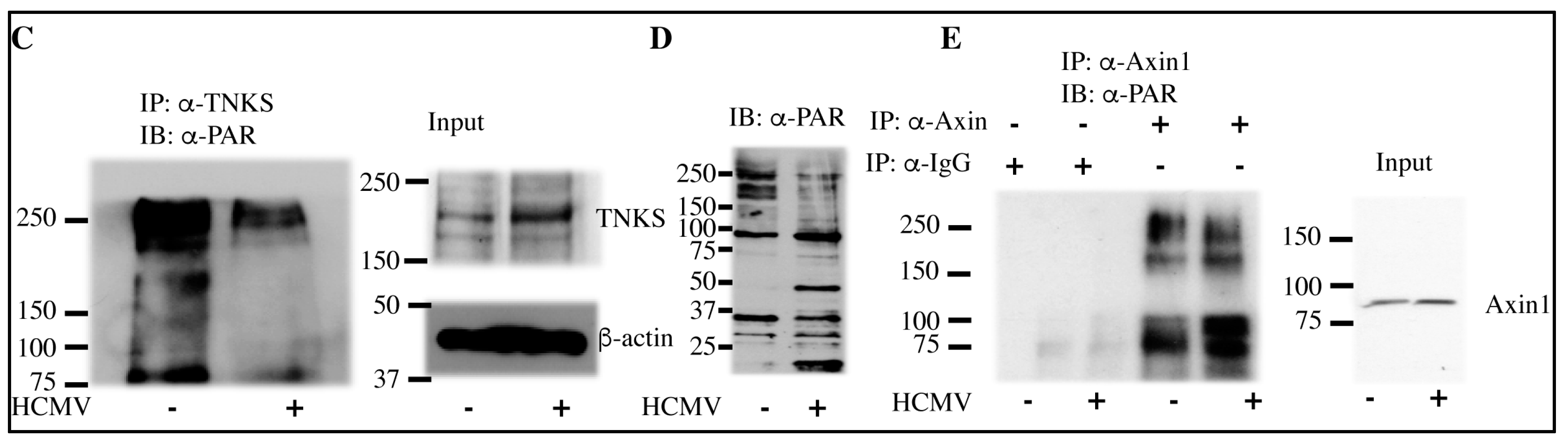
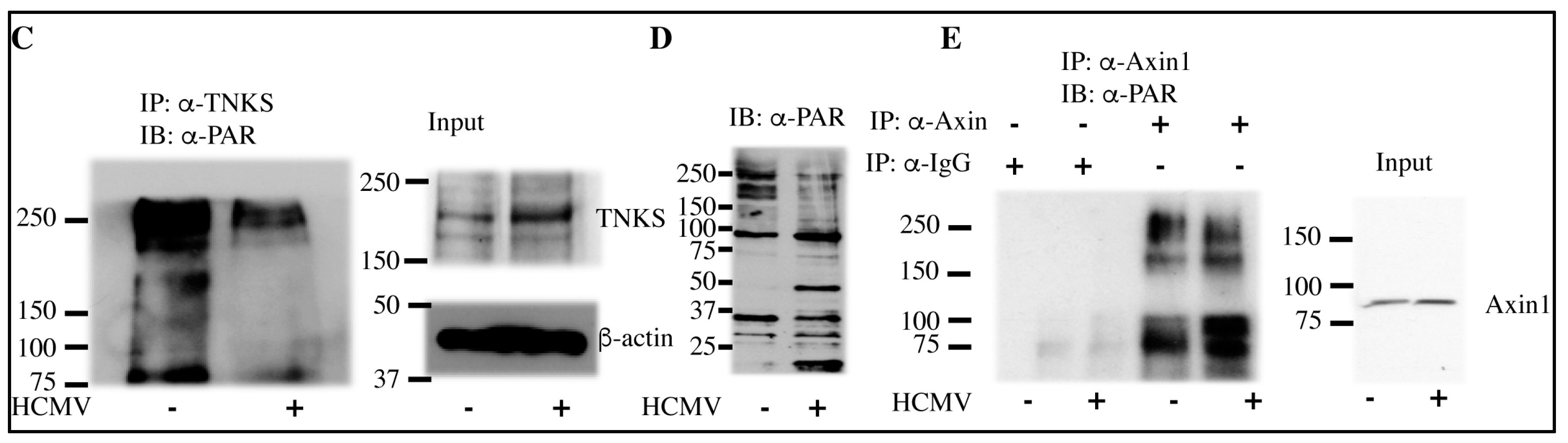
2.3. HCMV Infection Reduces PARsylation Activity of TNKS

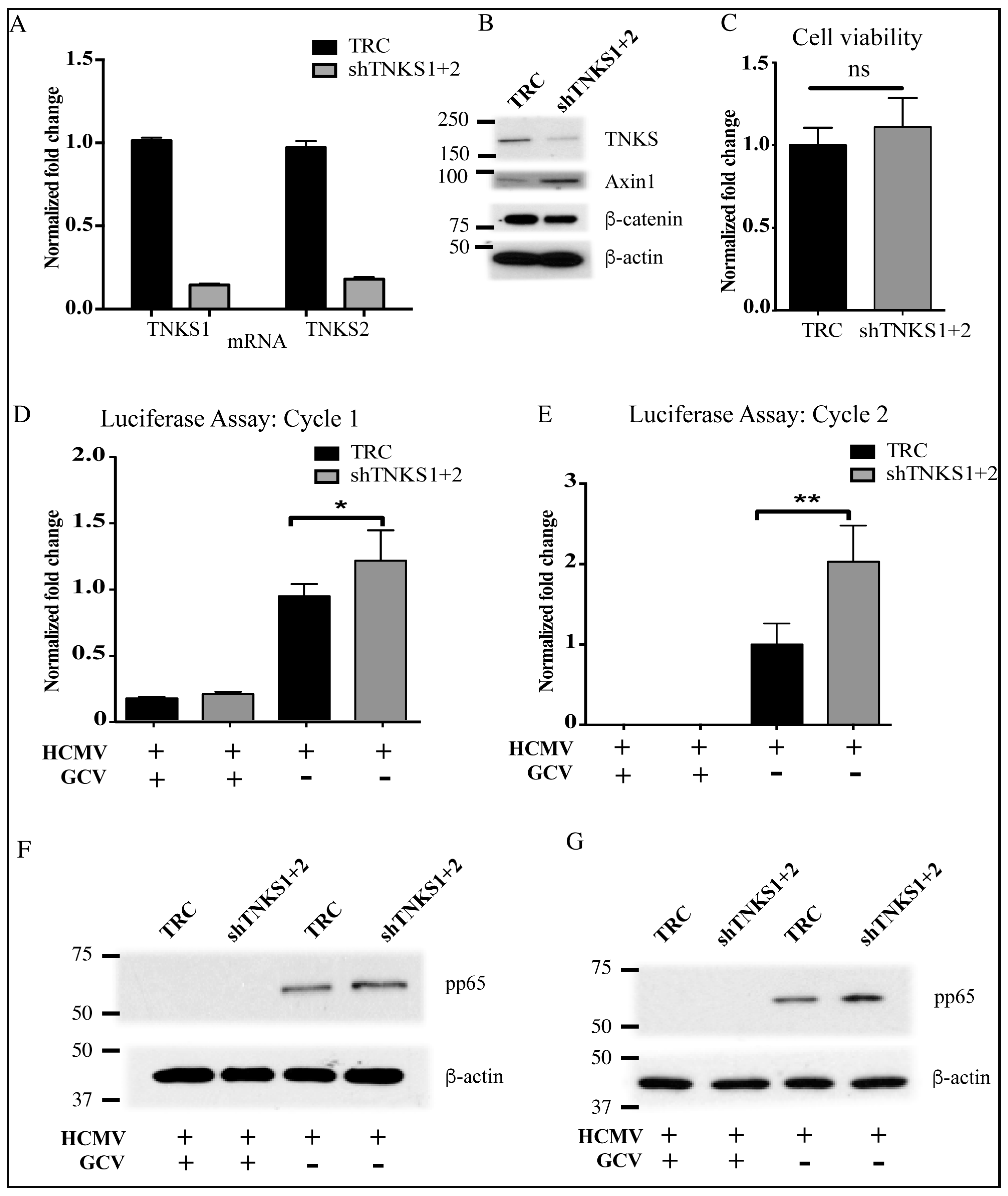
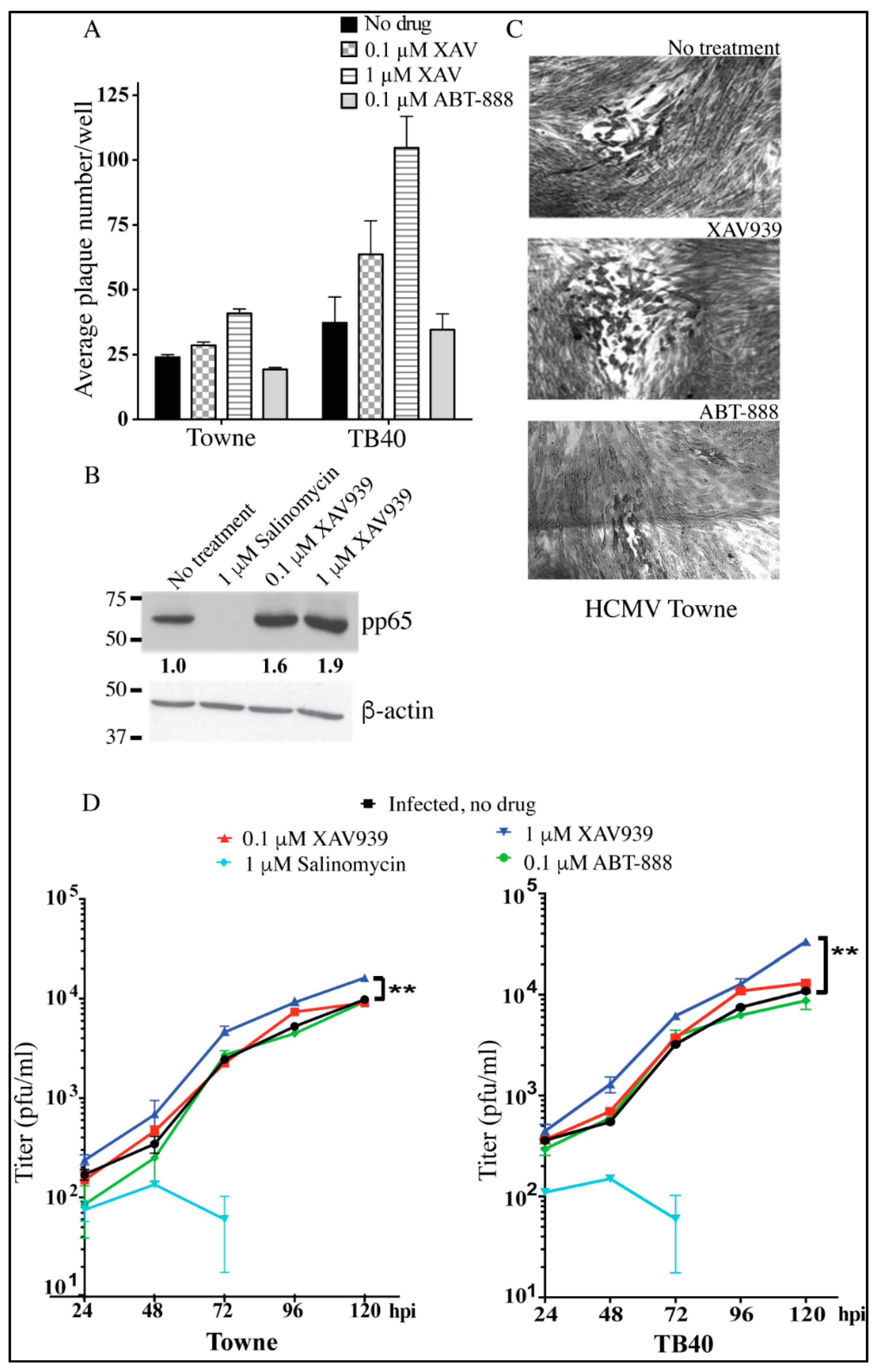
2.4. Inhibition of TNKS Activity Promotes HCMV Replication
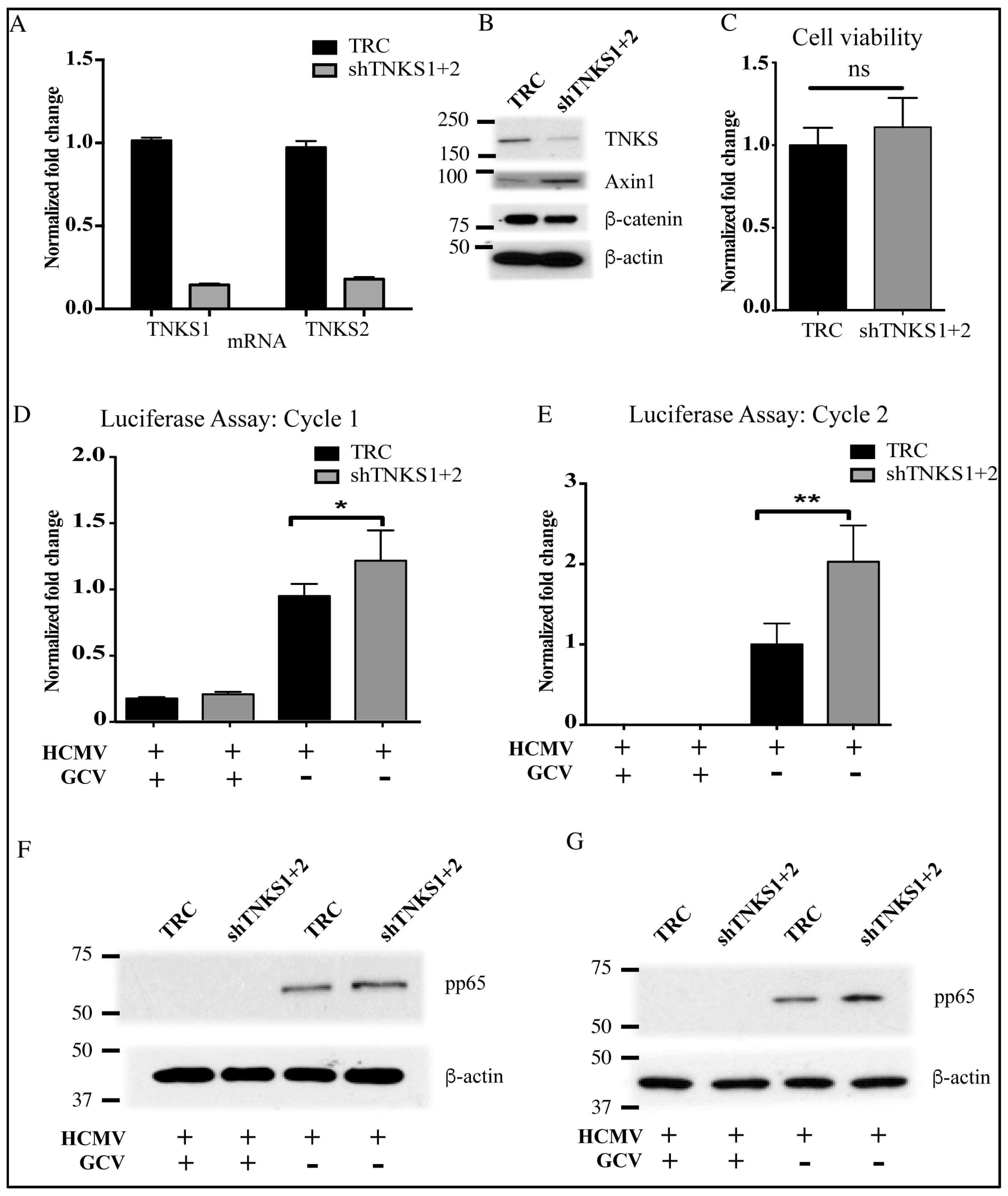
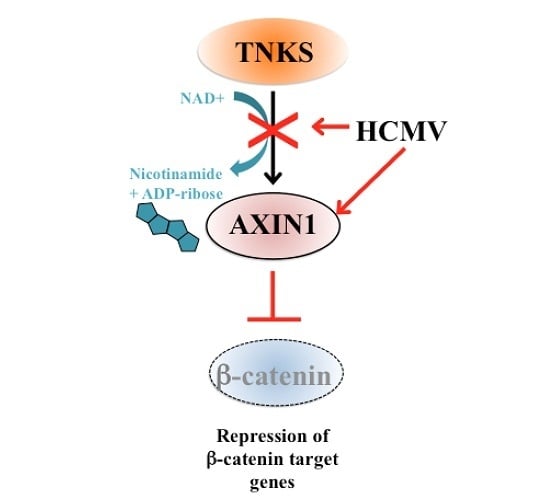
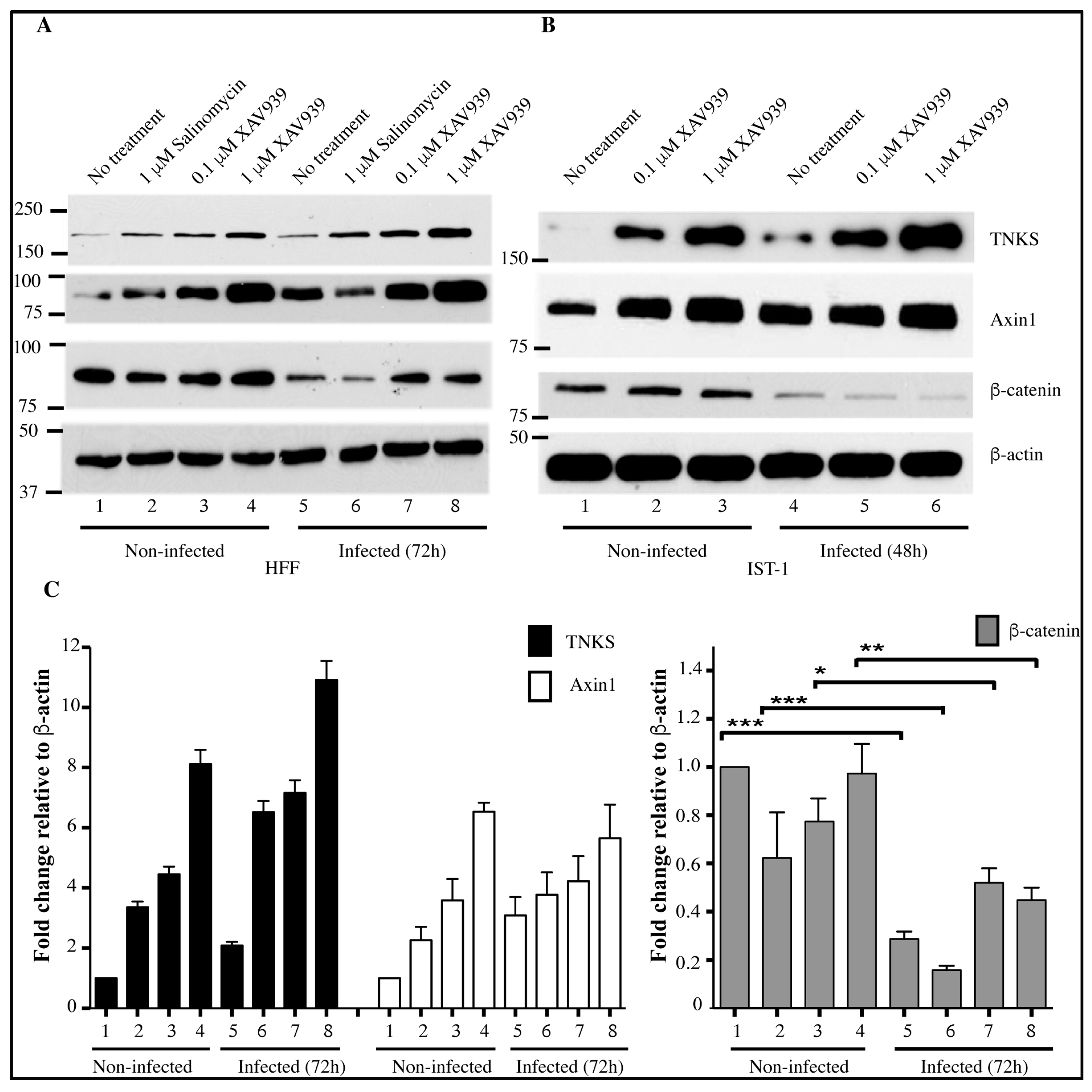
2.5. Accumulation of TNKS during Infection Correlates with Wnt Inhibition
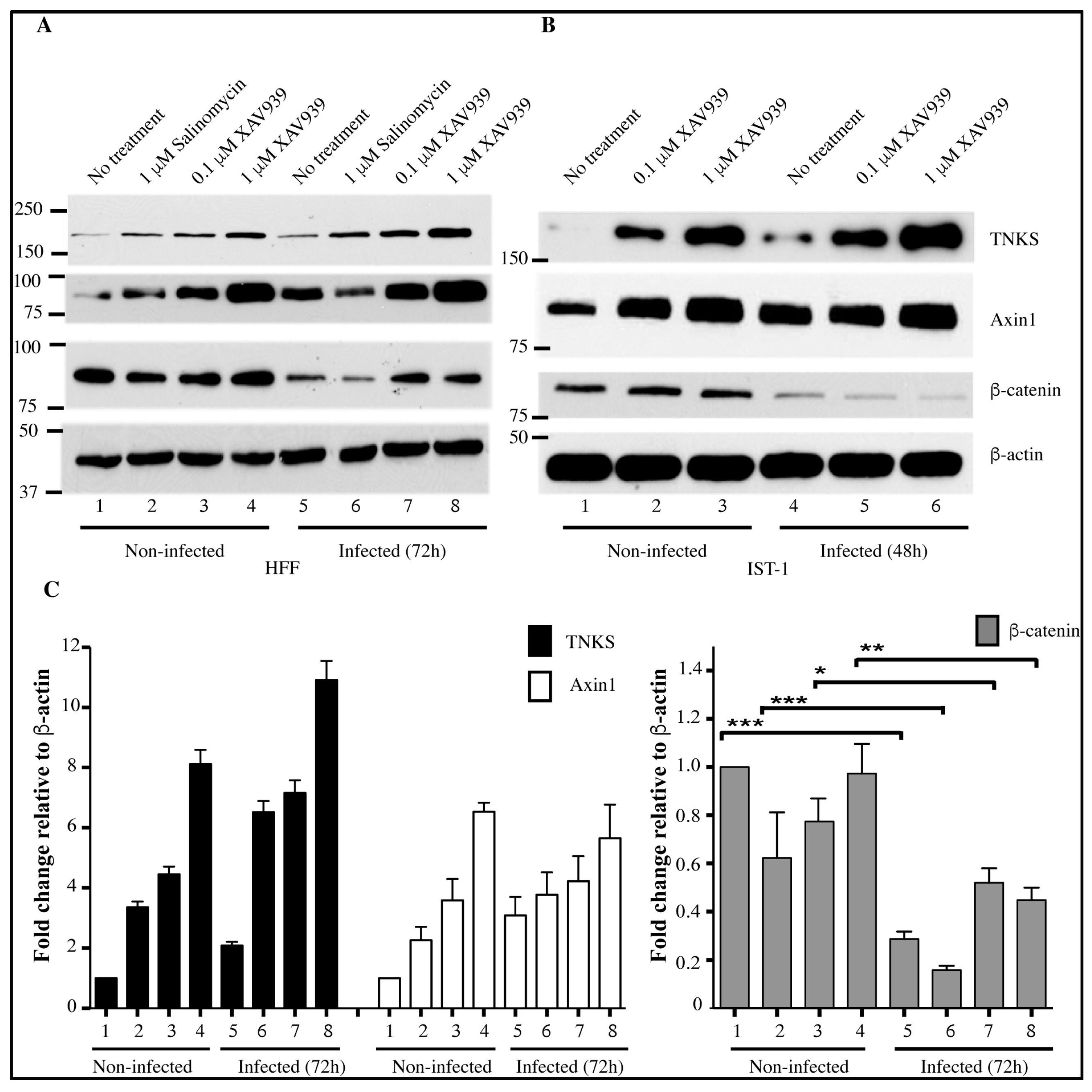
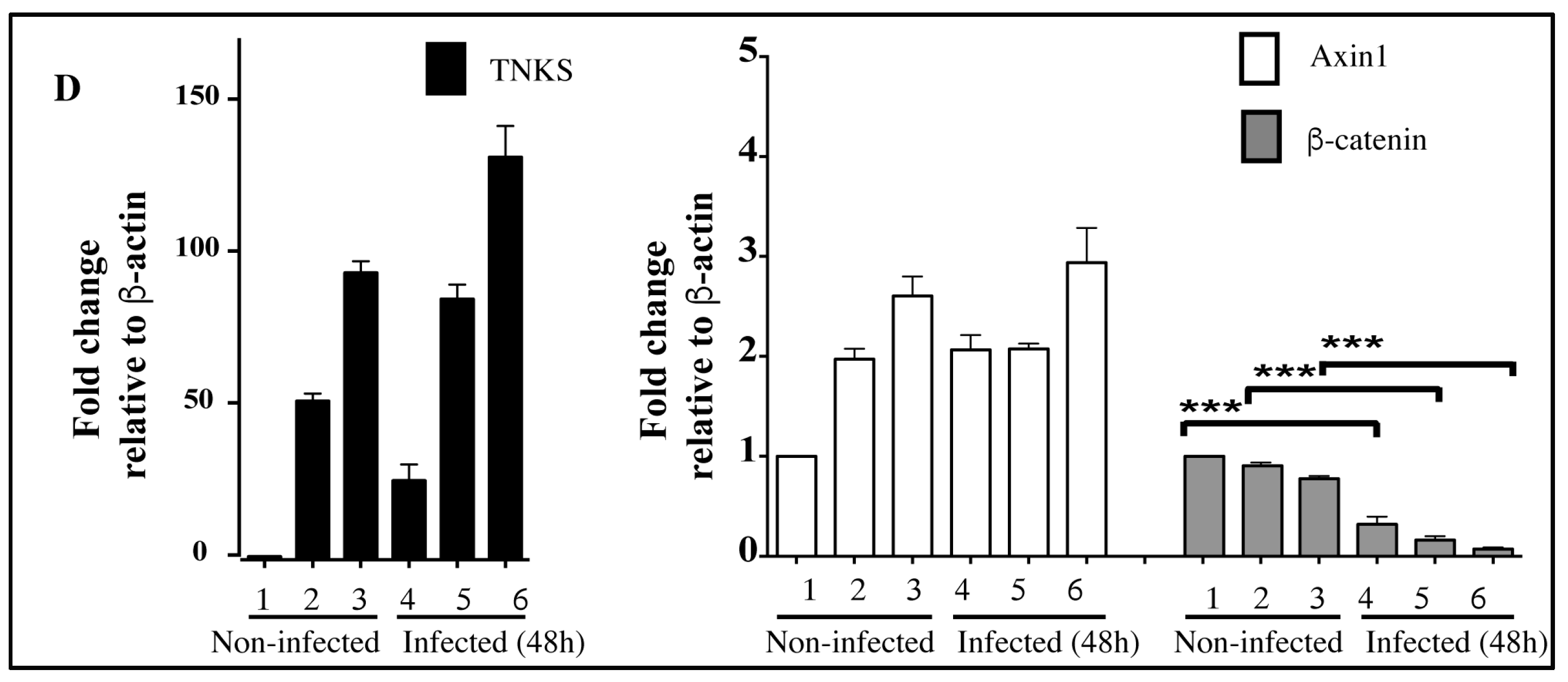
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Viruses
4.3. Cell Culture, Virus Infection and Anti-Viral Assays
4.4. SDS-PAGE and Immunoblot Analysis
4.5. Protein Immunoprecipitation (IP)
4.6. IP of TNKS and In Vitro PARsylation Assay
4.7. Determination of TNKS Degradation
4.8. Proteasome Assay
4.9. RNA Isolation and Real-Time Quantitative Reverse Transcriptase (qRT) PCR
4.10. Knockdown of TNKS
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Knipe, D.M.; Howley, P.M. Fields virology., 6th ed.; Wolters Kluwer/Lippincott Williams and Wilkins Health: Philadelphia, PA, USA, 2013. [Google Scholar]
- Roy, S.; Arav-Boger, R. New cell-signaling pathways for controlling cytomegalovirus replication. Am. J. Transplant. 2014, 14, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.D.; Liu, J.; Fujimuro, M. Notch and wnt signaling: Mimicry and manipulation by γ herpesviruses. Sci. STKE 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Jerchow, B.A.; Wurtele, M.; Grimm, J.; Asbrand, C.; Wirtz, R.; Kuhl, M.; Wedlich, D.; Birchmeier, W. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3β. Science 1998, 280, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Kishida, M.; Koyama, S.; Kishida, S.; Matsubara, K.; Nakashima, S.; Higano, K.; Takada, R.; Takada, S.; Kikuchi, A. Axin prevents Wnt-3a-induced accumulation of β-catenin. Oncogene 1999, 18, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Salic, A.; Kruger, R.; Heinrich, R.; Kirschner, M.W. The roles of APC and axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003, 1, E10. [Google Scholar] [CrossRef] [PubMed]
- Salic, A.; Lee, E.; Mayer, L.; Kirschner, M.W. Control of β-catenin stability: Reconstitution of the cytoplasmic steps of the wnt pathway in xenopus egg extracts. Mol. Cell 2000, 5, 523–532. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Zwezdaryk, K.; Ferris, M.; Shan, B.; Morris, C.A.; Sullivan, D.E. Human cytomegalovirus infection dysregulates the canonical Wnt/β-catenin signaling pathway. PLoS Pathog. 2012, 8, e1002959. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; He, R.; Venkatadri, R.; Forman, M.; Arav-Boger, R. Wnt modulating agents inhibit human cytomegalovirus replication. Antimicrob. Agents Chemother. 2013, 57, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yamauchi, Y.; Kamakura, M.; Murayama, T.; Goshima, F.; Kimura, H.; Nishiyama, Y. Herpes simplex virus requires poly(ADP-ribose) polymerase activity for efficient replication and induces extracellular signal-related kinase-dependent phosphorylation and ICP0-dependent nuclear localization of tankyrase 1. J. Virol. 2012, 86, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.Y.; Meyer, T.N.; Schwesinger, C.; Tsun, Z.Y.; Lee, R.M.; Chi, N.W. Tankyrase recruitment to the lateral membrane in polarized epithelial cells: Regulation by cell-cell contact and protein poly(ADP-ribosyl)ation. Biochem. J. 2006, 399, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Mickanin, C.; Feng, Y.; Charlat, O.; Michaud, G.A.; Schirle, M.; Shi, X.; Hild, M.; Bauer, A.; et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 2011, 13, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Bao, R.; Christova, T.; Song, S.; Angers, S.; Yan, X.; Attisano, L. Inhibition of tankyrases induces axin stabilization and blocks wnt signalling in breast cancer cells. PLoS ONE 2012, 7, e48670. [Google Scholar] [CrossRef] [PubMed]
- Morrone, S.; Cheng, Z.; Moon, R.T.; Cong, F.; Xu, W. Crystal structure of a tankyrase-axin complex and its implications for axin turnover and tankyrase substrate recruitment. Proc. Nat. Acad. Sci. USA 2012, 109, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Lehtio, L.; Chi, N.W.; Krauss, S. Tankyrases as drug targets. FEBS J. 2013, 280, 3576–3593. [Google Scholar] [CrossRef] [PubMed]
- Riffell, J.L.; Lord, C.J.; Ashworth, A. Tankyrase-targeted therapeutics: Expanding opportunities in the PARP family. Nat. Rev. Drug Discov. 2012, 11, 923–936. [Google Scholar] [CrossRef] [PubMed]
- De la Roche, M.; Ibrahim, A.E.; Mieszczanek, J.; Bienz, M. LEF1 and B9l shield β-catenin from inactivation by axin, desensitizing colorectal cancer cells to tankyrase inhibitors. Cancer Res. 2014, 74, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Salvant, B.S.; Fortunato, E.A.; Spector, D.H. Cell cycle dysregulation by human cytomegalovirus: Influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J. Virol. 1998, 72, 3729–3741. [Google Scholar] [PubMed]
- Casavant, N.C.; Luo, M.H.; Rosenke, K.; Winegardner, T.; Zurawska, A.; Fortunato, E.A. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J. Virol. 2006, 80, 8390–8401. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Kim, D.J.; Lee, J.M.; Choi, C.Y.; Ahn, B.Y.; Hayward, G.S.; Ahn, J.H. Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. J. Virol. 2004, 78, 6527–6542. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Doerr, H.W.; Cinatl, J. The story of human cytomegalovirus and cancer: Increasing evidence and open questions. Neoplasia 2009, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.P.; Tang, Q. Identification of cellular proteins that interact with human cytomegalovirus immediate-early protein 1 by protein array assay. Viruses 2014, 6, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, O.; Cohen, Y.; Shitrit, A.; Shani, O.; Le-Trilling, V.T.; Trilling, M.; Friedlander, G.; Tanenbaum, M.; Stern-Ginossar, N. The transcription and translation landscapes during human cytomegalovirus infection reveal novel host-pathogen interactions. PLoS Pathog. 2015, 11, e1005288. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Hayward, S.D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3β. J. Virol. 2003, 77, 8019–8030. [Google Scholar] [CrossRef] [PubMed]
- Callow, M.G.; Tran, H.; Phu, L.; Lau, T.; Lee, J.; Sandoval, W.N.; Liu, P.S.; Bheddah, S.; Tao, J.; Lill, J.R.; et al. Ubiquitin ligase RNF146 regulates tankyrase and axin to promote Wnt signaling. PLoS ONE 2011, 6, e22595. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L. The WWE domain: A common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem. Sci. 2001, 26, 273–275. [Google Scholar] [CrossRef]
- Child, S.J.; Jarrahian, S.; Harper, V.M.; Geballe, A.P. Complementation of vaccinia virus lacking the double-stranded RNA-binding protein gene E3L by human cytomegalovirus. J. Virol. 2002, 76, 4912–4918. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Sandford, G.; Hayward, G.S.; Burns, W.H.; Posner, G.H.; Forman, M.; Arav-Boger, R. Recombinant luciferase-expressing human cytomegalovirus (CMV) for evaluation of CMV inhibitors. Virol. J. 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.; Wang, T.; Wu, T.; Kurman, R.J.; Gearhart, J.D. Expression of Mel-CAM in implantation site intermediate trophoblastic cell line, IST-1, limits its migration on uterine smooth muscle cells. J. Cell Sci. 1998, 111, 2655–2664. [Google Scholar] [PubMed]
- E, X.; Pickering, M.T.; Debatis, M.; Castillo, J.; Lagadinos, A.; Wang, S.; Lu, S.; Kowalik, T.F. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011, 7, e1001342. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Giriat, I.; Schmitt, A.; de Lange, T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 1998, 282, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Dargusch, R.; Maher, P.A.; Schubert, D. Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer’s disease. J. Neurosci. 2013, 33, 10512–10525. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, S.; Liu, F.; Arav-Boger, R. Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase—A Potential Strategy for Suppression of the Wnt Pathway. Viruses 2016, 8, 8. https://doi.org/10.3390/v8010008
Roy S, Liu F, Arav-Boger R. Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase—A Potential Strategy for Suppression of the Wnt Pathway. Viruses. 2016; 8(1):8. https://doi.org/10.3390/v8010008
Chicago/Turabian StyleRoy, Sujayita, Fengjie Liu, and Ravit Arav-Boger. 2016. "Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase—A Potential Strategy for Suppression of the Wnt Pathway" Viruses 8, no. 1: 8. https://doi.org/10.3390/v8010008
APA StyleRoy, S., Liu, F., & Arav-Boger, R. (2016). Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase—A Potential Strategy for Suppression of the Wnt Pathway. Viruses, 8(1), 8. https://doi.org/10.3390/v8010008