
Phylogenetic Studies of the Three RNA Silencing Suppressor Genes of South American CTV Isolates Reveal the Circulation of a Novel Genetic Lineage

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Population Structure and Geographical Distribution of CTV Isolates in Uruguay
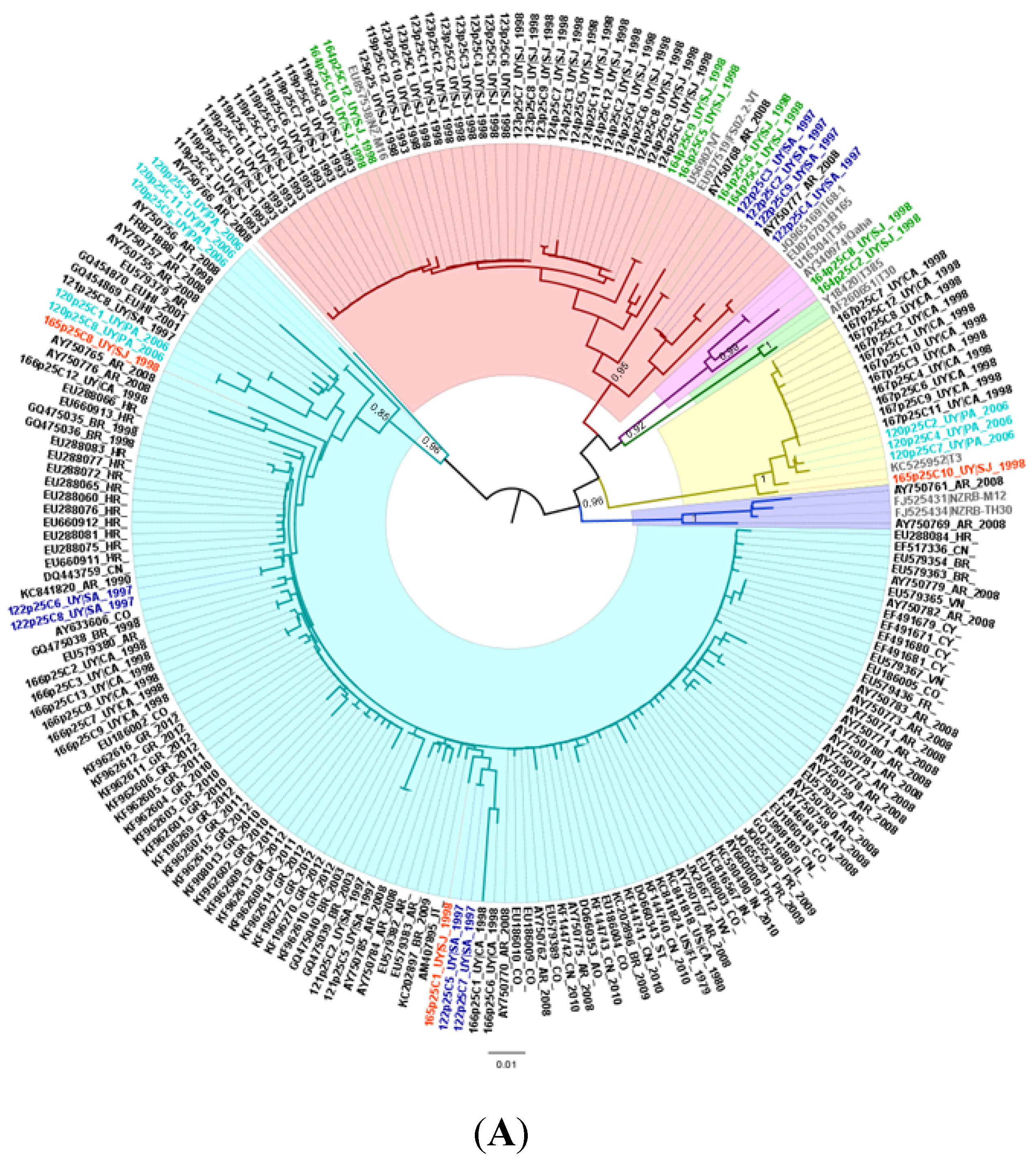

2.2. Genetic Divergence Analysis

{kind=link}
{kind=link}
{kind=link}
| Region | NC average | RB | T3 | T30 | T36 | VT | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nt | aa | nt | aa | nt | aa | nt | aa | nt | aa | nt | aa | |
| p25 | 98.8 ± 0.1 | 98.8 ± 0.2 | 90.6 ± 1.3 | 95.2 ± 1.2 | 89.4 ± 1.5 | 96.4 ± 1.2 | 89.1 ± 1.5 | 94.4 ± 1.5 | 89.1 ± 1.5 | 93.6 ± 1.6 | 90.2 ± 1.4 | 95.4 ± 1.2 |
| p20 | 98.9 ± 0.2 | 99.1 ± 0.3 | 84.4 ± 2.5 | 93.9 ± 2.1 | 82.1 ± 2.7 | 92.6 ± 2.2 | 81.8 ± 2.9 | 92.2 ± 2.4 | 88.6± 1.8 | 95.3 ± 1.8 | 84.1 ± 2.4 | 93.1 ± 2.2 |
| p20 | 97.5 ± 0.3 | 96.9 ± 0.7 | 87.3 ± 1.5 | 88.2 ± 2.2 | 90.5 ± 1.2 | 89.9 ± 2.0 | 89.9 ± 1.2 | 90.9 ± 2.0 | 89.3 ± 1.3 | 88.5 ± 2.3 | 89.3 ± 1.3 | 88.7 ± 2.1 |
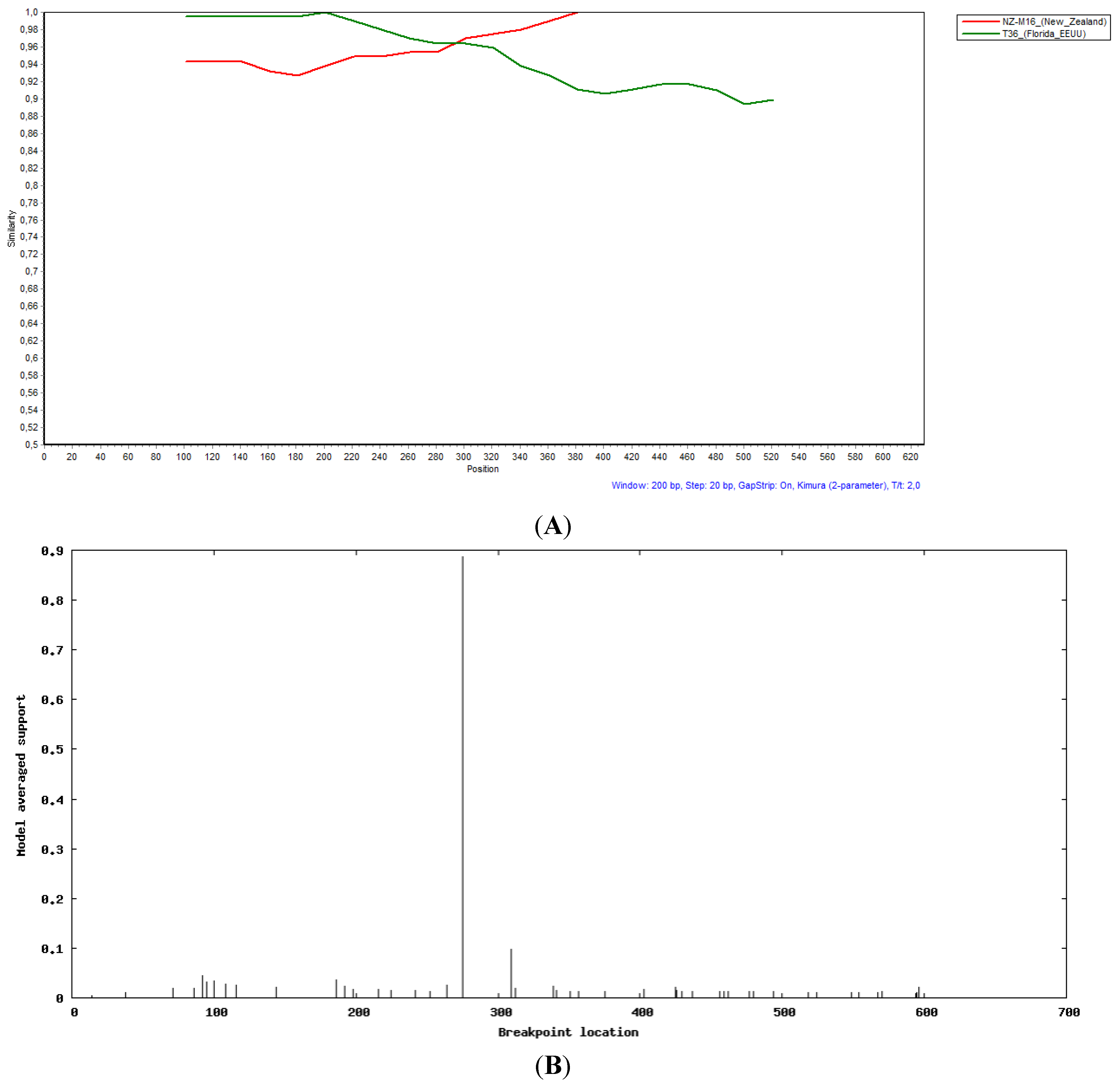
2.3. Recombination Analysis
3. Discussion
3.1. Composition of Viral Populations and Geographical Information
3.2. Phylogenetic Relationships among the Isolates
3.3. Genetic Divergence of CTV Populations in Uruguay
3.4. Evidence of Recombination
4. Materials and Methods
4.1. Virus Isolates
| Isolate | Host | Origin | Collection date | Aspect of the tree in the field | Stem Pitting | ELISA | Number of clones | ||
|---|---|---|---|---|---|---|---|---|---|
| p25 | p20 | p23 | |||||||
| 119 | Lemon | San José | 1993 | Stunted | NA | + | 12 | 9 | 11 |
| 120 | Washington Navel sweet orange | Paysandú | 2006 | Stunted | NA | + | 8 | 4 | 14 |
| 121 | Star Ruby grapefruit | Salto | 1997 | Stunted | + | - | 3 | 6 | NC |
| 122 | Star Ruby grapefruit | Salto | 1997 | Stunted | + | + | 8 | 12 | 12 |
| 123 | Lemon | San José | 1998 | Normal | ++ | + | 12 | 5 | 10 |
| 124 | Lemon | San José | 1998 | Normal | ++ | UND | 11 | 11 | 12 |
| 125 | Lemon | San José | 1998 | Stunted | +++ | - | 1 | 14 | 8 |
| 164 | Lemon | San José | 1998 | Stunted | +++ | + | 8 | 12 | 11 |
| 165 | Valencia sweet orange | San José | 1998 | Stunted | - | + | 3 | 13 | 8 |
| 166 | Lemon | Canelones | 1998 | Stunted | +++ | - | 9 | 9 | 10 |
| 167 | Lemon | Canelones | 1998 | Normal | ++ | + | 11 | 11 | 10 |
| 168 | Star Ruby grapefruit | Salto | 1998 | Stunted | - | UND | NC | 12 | NC |
| MIL | Satsuma Owari mandarin | Salto | 2012 | Normal | NA | NA | NC | 12 | 9 |
4.2. RNA Extraction, cDNA Synthesis, and PCR Amplification
| Name | Sequence | Genome Position* | Expected Size (bp) | Reference |
|---|---|---|---|---|
| p20F | 5' ACAATATGCGAGCTTACTTTA 3' | 17691–17710 | 555 | [18] |
| p20R | 5' AACCTACACGCAAGATGGA 3' | 18229–18246 | ||
| p23F | 5' GTCTCTCCATCTTGCGGTGTAG 3' | 18224–18244 | 733 | [30] |
| p23R | 5' CAATCAGATGAAGTGGTG3' | 18941–18957 | ||
| p25F | 5' TGAATTATGGACGACGAAAC 3' | 16079–16097 | 676 | |
| p25R | 5' TCAACGTGTGTTGAATTTCCC 3' | 16736–16755 |
4.3. Cloning
4.4. Nucleotide Sequences and Phylogenetic Analysis
4.5. Recombination Analysis
4.6. Nucleotide Sequence Accession Number
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interests
References
- Moreno, P.; Ambros, S.; Albiach-Marti, M.R.; Guerri, J.; Peña, L. Plant diseases that changed the world Citrus tristeza virus: A pathogen that changed the course of the citrus industry. Mol. Plant Pathol. 2008, 9, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Brlansky, R.H.; Damsteegt, V.D.; Howd, D.S.; Roy, A. Molecular analyses of citrus tristeza virus subisolates separated by aphid transmission. Plant Dis. 2003, 87, 397–401. [Google Scholar] [CrossRef]
- Bar-Joseph, M.; Marcus, R.; Lee, R.F. The continuous challenge of citrus tristeza virus control. Annu. Rev. Phytopathol. 1989, 27, 291–316. [Google Scholar] [CrossRef]
- Karasev, A.V.; Boyko, V.P.; Gowda, S.; Nikolaeva, O.V.; Hilf, M.E.; Koonin, E.V.; Niblett, C.L.; Cline, K.; Gumpf, D.J.; Lee, R.F.; et al. Complete Sequence of the Citrus Tristeza Virus RNA Genome. Virology 1995, 208, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Febres, V.J.; Ashoulin, L.; Mawassi, M.; Frank, A.; Bar-Joseph, M.; Manjunath, K.L.; Lee, R.F.; Niblett, C.L. The p27 protein is present at one end of citrus tristeza virus particles. Phytopathology 1996, 86, 1331–1335. [Google Scholar]
- Satyanarayana, T.; Gowda, S.; Ayllon, M.A.; Dawson, W.O. Closterovirus bipolar virion: Evidence for initiation of assembly by minor coat protein and its restriction to the genomic RNA 5′ region. Proc. Natl. Acad. Sci. USA 2004, 101, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Agranovsky, A.A. Principles of molecular organization, expression and evolution of closterovirus: Over the barriers. Adv. Virus Res. 1996, 17, 119–158. [Google Scholar]
- Hilf, M.; Karasev, A.; Pappu, H.; Gumpf, D.; Niblett, C.; Garnsey, S. Characterization of Citrus Tristeza Virus subgenomic RNAs in infected tissue. Virology 1995, 208, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Navas-Castillo, J.; Albiachi-Martí, M.; Gowda, S.; Hilf, M.; Garnsey, S.; Dawson, W. Kinetics of accumulation of citrus tristeza virus RNAs. Virology 1997, 228, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, T.; Gowda, S.; Mawassi, M.; Albiach-Martí, M.R.; Ayllón, M.A.; Robertson, C.; Garnsey, S.M.; Dawson, W.O. Closterovirus encoded HSP70 homolog and p61 in addition to both coat proteins function in efficient virion assembly. Virology 2000, 278, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Folimonov, A.; Shintaku, M.; Wan-Xiang, L.; Falk, B.W.; Dawson, W.O.; Ding, S. Three distinct suppressors of RNA silencing encoded by a 20-kb viral RNA genome. Proc. Natl. Acad. Sci. USA 2004, 101, 15742–15747. [Google Scholar] [CrossRef] [PubMed]
- Tatineni, S.; Robertson, C.J.; Garnsey, S.M.; Dawson, W.O. A plant virus evolved by acquiring multiple nonconserved genes to extend its host range. Proc. Natl. Acad. Sci. USA 2011, 108, 17366–17371. [Google Scholar] [CrossRef] [PubMed]
- Gowda, S.; Satyanarayana, T.; Davis, C.; Navas-Castillo, J.; Albiachi-Martí, M.; Mawassi, M.; Valkov, N.; Bar-Joseph, M.; Moreno, P.; Dawson, W. The p20 gene product of Citrus tristeza virus accumulates in the amorphous inclusion bodies. Virology 2000, 272, 246–254. [Google Scholar] [CrossRef] [PubMed]
- López, C.; Navas-Castillo, J.; Gowda, S.; Moreno, P.; Flores, R. The 23-kDa Protein Coded by the 3′-Terminal Gene of Citrus Tristeza Virus Is an RNA-Binding Protein. Virology 2000, 269, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, T.; Gowda, S.; Ayllon, M.A.; Albiach-Marti, M.R.; Rabindram, R.; Dawson, W.O. The p23 protein of Citrus tristeza virus controls asymmetrical RNA accumulation. J. Virol. 2002, 76, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Fagoaga, C.; López, C.; Moreno, P.; Navarro, L.; Flores, R.; Peña, L. Viral-Like Symptoms Induced by the Ectopic Expression of the p23 Gene of Citrus tristeza virus Are Citrus Specific and Do Not Correlate with the Pathogencity of the Virus Strain. Mol. Plant Microbe Interact. 2005, 18, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Rubio, L.; Polek, M.; Falk, B. Population structure and genetic diversity within California Citrus tristeza virus (CTV) isolates. Virus Genes 2000, 21, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.; Ayllon, M.A.; Kong, P.; Fernandez, A.; Polek, M.L.; Guerri, J.; Moreno, P.; Falk, B.W. Genetic variation of Citrus tristeza virus isolates from California and Spain: Evidence for mixed infections and recombination. J. Virol. 2001, 75, 8054–8062. [Google Scholar] [CrossRef] [PubMed]
- Roistacher, C.N.; da Graça, J.V.; Müller, G.W. Cross Protection Against Citrus Tristeza Virus a Review. In Proceedings of the 17th Conference of the International Organization of Citrus Virologists, 2010; Brlansky, R.H., Lee, R.F., Timmer, L.W., Eds.; IOCV: Riverside, CA, USA, 2010; pp. 1–27. [Google Scholar]
- Folimonova, S.Y. Developing an understanding of cross-protection by Citrus tristeza virus. Front. Microbiol. 2013, 4, e76. [Google Scholar] [CrossRef] [PubMed]
- Niblett, C.; Genc, H.; Cevik, B.; Halbert, S.; Brown, L.; Nolasco, G.; Bonacalza, B.; Manjunath, K.; Febres, V.; Pappu, H.; et al. Progress on strain differentiation of Citrus Tristeza Virus and its application to the epidemiology of citrus tristeza disease. Virus Res. 2000, 71, 97–106. [Google Scholar] [CrossRef]
- Hilf, M.E.; Garnsey, S.M. Characterization and classification of Citrus tristeza virus isolates by amplification of multiple molecular markers. In Proceedings of the 14th IOCV Conference, 2000; IOCV: Riverside, CA, USA, 2000; pp. 18–27. [Google Scholar]
- Ayllon, M.A.; Lopez, C.; Navas-Castillo, J.; Garnsey, S.M.; Guerri, J.; Flores, R.; Moreno, P. Polymorphism of the 5′ terminal region of Citrus tristeza virus (CTV) RNA: Incidence of three sequence types in isolates of different origin and pathogenicity. Arch. Virol. 2001, 146, 27–40. [Google Scholar] [PubMed]
- Nolasco, G.; Santos, C.; Silva, G.; Fonseca, F. Development of an asymmetric PCR-ELISA typing method for citrus tristeza virus based on the coat protein gene. J. Virol. Methods 2009, 155, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.J.; Borth, W.B.; Sether, D.M.; Ferreira, S.; Gonsalves, D.; Hu, J.S. Genetic diversity and evidence for recent modular recombination in Hawaiian Citrus tristeza virus. Virus Genes 2010, 40, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J. Citrus tristeza virus: Evolution of complex and varied genotypic groups. Front. Microbiol. 2013, 4, e93. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.P.; Nagata, T.; de Jesus, W.C., Jr.; Neto, C.R.; Pappas, G.J., Jr.; Martin, D.P. Genetic variation and recombination of RdRp and HSP 70h genes of Citrus Tristeza Virus isolates from orange trees showing symptoms of citrus sudden death disease. Virol. J. 2008, 16, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveros-Garay, O.; Martinez-Salazar, N.; Torres-Ruiz, Y.; Acosta, O. CPm gene diversity in field isolates of Citrus tristez virus from Colombia. Arch. Virol. 2009, 154, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Peroni, L.; Lorencini, M.; Ribeiro dos Reis, J.; Machado, M.; Stach-Machado, D. Differential diagnosis of Brazilian strains of Citrus tristeza virus by epitope mapping of coat protein using monoclonal antibodies. Virus Res. 2009, 145, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, N.G.; Gago-Zachert, S.P.; Robledo, G.; Costa, N.; Plata, M.I.; Vera, O.; Grau, O.; Semorile, L.C. Population structure of Citrus tristeza virus from field Argentinean isolates. Virus Genes 2008, 36, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Koch de Brotos, L.; Boasso, C. Lista de las Enfermedades de los Vegetalesen el Uruguay; Ministerio de Ganadería y Agricultura, Dirección de Agronomía, Laboratorio de Fisiología y Patología Vegetal: Montevideo, Uruguay, 1955; p. 65. [Google Scholar]
- Iglesias, N.; Marengo, J.; Riquelme, K.; Costa, N.; Plata, M.; Semorile, L. Characterization of the population structure of a grapefruit isolate of Citrus tristeza virus (CTV) selected for pre-immunization assays in Argentina. In Proceedings of the 16th Conference of the International Organization of Citrus Virologists, 2005; Hilf, M.E., Duran-Vila, N., Rocha-Peña, M.A., Eds.; IOCV: Riverside, CA, USA, 2005; pp. 150–158. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [PubMed]
- Kosakovsky Pond, S.; Posada, D.; Gravenor, M.; Woelk, H.; Frost, S. Automated Phylogenetic Detection of Recombination Using a Genetic Algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Hilf, M.; Karasev, A.; Albiach-Martí, M.; Dawson, W.; Garnsey, S. Two paths of sequence divergence in the Citrus Tristeza Virus complex. Phytopathology 1999, 89, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bozan, O.; Kwon, S.J.; Dang, T.; Rucker, T.; Yokomi, R.; Lee, R.; Folimonova, S.; Krueger, R.; Bash, J.; et al. Past and future of a century old Citrus tristeza virus collection: A California citrus germplasm tale. Front. Microbiol. 2013, 4, e366. [Google Scholar] [CrossRef] [PubMed]
- Hančević, K.; Černi, S.; Nolasco, G.; Radić, T.; Djelouah, K.; Škorić, D. Biological characterization of Citrus tristeza virus monophyletic isolates with respect to p25 gene. Physiol. Mol. Plant Pathol. 2013, 81, 45–53. [Google Scholar] [CrossRef]
- Černi, S.; Ruščić, J.; Nolasco, G.; Gatin, Ž.; Krajačić, M.; Škorić, D. Stem pitting and seedling yellows symptoms of Citrus tristeza virus infection may be determined by minor sequence variants. Virus Genes 2008, 36, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Černi, S.; Škorić, D.; Ruščić, J.; Krajačić, M.; Papic, T.; Djelouah, K.; Nolasco, G. East Adriatic: A reservoir region of severe Citrus tristeza virus strains. Eur. J. Plant Pathol. 2009, 124, 701–706. [Google Scholar] [CrossRef]
- Papayiannis, L.; Santos, C.; Kyriakou, A.; Kapari, T.; Nolasco, G. Molecular characterization of Citrus tristeza virus isolates from Cyprus on the basis of the coat protein gene. J. Plant Pathol. 2007, 89, 291–295. [Google Scholar]
- Hilf, M.E.; Mavrodieva, V.A.; Garnsey, S.M. Genetic marker analysis of a global collection of isolates of Citrus tristeza virus: Characterization and distribution of CTV genotypes and association with symptoms. Phytopathology 2005, 95, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Sambade, A.; Rubio, L.; Vives, M.C.; Moya, P.; Guerri, J.; Elena, S.F.; Moreno, P. Contribution of recombination and selection to molecular evolution of Citrus tristeza virus. J. Gen. Virol. 2009, 90, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Dawson, T.E.; Pearson, M.N. Isolates of Citrus tristeza virus that overcome Poncirus trifoliata resistance comprise a novel strain. Arch. Virol. 2010, 155, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Obbard, D.J.; Gordon, K.H.J.; Buck, A.H.; Jiggins, F.M. The evolution of RNAi as a defense against viruses and transposable elements. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Barthelson, R.; Gowda, S.; Hilf, M.E.; Dawson, W.O.; Galbraith, D.W.; Xiong, Z. Persistent infection and promiscuous recombination of multiple genotypes of an RNA virus within a single host generate extensive diversity. PLoS ONE 2007, 2, e917. [Google Scholar] [CrossRef] [PubMed]
- Swindell, S.; Plasterer, T. SEQMAN. Contig assembly. Methods Mol. Biol. 1997, 70, 75–89. [Google Scholar] [PubMed]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/genbank (accessed on 17 February 2015).
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benítez-Galeano, M.J.; Rubio, L.; Bertalmío, A.; Maeso, D.; Rivas, F.; Colina, R. Phylogenetic Studies of the Three RNA Silencing Suppressor Genes of South American CTV Isolates Reveal the Circulation of a Novel Genetic Lineage. Viruses 2015, 7, 4152-4168. https://doi.org/10.3390/v7072814
Benítez-Galeano MJ, Rubio L, Bertalmío A, Maeso D, Rivas F, Colina R. Phylogenetic Studies of the Three RNA Silencing Suppressor Genes of South American CTV Isolates Reveal the Circulation of a Novel Genetic Lineage. Viruses. 2015; 7(7):4152-4168. https://doi.org/10.3390/v7072814
Chicago/Turabian StyleBenítez-Galeano, María José, Leticia Rubio, Ana Bertalmío, Diego Maeso, Fernando Rivas, and Rodney Colina. 2015. "Phylogenetic Studies of the Three RNA Silencing Suppressor Genes of South American CTV Isolates Reveal the Circulation of a Novel Genetic Lineage" Viruses 7, no. 7: 4152-4168. https://doi.org/10.3390/v7072814
APA StyleBenítez-Galeano, M. J., Rubio, L., Bertalmío, A., Maeso, D., Rivas, F., & Colina, R. (2015). Phylogenetic Studies of the Three RNA Silencing Suppressor Genes of South American CTV Isolates Reveal the Circulation of a Novel Genetic Lineage. Viruses, 7(7), 4152-4168. https://doi.org/10.3390/v7072814