
Anti-HBV Drugs: Progress, Unmet Needs, and New Hope

Abstract
:1. Introduction
2. HBV
2.1. Morphology and Viral Proteins
2.2. Life Cycle
3. Current State of Anti-HBV Drugs
3.1. Immunoregulators
3.1.1. Interferon (IFN)
3.1.2. Thymosin-α1
3.1.3. Cytokines
3.2. Nucleos/Tide Analogues (NAs)
3.2.1. Lamivudine
3.2.2. Telbivudine
3.2.3. Entecavir
3.2.4. Adefovir
3.2.5. Tenofovir
3.3. Traditional Chinese Medicine
3.3.1. Sedum sarmentosum Granules

| Structures | Active Form Structures | EC50 | Therapeutic Target | |
|---|---|---|---|---|
| Lamivudine (EPIVIR-HBV [31]) | 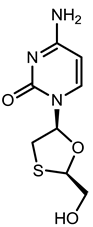 | 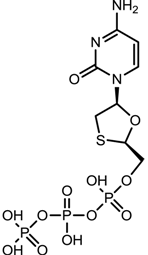 | Varies from 0.01 μM (2.3 ng/mL) to 5.6 μM (1.3 μg/mL) depending upon the duration of exposure of cells to lamivudine, the cell model system, and the protocol used | Inhibition of the RNA- and DNA-dependent polymerase activities of HBV reverse transcriptase |
| Telbivudine (Sebivo [32]) | 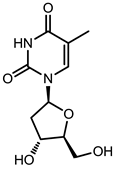 | 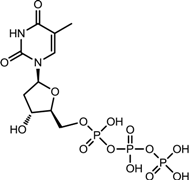 | HBV first strand (EC50 = 0.4–1.3 μM) and second strand (EC50 = 0.12–0.24 μM) | Inhibition of HBV DNA polymerase (reverse transcriptase) by competing with the natural substrate, thymidine 5ʹ-triphosphate |
| Entecavir (Baraclude [33]) | 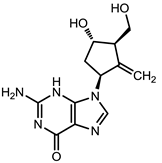 |  | 0.004 μM in human HepG2 cells transfected with wild-type HBV, 0.026 μM (range, 0.010–0.059 μM) against lamivudine resistance HBV (rtL180M and rtM204V) | Inhibition of (1) priming of the HBV polymerase; (2) reverse transcription of the negative strand DNA from the pregenomic messenger RNA; and (3) synthesis of the positive strand HBV DNA |
| Adefovir dipivoxil (SigmaPharm Laboratories, LLC [34]) |  |  | Ranges from 0.2 to 2.5 μM in HBV-transfected human hepatoma cell lines | Inhibition of HBV DNA polymerase (reverse transcriptase) by competing with the natural substrate deoxyadenosine triphosphate and by causing DNA chain termination after its incorporation into viral DNA |
| Tenofovir (VIREAD [35]) | 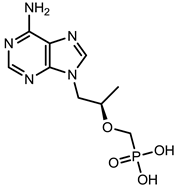 |  | Ranges from 0.04 μM to 8.5 μM | Inhibition of the activity of HBV reverse transcriptase by competing with the natural substrate deoxyadenosine 5ʹ-triphosphate and, after incorporation into DNA, by DNA chain termination |
| Oxymatrine | 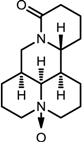 |  | Unknown | Unknown |
3.3.2. Oxymatrine
4. New Anti-HBV Drugs under Development and Evaluation
4.1. New Drugs that Target the Viral Components
4.1.1. MCC-478
4.1.2. cccDNA
4.1.3. HBsAg Gene
4.1.4. Chinese Herbal Medicines
4.2. New Drugs that Target Cellular Factors
4.2.1. HBV Receptors
4.2.2. Novel Target: La Protein Inhibitor (HBSC11)
4.2.3. Transforming Growth Factor-β (TGF-β)
4.2.4. MicroRNAs
4.3. Immune Checkpoints
5. Guidelines for Currently Approved Medications
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grimm, D.; Thimme, R.; Blum, H.E. HBV life cycle and novel drug targets. Hepatol. Int. 2011, 5, 644–653. [Google Scholar] [CrossRef]
- Stein, L.L.; Loomba, R. Drug targets in hepatitis B virus infection. Infect. Disord. Drug Targets 2009, 9, 105–116. [Google Scholar] [CrossRef]
- WHO. Guidelines for the Prevention, Care and Treatment of Persons with Chronic Hepatitis B Infection. Available online: http://www.who.int/hiv/pub/hepatitis/hepatitis-b-guidelines/en/ (accessed on 8 July 2015).
- Lu, F.M.; Zhuang, H. Management of hepatitis B in China. Chin. Med. J. 2009, 122, 3–4. [Google Scholar]
- Bhattacharya, D.; Thio, C.L. Review of hepatitis B therapeutics. Clin. Infect. Dis. 2010, 51, 1201–1208. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Chen, A.; Panjaworayan, T.T.N.; Brown, C.M. Prospects for inhibiting the post-transcriptional regulation of gene expression in hepatitis B virus. World J. Gastroenterol. 2014, 20, 7993–8004. [Google Scholar] [CrossRef]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef]
- Bharadwaj, M.; Roy, G.; Dutta, K.; Misbah, M.; Husain, M.; Hussain, S. Tackling hepatitis B virus-associated hepatocellular carcinoma—The future is now. Cancer Metastasis Rev. 2013, 32, 229–268. [Google Scholar] [CrossRef]
- Singer, G.A.; Zielsdorf, S.; Fleetwood, V.A.; Alvey, N.; Cohen, E.; Eswaran, S.; Shah, N.; Chan, E.Y.; Hertl, M.; Fayek, S.A. Limited hepatitis b immunoglobulin with potent nucleos(t)ide analogue is a cost-effective prophylaxis against hepatitis b virus after liver transplantation. Transplant. Proc. 2015, 47, 478–484. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis b virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Zhao, Z.M.; Jin, Y.; Gan, Y.; Zhu, Y.; Chen, T.Y.; Wang, J.B.; Sun, Y.; Cao, Z.G.; Qian, G.S.; Tu, H. Novel approach to identifying the hepatitis B virus pre-s deletions associated with hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 13573–13581. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and d virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Chua, P.K.; Wang, R.Y.; Lin, M.H.; Masuda, T.; Suk, F.M.; Shih, C. Reduced secretion of virions and hepatitis B virus (HBV) surface antigen of a naturally occurring HBV variant correlates with the accumulation of the small s envelope protein in the endoplasmic reticulum and golgi apparatus. J. Virol. 2005, 79, 13483–13496. [Google Scholar] [CrossRef]
- Gaggar, A.; Coeshott, C.; Apelian, D.; Rodell, T.; Armstrong, B.R.; Shen, G.; Subramanian, G.M.; McHutchison, J.G. Safety, tolerability and immunogenicity of gs-4774, a hepatitis B virus-specific therapeutic vaccine, in healthy subjects: A randomized study. Vaccine 2014, 32, 4925–4931. [Google Scholar] [CrossRef]
- Jiang, W. Blockade of b7-h1 enhances dendritic cell-mediated T cell response and antiviral immunity in HBV transgenic mice. Vaccine 2012, 30, 758–766. [Google Scholar] [CrossRef]
- Tian, Y.; Chen, W.L.; Ou, J.H. Effects of interferon-alpha/beta on HBV replication determined by viral load. PLoS Pathog. 2011, 7, e1002159. [Google Scholar] [CrossRef]
- Liaw, Y.F.; Kao, J.H.; Piratvisuth, T.; Chien, R.N. Asian-pacific consensus statement on the management of chronic hepatitis B: A 2012 update. Hepatol. Int. 2012, 6, 531–561. [Google Scholar] [CrossRef]
- Xiang, X.G.; Xie, Q. IL-35: A potential therapeutic target for controlling hepatitis B virus infection. J. Dig. Dis. 2015, 16, 1–6. [Google Scholar] [CrossRef]
- Lai, C.L.; Dienstag, J.; Schiff, E.; Leung, N.W.; Atkins, M.; Hunt, C.; Brown, N.; Woessner, M.; Boehme, R.; Condreay, L. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin. Infect. Dis. 2003, 36, 687–696. [Google Scholar] [CrossRef]
- Yao, G.B.; Zhu, M.; Cui, Z.Y.; Wang, B.E.; Yao, J.L.; Zeng, M.D. A 7-year study of lamivudine therapy for hepatitis B virus E antigen-positive chronic hepatitis B patients in china. J. Dig. Dis. 2009, 10, 131–137. [Google Scholar] [CrossRef]
- Hou, J.; Yin, Y.K.; Xu, D.; Tan, D.; Niu, J.; Zhou, X.; Wang, Y.; Zhu, L.; He, Y.; Ren, H.; et al. Telbivudine versus lamivudine in chinese patients with chronic hepatitis B: Results at 1 year of a randomized, double-blind trial. Hepatology 2008, 47, 447–454. [Google Scholar] [CrossRef]
- Liaw, Y.F.; Gane, E.; Leung, N.; Zeuzem, S.; Wang, Y.; Lai, C.L.; Heathcote, E.J.; Manns, M.; Bzowej, N.; Niu, J.; et al. 2-year globe trial results: Telbivudine is superior to lamivudine in patients with chronic hepatitis B. Gastroenterology 2009, 136, 486–495. [Google Scholar] [CrossRef]
- Papatheodoridis, G.; Buti, M.; Cornberg, M.; Janssen, H.; Mutimer, D. Easl clinical practice guidelines: Management of chronic hepatitis B virus infection. J. Hepatol. 2012, 57, 167–185. [Google Scholar]
- Zhang, Q.; Han, T.; Nie, C.Y.; Ha, F.S.; Liu, L.; Liu, H. Tenofovir rescue regimen following prior suboptimal response to entecavir and adefovir combination therapy in chronic hepatitis B patients exposed to multiple treatment failures. J. Med. Virol. 2015, 87, 1013–1021. [Google Scholar] [CrossRef]
- Yatsuji, H.; Suzuki, F.; Sezaki, H.; Akuta, N.; Suzuki, Y.; Kawamura, Y.; Hosaka, T.; Kobayashi, M.; Saitoh, S.; Arase, Y.; et al. Low risk of adefovir resistance in lamivudine-resistant chronic hepatitis B patients treated with adefovir plus lamivudine combination therapy: Two-year follow-up. J. Hepatol. 2008, 48, 923–931. [Google Scholar] [CrossRef]
- Lee, J.M.; Park, J.Y.; Kim do, Y.; Nguyen, T.; Hong, S.P.; Kim, S.O.; Chon, C.Y.; Han, K.H.; Ahn, S.H. Long-term adefovir dipivoxil monotherapy for up to 5 years in lamivudine-resistant chronic hepatitis B. Antivir. Ther. 2010, 15, 235–241. [Google Scholar] [CrossRef]
- Ninomiya, K.; Morikawa, T.; Zhang, Y.; Nakamura, S.; Matsuda, H.; Muraoka, O.; Yoshikawa, M. Bioactive constituents from chinese natural medicines. Xxiii. Absolute structures of new megastigmane glycosides, sedumosides a(4), a(5), a(6), H, and I, and hepatoprotective megastigmanes from sedum sarmentosum. Chem. Pharm. Bull. 2007, 55, 1185–1191. [Google Scholar] [CrossRef]
- Lian, L.H.; Jin, X.; Wu, Y.L.; Cai, X.F.; Lee, J.J.; Nan, J.X. Hepatoprotective effects of sedum sarmentosum on d-galactosamine/lipopolysaccharide-induced murine fulminant hepatic failure. J. Pharmacol. Sci. 2010, 114, 147–157. [Google Scholar] [CrossRef]
- He, A.; Wang, M.; Hao, H.; Zhang, D.; Lee, K.H. Hepatoprotective triterpenes from sedum sarmentosum. Phytochemistry 1998, 49, 2607–2610. [Google Scholar] [CrossRef]
- Epivir-hbv Prescribing Information. GlaxoSmithKline. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021003s015,021004s015lbl.pdf. 20-Dec-2013 (accessed on 16 August 2015).
- Sebivo prescribing information. Novartis Pharmaceuticals UK Ltd. Available online: http://www.medicines.org.uk/emc/medicine/19740. 02-Feb-2015 (accessed on 16 August 2015).
- Baraclude Prescribing Information. Bristol-Myers Squibb Pharmaceutical Limited. Available online: http://www.medicines.org.uk/emc/medicine/18377 (accessed on 16 August 2015).
- Adefovir Dipivoxil Prescribing Information. SigmaPharm Laboratories, LLC. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202051Orig1s000lbl.pdf (accessed on 16 August 2015).
- Viread Prescribing Information. Gilead Sciences, Inc. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021356s049,022577s007lbl.pdf (accessed on 16 August 2015).
- Chen, X.S.; Wang, G.J.; Cai, X.; Yu, H.Y.; Hu, Y.P. Inhibition of hepatitis b virus by oxymatrine in vivo. World J. Gastroenterol. 2001, 7, 49–52. [Google Scholar]
- Lu, L.G.; Zeng, M.D.; Mao, Y.M.; Fang, J.Y.; Song, Y.L.; Shen, Z.H.; Cao, A.P. Inhibitory effect of oxymatrine on serum hepatitis B virus DNA in HBV transgenic mice. World J. Gastroenterol. 2004, 10, 1176–1179. [Google Scholar]
- Lu, L.G.; Zeng, M.D.; Mao, Y.M.; Li, J.Q.; Wan, M.B.; Li, C.Z.; Chen, C.W.; Fu, Q.C.; Wang, J.Y.; She, W.M.; et al. Oxymatrine therapy for chronic hepatitis B: A randomized double-blind and placebo-controlled multi-center trial. World J. Gastroenterol. 2003, 9, 2480–2483. [Google Scholar]
- Wang, Y.P.; Zhao, W.; Xue, R.; Zhou, Z.X.; Liu, F.; Han, Y.X.; Ren, G.; Peng, Z.G.; Cen, S.; Chen, H.S.; et al. Oxymatrine inhibits hepatitis b infection with an advantage of overcoming drug-resistance. Antivir. Res. 2011, 89, 227–231. [Google Scholar] [CrossRef]
- Xu, W.S.; Zhao, K.K.; Miao, X.H.; Ni, W.; Cai, X.; Zhang, R.Q.; Wang, J.X. Effect of oxymatrine on the replication cycle of hepatitis B virus in vitro. World J. Gastroenterol. 2010, 16, 2028–2037. [Google Scholar] [CrossRef]
- Wang, Y.P.; Liu, F.; He, H.W.; Han, Y.X.; Peng, Z.G.; Li, B.W.; You, X.F.; Song, D.Q.; Li, Z.R.; Yu, L.Y.; et al. Heat stress cognate 70 host protein as a potential drug target against drug resistance in hepatitis B virus. Antimicrob. Agents Chemother. 2010, 54, 2070–2077. [Google Scholar] [CrossRef]
- Chinese Society of Hepatology and Chinese Society of Infectious Diseases, Chinese Medical Association. The guidelines of prevention and treatment for chronic hepatitis B. Zhonghua Gan Zang Bing Za Zhi 2005, 13, 881–891. (In Chinese) [Google Scholar]
- Soon, D.K.; Lowe, S.L.; Teng, C.H.; Yeo, K.P.; McGill, J.; Wise, S.D. Safety and efficacy of alamifovir in patients with chronic hepatitis b virus infection. J. Hepatol. 2004, 41, 852–858. [Google Scholar] [CrossRef]
- Chan, C.; Abu-Raddad, E.; Golor, G.; Watanabe, H.; Sasaki, A.; Yeo, K.P.; Soon, D.; Sinha, V.P.; Flanagan, S.D.; He, M.M.; et al. Clinical pharmacokinetics of alamifovir and its metabolites. Antimicrob. Agents Chemother. 2005, 49, 1813–1822. [Google Scholar] [CrossRef]
- Ono-Nita, S.K.; Kato, N.; Shiratori, Y.; Carrilho, F.J.; Omata, M. Novel nucleoside analogue mcc-478 (ly582563) is effective against wild-type or lamivudine-resistant hepatitis B virus. Antimicrob. Agents Chemother. 2002, 46, 2602–2605. [Google Scholar] [CrossRef]
- Kamiya, N.; Kubota, A.; Iwase, Y.; Sekiya, K.; Ubasawa, M.; Yuasa, S. Antiviral activities of mcc-478, a novel and specific inhibitor of hepatitis B virus. Antimicrob. Agents Chemother. 2002, 46, 2872–2877. [Google Scholar] [CrossRef]
- Wu, T.T.; Coates, L.; Aldrich, C.E.; Summers, J.; Mason, W.S. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 1990, 175, 255–261. [Google Scholar] [CrossRef]
- Locarnini, S.A.; Yuen, L. Molecular genesis of drug-resistant and vaccine-escape HBV mutants. Antivir. Ther. 2010, 15, 451–461. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccdna. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Thomas, H.C.; Karayiannis, P.; Brook, G. Treatment of hepatitis b virus infection with interferon. Factors predicting response to interferon. J. Hepatol. 1991, 1, S4–S7. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, G. A review of non-nucleoside anti-hepatitis B virus agents. Eur. J. Med. Chem. 2014, 75, 267–281. [Google Scholar] [CrossRef]
- Fattovich, G.; Giustina, G.; Favarato, S.; Ruol, A. A survey of adverse events in 11,241 patients with chronic viral hepatitis treated with Alfa interferon. J. Hepatol. 1996, 24, 38–47. [Google Scholar] [CrossRef]
- Muckenfuss, H.; Hamdorf, M.; Held, U.; Perkovic, M.; Lower, J.; Cichutek, K.; Flory, E.; Schumann, G.G.; Munk, C. Apobec3 proteins inhibit human line-1 retrotransposition. J. Biol. Chem. 2006, 281, 22161–22172. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Burns, M.B.; Li, M.; Lengyel, J.; Harris, R.S. Apobec3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010, 17, 222–229. [Google Scholar] [CrossRef]
- Carpenter, M.A.; Li, M.; Rathore, A.; Lackey, L.; Law, E.K.; Land, A.M.; Leonard, B.; Shandilya, S.M.; Bohn, M.F.; Schiffer, C.A.; et al. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme apobec3a. J. Biol. Chem. 2012, 287, 34801–34808. [Google Scholar] [CrossRef]
- Krebs, K.; Böttinger, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis b virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef]
- Ahmed, M.; Wang, F.; Levin, A.; Le, C.; Eltayebi, Y.; Houghton, M.; Tyrrell, L.; Barakat, K. Targeting the achilles heel of the hepatitis b virus: A review of current treatments against covalently closed circular DNA. Drug Discov. Today 2015, 20, 548–561. [Google Scholar] [CrossRef]
- Jagya, N.; Varma, S.P.; Thakral, D.; Joshi, P.; Durgapal, H.; Panda, S.K. Rna-seq based transcriptome analysis of hepatitis e virus (HEV) and hepatitis b virus (HBV) replicon transfected huh-7 cells. PLoS ONE 2014, 9, e87835. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Ying, C.X.; Leung, C.H.; Li, Y. New targets and inhibitors of HBV replication to combat drug resistance. J. Clin. Virol. 2005, 34, S147–S150. [Google Scholar] [CrossRef]
- Ying, C.; Li, Y.; Leung, C.H.; Robek, M.D.; Cheng, Y.C. Unique antiviral mechanism discovered in anti-hepatitis B virus research with a natural product analogue. Proc. Natl. Acad. Sci. USA 2007, 104, 8526–8531. [Google Scholar] [CrossRef]
- Janmanchi, D.; Tseng, Y.P.; Wang, K.C.; Huang, R.L.; Lin, C.H.; Yeh, S.F. Synthesis and the biological evaluation of arylnaphthalene lignans as anti-hepatitis B virus agents. Bioorg. Med. Chem. 2010, 18, 1213–1226. [Google Scholar] [CrossRef]
- Janmanchi, D.; Lin, C.H.; Hsieh, J.Y.; Tseng, Y.P.; Chen, T.A.; Jhuang, H.J.; Yeh, S.F. Synthesis and biological evaluation of helioxanthin analogues. Bioorg. Med. Chem. 2013, 21, 2163–2176. [Google Scholar] [CrossRef]
- Tseng, Y.P.; Kuo, Y.H.; Hu, C.P.; Jeng, K.S.; Janmanchi, D.; Lin, C.H.; Chou, C.K.; Yeh, S.F. The role of helioxanthin in inhibiting human hepatitis B viral replication and gene expression by interfering with the host transcriptional machinery of viral promoters. Antivir. Res. 2008, 77, 206–214. [Google Scholar] [CrossRef]
- Pang, R.; Tao, J.Y.; Zhang, S.L.; Chen, K.L.; Zhao, L.; Yue, X.; Wang, Y.F.; Ye, P.; Zhu, Y.; Wu, J.G. Ethanol extract from ampelopsis sinica root exerts anti-hepatitis B virus activity via inhibition of p53 pathway in vitro. Evid. Based Complement. Altern. Med. 2011, 2011, e939205. [Google Scholar] [CrossRef]
- Le Seyec, J.; Chouteau, P.; Cannie, I.; Guguen-Guillouzo, C.; Gripon, P. Infection process of the hepatitis b virus depends on the presence of a defined sequence in the pre-s1 domain. J. Virol. 1999, 73, 2052–2057. [Google Scholar]
- Blanchet, M.; Sureau, C. Infectivity determinants of the hepatitis b virus pre-s domain are confined to the N-terminal 75 amino acid residues. J. Virol. 2007, 81, 5841–5849. [Google Scholar] [CrossRef]
- Volz, T.; Allweiss, L.; ḾBarek, M.B.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lütgehetmann, M.; et al. The entry inhibitor myrcludex-b efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis b virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]
- Yan, H.; Peng, B.; Liu, Y.; Xu, G.; He, W.; Ren, B.; Jing, Z.; Sui, J.; Li, W. Viral entry of hepatitis b and d viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J. Virol. 2014, 88, 3273–3284. [Google Scholar] [CrossRef]
- Slijepcevic, D.; Kaufman, C.; Wichers, C.G.; Gilglioni, E.H.; Lempp, F.A.; Duijst, S.; de Waart, D.R.; Elferink, R.P.; Mier, W.; Stieger, B.; et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+) -taurocholate cotransporting polypeptide knockout mice. Hepatology 2015, 62, 207–219. [Google Scholar] [CrossRef]
- Tsukuda, S.; Watashi, K.; Iwamoto, M.; Suzuki, R.; Aizaki, H.; Okada, M.; Sugiyama, M.; Kojima, S.; Tanaka, Y.; Mizokami, M.; et al. Dysregulation of retinoic acid receptor diminishes hepatocyte permissiveness to hepatitis b virus infection through modulation of sodium taurocholate cotransporting polypeptide (NTCP) expression. J. Biol. Chem. 2015, 290, 5673–5684. [Google Scholar] [CrossRef]
- Bouezzedine, F.; Fardel, O.; Gripon, P. Interleukin 6 inhibits HBV entry through NTCP down regulation. Virology 2015, 481, 34–42. [Google Scholar] [CrossRef]
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.V. Toll-like receptor signaling inhibits hepatitis b virus replication in vivo. J. Virol. 2005, 79, 7269–7272. [Google Scholar] [CrossRef]
- Lopatin, U.; Wolfgang, G.; Tumas, D.; Frey, C.R.; Ohmstede, C.; Hesselgesser, J.; Kearney, B.; Moorehead, L.; Subramanian, G.M.; McHutchison, J.G. Safety, pharmacokinetics and pharmacodynamics of gs-9620, an oral toll-like receptor 7 agonist. Antivir. Ther. 2013, 18, 409–418. [Google Scholar] [CrossRef]
- Lawitz, E.; Gruener, D.; Marbury, T.; Hill, J.; Webster, L.; Hassman, D.; Nguyen, A.H.; Pflanz, S.; Mogalian, E.; Gaggar, A.; et al. Safety, pharmacokinetics and pharmacodynamics of the oral toll-like receptor 7 agonist gs-9620 in treatment-naive patients with chronic hepatitis C. Antivir. Ther. 2014. [Google Scholar] [CrossRef]
- Gane, E.J.; Lim, Y.S.; Gordon, S.C.; Visvanathan, K.; Sicard, E.; Fedorak, R.N.; Roberts, S.; Massetto, B.; Ye, Z.; Pflanz, S.; et al. The oral toll-like receptor-7 agonist gs-9620 in patients with chronic hepatitis b virus infection. J. Hepatol. 2015, 63, 320–328. [Google Scholar] [CrossRef]
- Zhang, E.; Lu, M. Toll-like receptor (tlr)-mediated innate immune responses in the control of hepatitis b virus (HBV) infection. Med. Microbiol. Immunol. 2015, 204, 11–20. [Google Scholar] [CrossRef]
- Kapoor, R.; Kottilil, S. Strategies to eliminate hbv infection. Future Virol. 2014, 9, 565–585. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, Z.H.; Huang, M.; Heise, T.; Zhang, J.; Liu, G.L. Phosphorylation of human la protein at ser 366 by casein kinase II contributes to hepatitis B virus replication and expression in vitro. J. Viral Hepat. 2013, 20, 24–33. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, Z.H.; Liu, G.L. A systematic analysis of the predicted human la protein targets identified a hepatitis b virus infection signature. J. Viral Hepat. 2013, 20, 12–23. [Google Scholar] [CrossRef]
- Heise, T.; Guidotti, L.G.; Chisari, F.V. Characterization of nuclear rnases that cleave hepatitis B virus RNA near the la protein binding site. J. Virol. 2001, 75, 6874–6883. [Google Scholar] [CrossRef]
- Tang, J.; Huang, Z.M.; Chen, Y.Y.; Zhang, Z.H.; Liu, G.L.; Zhang, J. A novel inhibitor of human la protein with anti-HBV activity discovered by structure-based virtual screening and in vitro evaluation. PLoS ONE 2012, 7, e36363. [Google Scholar] [CrossRef]
- Yu, X.; Guo, R.; Ming, D.; Deng, Y.; Su, M.; Lin, C.; Li, J.; Lin, Z.; Su, Z. The tgf-beta1/il-31 pathway is up-regulated in patients with acute-on-chronic hepatitis b liver failure and is associated with disease severity and survival. Clin. Vaccine Immunol. 2015, 22, 484–492. [Google Scholar] [CrossRef]
- Qi, P.; Chen, Y.M.; Wang, H.; Fang, M.; Ji, Q.; Zhao, Y.P.; Sun, X.J.; Liu, Y.; Gao, C.F. −509c > t polymorphism in the tgf-beta1 gene promoter, impact on the hepatocellular carcinoma risk in Chinese patients with chronic hepatitis b virus infection. Cancer Immunol. Immunother. 2009, 58, 1433–1440. [Google Scholar] [CrossRef]
- Wu, Y.; Zhao, J.; He, M. Correlation between tgf-beta1 gene 29 t > c single nucleotide polymorphism and clinicopathological characteristics of osteosarcoma. Tumour Biol. 2015, 36, 5149–5156. [Google Scholar] [CrossRef]
- Yin, W.; Zhao, Y.; Ji, Y.J.; Tong, L.P.; Liu, Y.; He, S.X.; Wang, A.Q. Serum/plasma micrornas as biomarkers for HBV-related hepatocellular carcinoma in China. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Webster, R.M. The immune checkpoint inhibitors: Where are we now? Nat. Rev. Drug Discov. 2014, 13, 883–884. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Barakat, K. Immune checkpoints: The search for a single antiviral-anticancer magic bullet. J. Pharma Care Health Syst. 2015, 2, e125. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. Tim-3 expression characterizes regulatory t cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef]
- Barakat, K. Do we need small molecule inhibitors for the immune checkpoints? J. Pharma Care Health Syst. 2014, 1. [Google Scholar] [CrossRef]
- Viricel, C.; Ahmed, M.; Barakat, K. Human pd-1 binds differently to its human ligands: A comprehensive modeling study. J. Mol. Graph. Model. 2015, 57, 131–142. [Google Scholar] [CrossRef]
- Barakat, K. Computer-aided drug design. J. Pharma Care Health Syst. 2014, 1. [Google Scholar] [CrossRef]
- Barakat, K.H.; Jordheim, L.P.; Perez-Pineiro, R.; Wishart, D.; Dumontet, C.; Tuszynski, J.A. Virtual screening and biological evaluation of inhibitors targeting the xpa-ercc1 interaction. PLoS ONE 2012, 7, e51329. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef]
- Chang, T.T.; Lai, C.L.; Kew Yoon, S.; Lee, S.S.; Coelho, H.S.; Carrilho, F.J.; Poordad, F.; Halota, W.; Horsmans, Y.; Tsai, N.; et al. Entecavir treatment for up to 5 years in patients with hepatitis b e antigen-positive chronic hepatitis B. Hepatology 2010, 51, 422–430. [Google Scholar] [CrossRef]
- Heathcote, E.J.; Marcellin, P.; Buti, M.; Gane, E.; De Man, R.A.; Krastev, Z.; Germanidis, G.; Lee, S.S.; Flisiak, R.; Kaita, K.; et al. Three-year efficacy and safety of tenofovir disoproxil fumarate treatment for chronic hepatitis B. Gastroenterology 2011, 140, 132–143. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, L.; Pan, J.; Wu, J.; Hu, J.; Sun, Q.; Tang, J. Anti-HBV Drugs: Progress, Unmet Needs, and New Hope. Viruses 2015, 7, 4960-4977. https://doi.org/10.3390/v7092854
Kang L, Pan J, Wu J, Hu J, Sun Q, Tang J. Anti-HBV Drugs: Progress, Unmet Needs, and New Hope. Viruses. 2015; 7(9):4960-4977. https://doi.org/10.3390/v7092854
Chicago/Turabian StyleKang, Lei, Jiaqian Pan, Jiaofen Wu, Jiali Hu, Qian Sun, and Jing Tang. 2015. "Anti-HBV Drugs: Progress, Unmet Needs, and New Hope" Viruses 7, no. 9: 4960-4977. https://doi.org/10.3390/v7092854
APA StyleKang, L., Pan, J., Wu, J., Hu, J., Sun, Q., & Tang, J. (2015). Anti-HBV Drugs: Progress, Unmet Needs, and New Hope. Viruses, 7(9), 4960-4977. https://doi.org/10.3390/v7092854