
Viral Membrane Channels: Role and Function in the Virus Life Cycle

Abstract
:1. Introduction

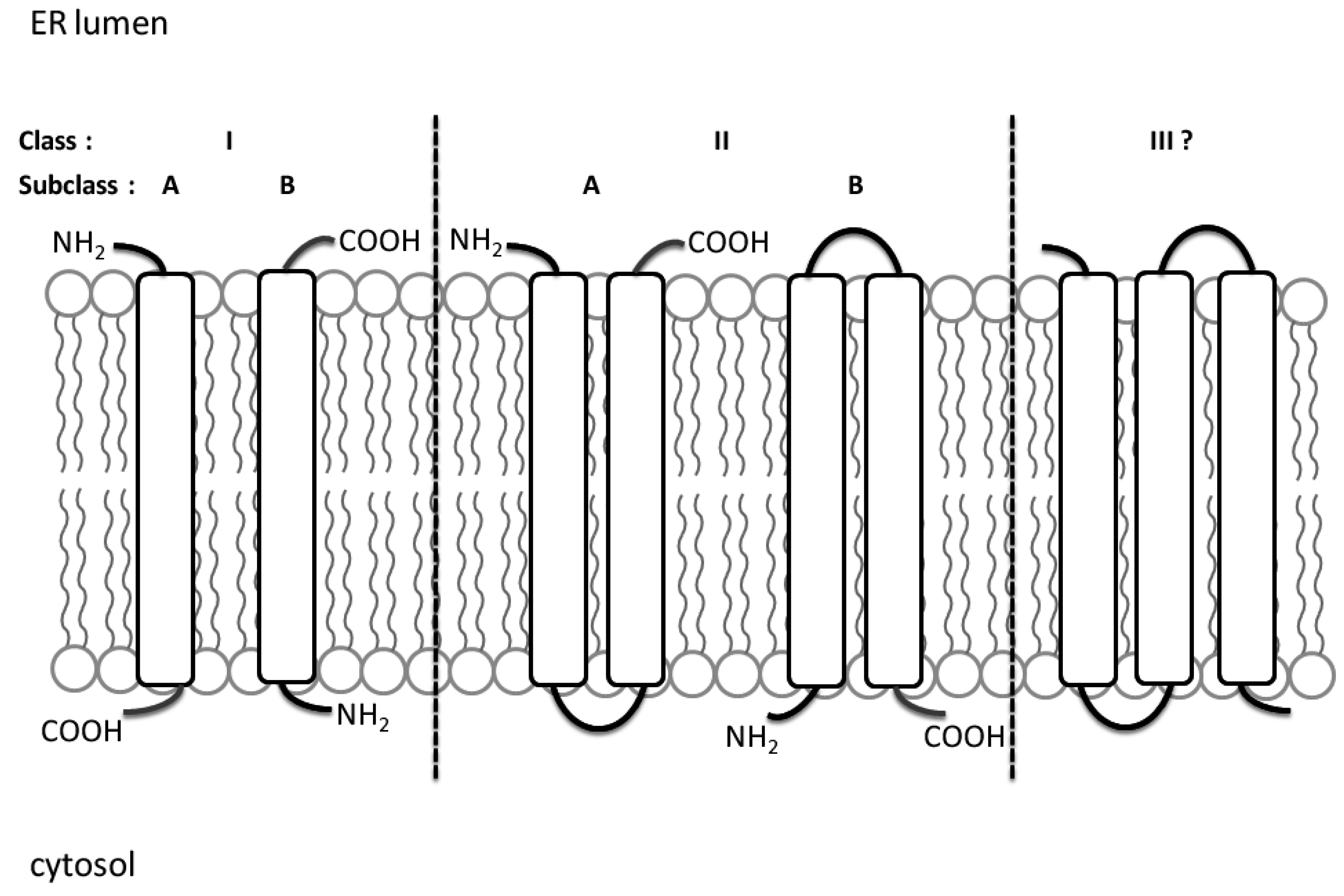{kind=link}
| Virus | Viroporin | Amino Acid | Function in Viral Life Cycle | References |
|---|---|---|---|---|
| IAV | M2 | 97 | Genome uncoating Glycoprotein processing/trafficking Delay protein trafficking through TGN Viral release | [8,9,10,11,12,13,14,15,16] |
| HIV-1 | Vpu | 77–86 | Degradation of CD4 and trafficking of Env proteins Viral release | [17,18,19,20,21,22,23] |
| HCV | P7 | 63 | Viral morphogenesis Viral polyprotein processing | [24,25,26,27] |
| CoV | E 3A | 76 274 | Viral morphogenesis and assembly Viral release | [28,29,30,31,32] |
| Poliovirus | 2B 3A | 97 87 | Blocks ER-Golgi traffic/host protein secretion | [33,34] |
| Alphavirus/Sindbis virus | 6K | 60 | Viral release | [35,36,37] |
| Coxsakievirus | 2B | 99 | Inhibit protein trafficking through Golgi Induce apoptosis for viral release | [38,39,40,41] |
| Rotavirus | NSP4 | 175 | Induce autophagy for viral protein transport | [42,43] |
| SV40 | VP2 VP3 VP4 | 352 234 125 | Translocation of DNA genome from ER to cytosol Viral release | [44,45,46,47,48] |
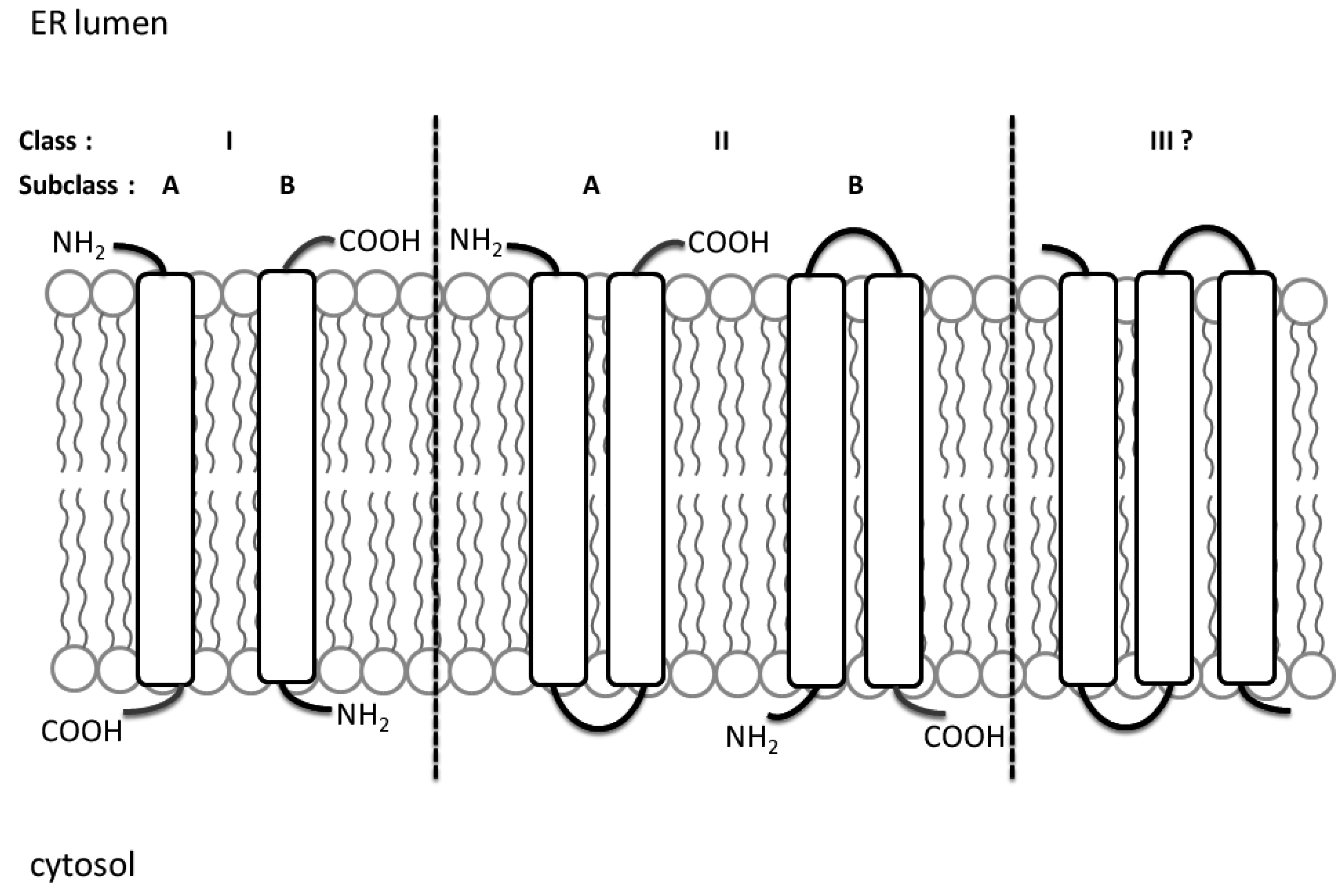
2. Viroporin and the Viral Life Cycle
2.1. Viral Entry and Uncoating
2.2. Viral Replication and Assembly
2.3. Viral Release
3. Viroporin-Induced Host Response
3.1. Apoptosis/Autophagy
3.2. Inflammasome Activation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, K.; Xie, S.; Sun, B. Viral proteins function as ion channels. Biochim. Biophys. Acta 2011, 1808, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nature Rev. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.B.; Wang, Y.T.; Schindler, C.; Chen, C.P. Mechanism of function of viral channel proteins and implications for drug development. Int. Rev. Cell Mol. Biol. 2012, 294, 259–321. [Google Scholar] [PubMed]
- Giorda, K.M.; Hebert, D.N. Viroporins customize host cells for efficient viral propagation. DNA Cell Biol. 2013, 32, 557–564. [Google Scholar] [CrossRef] [PubMed]
- OuYang, B.; Chou, J.J. The minimalist architectures of viroporins and their therapeutic implications. Biochim. Biophys. Acta 2014, 1838, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D. Viral miniproteins. Annu. Rev. Microbiol. 2014, 68, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Martin, K.; Helenius, A. Nuclear transport of influenza virus ribonucleoproteins: The viral matrix protein (M1) promotes export and inhibits import. Cell 1991, 67, 117–130. [Google Scholar] [CrossRef]
- Helenius, A. Unpacking the incoming influenza virus. Cell 1992, 69, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef] [PubMed]
- Grambas, S.; Hay, A.J. Maturation of influenza A virus hemagglutinin—Estimates of the pH encountered during transport and its regulation by the M2 protein. Virology 1992, 190, 11–18. [Google Scholar] [CrossRef]
- Henkel, J.R.; Weisz, O.A. Influenza virus M2 protein slows traffic along the secretory pathway. pH perturbation of acidified compartments affects early Golgi transport steps. J. Biol. Chem. 1998, 273, 6518–6524. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, T.; Leser, G.P.; Lamb, R.A. The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J. Cell Biol. 1996, 133, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.R.; Gibson, G.A.; Poland, P.A.; Ellis, M.A.; Hughey, R.P.; Weisz, O.A. Influenza M2 proton channel activity selectively inhibits trans-Golgi network release of apical membrane and secreted proteins in polarized Madin-Darby canine kidney cells. J. Cell Biol. 2000, 148, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Balannik, V.; Pinto, L.H.; Lamb, R.A. Influenza virus m2 ion channel protein is necessary for filamentous virion formation. J. Virol. 2010, 84, 5078–5088. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Maldarelli, F.; Karczewski, M.K.; Willey, R.L.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces degradation of CD4 in vitro: The cytoplasmic domain of CD4 contributes to Vpu sensitivity. J. Virol. 1993, 67, 3877–3884. [Google Scholar] [PubMed]
- Lenburg, M.E.; Landau, N.R. Vpu-induced degradation of CD4: Requirement for specific amino acid residues in the cytoplasmic domain of CD4. J. Virol. 1993, 67, 7238–7245. [Google Scholar] [PubMed]
- Vincent, M.J.; Raja, N.U.; Jabbar, M.A. Human immunodeficiency virus type 1 Vpu protein induces degradation of chimeric envelope glycoproteins bearing the cytoplasmic and anchor domains of CD4: Role of the cytoplasmic domain in Vpu-induced degradation in the endoplasmic reticulum. J. Virol. 1993, 67, 5538–5549. [Google Scholar] [PubMed]
- Willey, R.L.; Buckler-White, A.; Strebel, K. Sequences present in the cytoplasmic domain of CD4 are necessary and sufficient to confer sensitivity to the human immunodeficiency virus type 1 Vpu protein. J. Virol. 1994, 68, 1207–1212. [Google Scholar] [PubMed]
- Levesque, K.; Zhao, Y.S.; Cohen, E.A. Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J. Biol. Chem. 2003, 278, 28346–28353. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Bego, M.G.; Paquay, C.; Cohen, E.A. Modulation of HIV-1-host interaction: Role of the Vpu accessory protein. Retrovirology 2010, 7, e114. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.; Han, J.; Shinlapawittayatorn, K.; Deschenes, I.; Marban, E. Membrane potential depolarization as a triggering mechanism for Vpu-mediated HIV-1 release. Biophys. J. 2010, 99, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef] [PubMed]
- Tedbury, P.; Welbourn, S.; Pause, A.; King, B.; Griffin, S.; Harris, M. The subcellular localization of the hepatitis C virus non-structural protein NS2 is regulated by an ion channel-independent function of the p7 protein. J. Gen. Virol. 2011, 92, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Gentzsch, J.; Brohm, C.; Steinmann, E.; Friesland, M.; Menzel, N.; Vieyres, G.; Perin, P.M.; Frentzen, A.; Kaderali, L.; Pietschmann, T. Hepatitis c Virus p7 is critical for capsid assembly and envelopment. PLoS Pathog. 2013, 9, e1003355. [Google Scholar] [CrossRef] [PubMed]
- Brohm, C.; Steinmann, E.; Friesland, M.; Lorenz, I.C.; Patel, A.; Penin, F.; Bartenschlager, R.; Pietschmann, T. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. J. Virol. 2009, 83, 11682–11693. [Google Scholar] [CrossRef] [PubMed]
- Vennema, H.; Godeke, G.J.; Rossen, J.W.; Voorhout, W.F.; Horzinek, M.C.; Opstelten, D.J.; Rottier, P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996, 15, 2020–2028. [Google Scholar] [PubMed]
- Wilson, L.; Gage, P.; Ewart, G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 2006, 353, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Hogue, B.G. Role of the coronavirus E viroporin protein transmembrane domain in virus assembly. J. Virol. 2007, 81, 3597–3607. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Yang, R.F.; Shi, M.D.; Jiang, M.R.; Xie, Y.H.; Ruan, H.Q.; Jiang, X.S.; Shi, L.; Zhou, H.; Zhang, L.; et al. Characterization of the 3a protein of SARS-associated coronavirus in infected vero E6 cells and SARS patients. J. Mol. Biol. 2004, 341, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [PubMed]
- Doedens, J.R.; Kirkegaard, K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995, 14, 894–907. [Google Scholar] [PubMed]
- Sandoval, I.V.; Carrasco, L. Poliovirus infection and expression of the poliovirus protein 2B provoke the disassembly of the Golgi complex, the organelle target for the antipoliovirus drug Ro-090179. J. Virol. 1997, 71, 4679–4693. [Google Scholar] [PubMed]
- Sanz, M.A.; Perez, L.; Carrasco, L. Semliki Forest virus 6K protein modifies membrane permeability after inducible expression in Escherichia coli cells. J. Biol. Chem. 1994, 269, 12106–12110. [Google Scholar] [PubMed]
- Loewy, A.; Smyth, J.; von Bonsdorff, C.H.; Liljestrom, P.; Schlesinger, M.J. The 6-kilodalton membrane protein of Semliki Forest virus is involved in the budding process. J. Virol. 1995, 69, 469–475. [Google Scholar] [PubMed]
- Sanz, M.A.; Madan, V.; Carrasco, L.; Nieva, J.L. Interfacial domains in Sindbis virus 6K protein. Detection and functional characterization. J. Biol. Chem. 2003, 278, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.S.; Visch, H.J.; de Mattia, F.; van Dommelen, M.M.; Swarts, H.G.; Luyten, T.; Callewaert, G.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and Golgi, thereby inhibiting protein trafficking through the Golgi. J. Biol. Chem. 2006, 281, 14144–14150. [Google Scholar] [CrossRef] [PubMed]
- Van Kuppeveld, F.J.; Hoenderop, J.G.; Smeets, R.L.; Willems, P.H.; Dijkman, H.B.; Galama, J.M.; Melchers, W.J. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 1997, 16, 3519–3532. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Frey, T.K.; Yang, J.J. Viral calciomics: Interplays between Ca2+ and virus. Cell Calcium 2009, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ao, D.; Sun, S.Q.; Guo, H.C. Topology and biological function of enterovirus non-structural protein 2B as a member of the viroporin family. Vet. Res. 2014, 45, e87. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Estes, M.K. Viroporin-mediated calcium-activated autophagy. Autophagy 2013, 9, 797–798. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Daniels, R.; Rusan, N.M.; Wadsworth, P.; Hebert, D.N. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: Implications for DNA translocation out of the ER. Mol. Cell 2006, 24, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Giorda, K.M.; Raghava, S.; Zhang, M.W.; Hebert, D.N. The viroporin activity of the minor structural proteins VP2 and VP3 is required for SV40 propagation. J. Biol. Chem. 2013, 288, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Raghava, S.; Giorda, K.M.; Romano, F.B.; Heuck, A.P.; Hebert, D.N. SV40 late protein VP4 forms toroidal pores to disrupt membranes for viral release. Biochemistry 2013, 52, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Raghava, S.; Giorda, K.M.; Romano, F.B.; Heuck, A.P.; Hebert, D.N. The SV40 late protein VP4 is a viroporin that forms pores to disrupt membranes for viral release. PLoS Pathog. 2011, 7, e1002116. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Collinson-Pautz, M.R.; Utama, B.; Estes, M.K. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. MBIO 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Schnell, J.R.; Chou, J.J. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA 2009, 106, 7379–7384. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Chou, J.J. Solution NMR structure of the V27A drug resistant mutant of influenza A M2 channel. Biochem. Biophys. Res. Commun. 2010, 401, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Chou, J.J. Kinetic analysis of the M2 proton conduction of the influenza virus. J. Am. Chem. Soc. 2010, 132, 17695–17697. [Google Scholar] [CrossRef] [PubMed]
- Cook, G.A.; Zhang, H.; Park, S.H.; Wang, Y.; Opella, S.J. Comparative NMR studies demonstrate profound differences between two viroporins: p7 of HCV and Vpu of HIV-1. Biochim. Biophys. Acta 2011, 1808, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qin, H.; Gao, F.P.; Cross, T.A. Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim. Biophys. Acta 2007, 1768, 3162–3170. [Google Scholar] [CrossRef] [PubMed]
- Stouffer, A.L.; Ma, C.; Cristian, L.; Ohigashi, Y.; Lamb, R.A.; Lear, J.D.; Pinto, L.H.; DeGrado, W.F. The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure 2008, 16, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qiu, J.X.; Soto, C.; DeGrado, W.F. Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr. Opin. Struct. Biol. 2011, 21, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Luik, P.; Chew, C.; Aittoniemi, J.; Chang, J.; Wentworth, P., Jr.; Dwek, R.A.; Biggin, P.C.; Venien-Bryan, C.; Zitzmann, N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 12712–12716. [Google Scholar] [CrossRef] [PubMed]
- OuYang, B.; Xie, S.; Berardi, M.J.; Zhao, X.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Griffin, S.; Beales, L.; Gelais, C.S.; Burgess, S.; Harris, M.; Rowlands, D. Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro. J. Biol. Chem. 2006, 281, 37057–37068. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.E.; Penin, F.; Schulten, K.; Chipot, C. The p7 protein of hepatitis C virus forms structurally plastic, minimalist ion channels. PLoS Comput. Biol. 2012, 8, e1002702. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.W.; Tan, E.; Lin, X.; Yu, D.; Wang, J.; Tan, G.M.; Vararattanavech, A.; Yeo, C.Y.; Soon, C.H.; Soong, T.W.; et al. The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J. Biol. Chem. 2012, 287, 24671–24689. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lamb, R.A.; Pinto, L.H. Activation of the M2 ion channel of influenza virus: A role for the transmembrane domain histidine residue. Biophys. J. 1995, 69, 1363–1371. [Google Scholar] [CrossRef]
- Strebel, K.; Klimkait, T.; Martin, M.A. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science 1988, 241, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, E.F.; Godin, B.; Sodroski, J.G.; Haseltine, W.A. Construction and use of a replication-competent human immunodeficiency virus (HIV-1) that expresses the chloramphenicol acetyltransferase enzyme. Proc. Natl. Acad. Sci. USA 1989, 86, 3857–3861. [Google Scholar] [CrossRef] [PubMed]
- Klimkait, T.; Strebel, K.; Hoggan, M.D.; Martin, M.A.; Orenstein, J.M. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 1990, 64, 621–629. [Google Scholar] [PubMed]
- Jabbar, M.A.; Nayak, D.P. Intracellular interaction of human immunodeficiency virus type 1 (ARV-2) envelope glycoprotein gp160 with CD4 blocks the movement and maturation of CD4 to the plasma membrane. J. Virol. 1990, 64, 6297–6304. [Google Scholar] [PubMed]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J. Virol. 1992, 66, 226–234. [Google Scholar] [PubMed]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [PubMed]
- Kimura, T.; Nishikawa, M.; Ohyama, A. Intracellular membrane traffic of human immunodeficiency virus type 1 envelope glycoproteins: Vpu liberates Golgi-targeted gp160 from CD4-dependent retention in the endoplasmic reticulum. J. Biochem. 1994, 115, 1010–1020. [Google Scholar] [PubMed]
- Wozniak, A.L.; Griffin, S.; Rowlands, D.; Harris, M.; Yi, M.; Lemon, S.M.; Weinman, S.A. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 2010, 6, e1001087. [Google Scholar] [CrossRef] [PubMed]
- Sakai, A.; Claire, M.S.; Faulk, K.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H.; Bukh, J. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl. Acad. Sci. USA 2003, 100, 11646–11651. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, D.; Neville, D.C.; Argaud, O.; Blumberg, B.; Dwek, R.A.; Fischer, W.B.; Zitzmann, N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc. Natl. Acad. Sci. USA 2003, 100, 6104–6108. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, A.; Wilson, L.; Ewart, G.D.; Gage, P.W. Cation-selective ion channels formed by p7 of hepatitis C virus are blocked by hexamethylene amiloride. FEBS Lett. 2004, 557, 99–103. [Google Scholar] [CrossRef]
- Steinmann, E.; Whitfield, T.; Kallis, S.; Dwek, R.A.; Zitzmann, N.; Pietschmann, T.; Bartenschlager, R. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 2007, 46, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, B.D.; Cheng, V.; Donahue, D.A.; Sloan, R.D.; Liang, C.; Wilkinson, J.; Wainberg, M.A. The HIV-1 Vpu viroporin inhibitor BIT225 does not affect Vpu-mediated tetherin antagonism. PLoS ONE 2011, 6, e27660. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Thompson, G.S.; Kalverda, A.P.; Kankanala, J.; Bentham, M.; Wetherill, L.F.; Thompson, J.; Barker, A.M.; Clarke, D.; Noerenberg, M.; et al. Structure-guided design affirms inhibitors of hepatitis C virus p7 as a viable class of antivirals targeting virion release. Hepatology 2014, 59, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. 2004, 2, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B. Penetration of nonenveloped viruses into the cytoplasm. Annu. Rev. Cell Dev. Biol. 2007, 23, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Edinger, T.O.; Pohl, M.O.; Stertz, S. Entry of influenza A virus: Host factors and antiviral targets. J. Gen. Virol. 2014, 95, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Cady, S.D.; Luo, W.; Hu, F.; Hong, M. Structure and function of the influenza A M2 proton channel. Biochemistry 2009, 48, 7356–7364. [Google Scholar] [CrossRef] [PubMed]
- Balannik, V.; Carnevale, V.; Fiorin, G.; Levine, B.G.; Lamb, R.A.; Klein, M.L.; Degrado, W.F.; Pinto, L.H. Functional studies and modeling of pore-lining residue mutants of the influenza a virus M2 ion channel. Biochemistry 2010, 49, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Polishchuk, A.L.; Ohigashi, Y.; Stouffer, A.L.; Schon, A.; Magavern, E.; Jing, X.; Lear, J.D.; Freire, E.; Lamb, R.A.; et al. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc. Natl. Acad. Sci. USA 2009, 106, 12283–12288. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Watanabe, S.; Ito, H.; Kida, H.; Kawaoka, Y. Influenza A virus can undergo multiple cycles of replication without M2 ion channel activity. J. Virol. 2001, 75, 5656–2662. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Pekosz, A.; Shuck, K.; Pinto, L.H.; Lamb, R.A. Influenza a virus M2 ion channel activity is essential for efficient replication in tissue culture. J. Virol. 2002, 76, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.; Sadowicz, D.; Hebert, D.N. A very late viral protein triggers the lytic release of SV40. PLoS Pathog. 2007, 3, e98. [Google Scholar] [CrossRef] [PubMed]
- Giorda, K.M.; Raghava, S.; Hebert, D.N. The Simian virus 40 late viral protein VP4 disrupts the nuclear envelope for viral release. J. Virol. 2012, 86, 3180–3192. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anderson, H.A.; Chen, Y.; Norkin, L.C. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell 1996, 7, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Qian, M. Cellular entry of polyomaviruses. Curr. Top. Microbiol. Immunol. 2010, 343, 177–194. [Google Scholar] [PubMed]
- Cohen, E.A.; Terwilliger, E.F.; Sodroski, J.G.; Haseltine, W.A. Identification of a protein encoded by the vpu gene of HIV-1. Nature 1988, 334, 532–534. [Google Scholar] [CrossRef] [PubMed]
- Cordes, F.S.; Tustian, A.D.; Sansom, M.S.; Watts, A.; Fischer, W.B. Bundles consisting of extended transmembrane segments of Vpu from HIV-1: Computer simulations and conductance measurements. Biochemistry 2002, 41, 7359–7365. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, I.; Koga, Y.; Oh-Hori, N.; Onodera, K.; Kimura, G.; Nomoto, K. Depletion of the surface CD4 molecule by the envelope protein of human immunodeficiency virus expressed in a human CD4+ monocytoid cell line. J. Virol. 1989, 63, 3748–3754. [Google Scholar] [PubMed]
- Crise, B.; Buonocore, L.; Rose, J.K. CD4 is retained in the endoplasmic reticulum by the human immunodeficiency virus type 1 glycoprotein precursor. J. Virol. 1990, 64, 5585–5593. [Google Scholar] [PubMed]
- Bour, S.; Boulerice, F.; Wainberg, M.A. Inhibition of gp160 and CD4 maturation in U937 cells after both defective and productive infections by human immunodeficiency virus type 1. J. Virol. 1991, 65, 6387–6396. [Google Scholar] [PubMed]
- Bour, S.; Schubert, U.; Strebel, K. The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: Implications for the mechanism of degradation. J. Virol. 1995, 69, 1510–1520. [Google Scholar] [PubMed]
- Margottin, F.; Benichou, S.; Durand, H.; Richard, V.; Liu, L.X.; Gomas, E.; Benarous, R. Interaction between the cytoplasmic domains of HIV-1 Vpu and CD4: Role of Vpu residues involved in CD4 interaction and in vitro CD4 degradation. Virology 1996, 223, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Bour, S.; Ferrer-Montiel, A.V.; Montal, M.; Maldarell, F.; Strebel, K. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 1996, 70, 809–819. [Google Scholar] [PubMed]
- Schubert, U.; Strebel, K. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J. Virol. 1994, 68, 2260–2271. [Google Scholar] [PubMed]
- Friborg, J.; Ladha, A.; Gottlinger, H.; Haseltine, W.A.; Cohen, E.A. Functional analysis of the phosphorylation sites on the human immunodeficiency virus type 1 Vpu protein. J. Acquir. Immun. Defic. Syndr. Hum. Retrovirol. 1995, 8, 10–22. [Google Scholar] [CrossRef]
- Magadan, J.G.; Bonifacino, J.S. Transmembrane domain determinants of CD4 Downregulation by HIV-1 Vpu. J. Virol. 2012, 86, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Deitz, S.B.; Dodd, D.A.; Cooper, S.; Parham, P.; Kirkegaard, K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 2000, 97, 13790–13795. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J. Virol. 2001, 75, 8158–8165. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.F. The involvement of calcium in transport of secretory proteins from the endoplasmic reticulum. Cell 1990, 61, 197–199. [Google Scholar] [CrossRef]
- Dinter, A.; Berger, E.G. Golgi-disturbing agents. Histochem. Cell Biol. 1998, 109, 571–590. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D.; Clague, M.J. Calcium and calmodulin in membrane fusion. Biochim. Biophys. Acta 2003, 1641, 137–143. [Google Scholar] [CrossRef]
- Hay, J.C. Calcium: A fundamental regulator of intracellular membrane fusion? EMBO Rep. 2007, 8, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Utama, B.; Crawford, S.E.; Broughman, J.R.; Estes, M.K. Activation of the endoplasmic reticulum calcium sensor STIM1 and store-operated calcium entry by rotavirus requires NSP4 viroporin activity. J. Virol. 2013, 87, 13579–13588. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Michelangeli, F.; Ruiz, M.C.; del Castillo, J.R.; Ludert, J.E.; Liprandi, F. Effect of rotavirus infection on intracellular calcium homeostasis in cultured cells. Virology 1991, 181, 520–527. [Google Scholar] [CrossRef]
- Tian, P.; Hu, Y.; Schilling, W.P.; Lindsay, D.A.; Eiden, J.; Estes, M.K. The nonstructural glycoprotein of rotavirus affects intracellular calcium levels. J. Virol. 1994, 68, 251–257. [Google Scholar] [PubMed]
- Tian, P.; Estes, M.K.; Hu, Y.; Ball, J.M.; Zeng, C.Q.; Schilling, W.P. The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. J. Virol. 1995, 69, 5763–5772. [Google Scholar] [PubMed]
- Zambrano, J.L.; Diaz, Y.; Pena, F.; Vizzi, E.; Ruiz, M.C.; Michelangeli, F.; Liprandi, F.; Ludert, J.E. Silencing of rotavirus NSP4 or VP7 expression reduces alterations in Ca2+ homeostasis induced by infection of cultured cells. J. Virol. 2008, 82, 5815–5824. [Google Scholar] [CrossRef] [PubMed]
- Ousingsawat, J.; Mirza, M.; Tian, Y.; Roussa, E.; Schreiber, R.; Cook, D.I.; Kunzelmann, K. Rotavirus toxin NSP4 induces diarrhea by activation of TMEM16A and inhibition of Na+ absorption. Pflug. Arch. 2011, 461, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Mirazimi, A.; Nilsson, M.; Svensson, L. The molecular chaperone calnexin interacts with the NSP4 enterotoxin of rotavirus in vivo and in vitro. J. Virol. 1998, 72, 8705–8709. [Google Scholar] [PubMed]
- Xu, A.; Bellamy, A.R.; Taylor, J.A. Immobilization of the early secretory pathway by a virus glycoprotein that binds to microtubules. EMBO J. 2000, 19, 6465–6474. [Google Scholar] [CrossRef] [PubMed]
- Boshuizen, J.A.; Rossen, J.W.; Sitaram, C.K.; Kimenai, F.F.; Simons-Oosterhuis, Y.; Laffeber, C.; Buller, H.A.; Einerhand, A.W. Rotavirus enterotoxin NSP4 binds to the extracellular matrix proteins laminin-beta3 and fibronectin. J. Virol. 2004, 78, 10045–10053. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.D.; Storey, S.M.; Mitchell, D.M.; McIntosh, A.L.; Zhou, M.; Mir, K.D.; Ball, J.M. The rotavirus enterotoxin NSP4 directly interacts with the caveolar structural protein caveolin-1. J. Virol. 2006, 80, 2842–2854. [Google Scholar] [CrossRef] [PubMed]
- Seo, N.S.; Zeng, C.Q.; Hyser, J.M.; Utama, B.; Crawford, S.E.; Kim, K.J.; Hook, M.; Estes, M.K. Integrins alpha1beta1 and alpha2beta1 are receptors for the rotavirus enterotoxin. Proc. Natl. Acad. Sci. USA 2008, 105, 8811–8818. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, N.; Maurice, M.; Delautier, D.; Quero, A.M.; Servin, A.L.; Trugnan, G. Rotavirus is released from the apical surface of cultured human intestinal cells through nonconventional vesicular transport that bypasses the Golgi apparatus. J. Virol. 1997, 71, 8268–8278. [Google Scholar] [PubMed]
- Silvestri, L.S.; Tortorici, M.A.; Vasquez-Del Carpio, R.; Patton, J.T. Rotavirus glycoprotein NSP4 is a modulator of viral transcription in the infected cell. J. Virol. 2005, 79, 15165–15174. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.; Camacho, M.; Zayas, M.; Najera, R.; Sanchez, R.; Arias, C.F.; Lopez, S. Silencing the morphogenesis of rotavirus. J. Virol. 2005, 79, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; McKinlay, C.; Gage, P.; Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Garcia Mde, J.; Sanz, M.A.; Carrasco, L. Viroporin activity of murine hepatitis virus E protein. FEBS Lett. 2005, 579, 3607–3612. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Yuan, Q.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology 2006, 349, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Corse, E.; Machamer, C.E. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 2000, 74, 4319–4326. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Repass, J.F.; Maeda, A.; Makino, S. Membrane topology of coronavirus E protein. Virology 2001, 281, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Liao, Y.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical evidence for the presence of mixed membrane topologies of the severe acute respiratory syndrome coronavirus envelope protein expressed in mammalian cells. FEBS Lett. 2006, 580, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Dediego, M.L.; Alvarez, E.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Llorente, M.; Kremer, L.; Shuo, S.; Enjuanes, L. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 2011, 415, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Stegen, C.F.; Masters, P.S.; Samsonoff, W.A. Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J. Virol. 1998, 72, 7885–7894. [Google Scholar] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Luo, G. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J. Gen. Virol. 2003, 84, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Boson, B.; Granio, O.; Bartenschlager, R.; Cosset, F.L. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog. 2011, 7, e1002144. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.; Bayer, K.; Rosch, K.; Schindler, M. The intraviral protein interaction network of hepatitis C virus. MCP 2014, 13, 1676–1689. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Yi, M. Efficiency of E2-p7 processing modulates production of infectious hepatitis C virus. J. Virol. 2013, 87, 11255–11266. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.D.; Beales, L.P.; Clarke, D.S.; Worsfold, O.; Evans, S.D.; Jaeger, J.; Harris, M.P.; Rowlands, D.J. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003, 535, 34–38. [Google Scholar] [CrossRef]
- Griffin, S.; Stgelais, C.; Owsianka, A.M.; Patel, A.H.; Rowlands, D.; Harris, M. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 2008, 48, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Verow, M.; Wozniak, A.L.; Bentham, M.J.; Thompson, J.; Atkins, E.; Weinman, S.A.; Fishwick, C.; Foster, R.; Harris, M.; et al. Resistance mutations define specific antiviral effects for inhibitors of the hepatitis C virus p7 ion channel. Hepatology 2011, 54, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Melton, J.V.; Ewart, G.D.; Weir, R.C.; Board, P.G.; Lee, E.; Gage, P.W. Alphavirus 6K proteins form ion channels. J. Biol. Chem. 2002, 277, 46923–46931. [Google Scholar] [CrossRef] [PubMed]
- McCown, M.F.; Pekosz, A. Distinct domains of the influenza a virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J. Virol. 2006, 80, 8178–8189. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Leser, G.P.; Ma, C.; Lamb, R.A. The amphipathic helix of influenza A virus M2 protein is required for filamentous bud formation and scission of filamentous and spherical particles. J. Virol. 2013, 87, 9973–9982. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 2009, 284, 35060–35072. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Neil, S.J. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Pathog. 2012, 8, e1002609. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Fruh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.T.; Serra-Moreno, R.; Singh, R.K.; Guatelli, J.C. BST-2/tetherin: A new component of the innate immune response to enveloped viruses. Trends Microbiol. 2010, 18, 388–396. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Weber, E.; Tokarev, A.; Lewinski, M.; Rizk, M.; Suarez, M.; Guatelli, J.; Xiong, Y. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife 2014, 3, e02362. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.D.; Harvey, R.; Clarke, D.S.; Barclay, W.S.; Harris, M.; Rowlands, D.J. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J. Gen. Virol. 2004, 85, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Castello, A.; Carrasco, L. Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell. Microbiol. 2008, 10, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Halder, U.C.; Chattopadhyay, S.; Chanda, S.; Nandi, S.; Bagchi, P.; Nayak, M.K.; Chakrabarti, O.; Kobayashi, N.; Chawla-Sarkar, M. Rotaviral enterotoxin nonstructural protein 4 targets mitochondria for activation of apoptosis during infection. J. Biol. Chem. 2012, 287, 35004–35020. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, I.; Kirchhoff, S.; Krammer, P.H. Regulation of death receptor-mediated apoptosis pathways. Int. J. Biochem. Cell Biol. 2000, 32, 1123–1136. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef] [PubMed]
- Aweya, J.J.; Mak, T.M.; Lim, S.G.; Tan, Y.J. The p7 protein of the hepatitis C virus induces cell death differently from the influenza A virus viroporin M2. Virus Res. 2013, 172, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Akari, H.; Bour, S.; Kao, S.; Adachi, A.; Strebel, K. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor kappaB-dependent expression of antiapoptotic factors. J. Exp. Med. 2001, 194, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Bour, S.; Perrin, C.; Akari, H.; Strebel, K. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J. Biol. Chem. 2001, 276, 15920–15928. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F. Membrane origin for autophagy. Curr. Top. Dev. Biol. 2006, 74, 1–30. [Google Scholar] [PubMed]
- Longatti, A.; Tooze, S.A. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009, 16, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Klionsky, D.J. Physiological functions of Atg6/Beclin 1: A unique autophagy-related protein. Cell Res. 2007, 17, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.W.; Teterina, N.; Egger, D.; Bienz, K.; Ehrenfeld, E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 1994, 202, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Kondratova, A.A.; Neznanov, N.; Kondratov, R.V.; Gudkov, A.V. Poliovirus protein 3A binds and inactivates LIS1, causing block of membrane protein trafficking and deregulation of cell division. Cell Cycle 2005, 4, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Burgon, T.B.; Kirkegaard, K.; Jackson, W.T. Role of microtubules in extracellular release of poliovirus. J. Virol. 2009, 83, 6599–6609. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Chen, I.Y.; Ichinohe, T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Triantafilou, M. Ion flux in the lung: Virus-induced inflammasome activation. Trends Microbiol. 2014, 22, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [PubMed]
- Pirhonen, J.; Sareneva, T.; Kurimoto, M.; Julkunen, I.; Matikainen, S. Virus infection activates IL-1 beta and IL-18 production in human macrophages by a caspase-1-dependent pathway. J. Immunol. 1999, 162, 7322–7329. [Google Scholar] [PubMed]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Kar, S.; Vakakis, E.; Kotecha, S.; Triantafilou, M. Human respiratory syncytial virus viroporin SH: A viral recognition pathway used by the host to signal inflammasome activation. Thorax 2013, 68, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Aldabe, R.; Barco, A.; Carrasco, L. Membrane permeabilization by poliovirus proteins 2B and 2BC. J. Biol. Chem. 1996, 271, 23134–23137. [Google Scholar] [PubMed]
- Ito, M.; Yanagi, Y.; Ichinohe, T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 2012, 8, e1002857. [Google Scholar] [CrossRef] [PubMed]
- Davies, W.L.; Grunert, R.R.; Haff, R.F.; McGahen, J.W.; Neumayer, E.M.; Paulshock, M.; Watts, J.C.; Wood, T.R.; Hermann, E.C.; Hoffmann, C.E. Antiviral Activity of 1-Adamantanamine (Amantadine). Science 1964, 144, 862–863. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.J.; Wolstenholme, A.J.; Skehel, J.J.; Smith, M.H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985, 4, 3021–3024. [Google Scholar] [PubMed]
- Wang, C.; Takeuchi, K.; Pinto, L.H.; Lamb, R.A. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J. Virol. 1993, 67, 5585–5594. [Google Scholar] [PubMed]
- Khoury, G.; Ewart, G.; Luscombe, C.; Miller, M.; Wilkinson, J. Antiviral efficacy of the novel compound BIT225 against HIV-1 release from human macrophages. Antimicrob. Agents Chemother. 2010, 54, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, C.A.; Huang, Z.; Murray, M.G.; Miller, M.; Wilkinson, J.; Ewart, G.D. A novel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viral diarrhea virus in vitro and shows synergism with recombinant interferon-alpha-2b and nucleoside analogues. Antivir. Res. 2010, 86, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; To, J.; Verdia-Baguena, C.; Dossena, S.; Surya, W.; Huang, M.; Paulmichl, M.; Liu, D.X.; Aguilella, V.M.; Torres, J. Inhibition of the human respiratory syncytial virus small hydrophobic protein and structural variations in a bicelle environment. J. Virol. 2014, 88, 11899–11914. [Google Scholar] [CrossRef] [PubMed]
- Meredith, L.W.; Zitzmann, N.; McKeating, J.A. Differential effect of p7 inhibitors on hepatitis C virus cell-to-cell transmission. Antivir. Res. 2013, 100, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Cady, S.D.; Schmidt-Rohr, K.; Wang, J.; Soto, C.S.; Degrado, W.F.; Hong, M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Bukrinskaya, A.G.; Vorkunova, N.K.; Kornilayeva, G.V.; Narmanbetova, R.A.; Vorkunova, G.K. Influenza virus uncoating in infected cells and effect of rimantadine. J. Gen. Virol. 1982, 60, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Bukrinskaya, A.G.; Vorkunova, N.K.; Pushkarskaya, N.L. Uncoating of a rimantadine-resistant variant of influenza virus in the presence of rimantadine. J. Gen. Virol. 1982, 60, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.A.; Shay, D.K.; Shu, B.; Cox, N.J.; Klimov, A.I. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA 2006, 295, 891–894. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sze, C.; Tan, Y.-J. Viral Membrane Channels: Role and Function in the Virus Life Cycle. Viruses 2015, 7, 3261-3284. https://doi.org/10.3390/v7062771
Sze C, Tan Y-J. Viral Membrane Channels: Role and Function in the Virus Life Cycle. Viruses. 2015; 7(6):3261-3284. https://doi.org/10.3390/v7062771
Chicago/Turabian StyleSze, ChingWooen, and Yee-Joo Tan. 2015. "Viral Membrane Channels: Role and Function in the Virus Life Cycle" Viruses 7, no. 6: 3261-3284. https://doi.org/10.3390/v7062771
APA StyleSze, C., & Tan, Y.-J. (2015). Viral Membrane Channels: Role and Function in the Virus Life Cycle. Viruses, 7(6), 3261-3284. https://doi.org/10.3390/v7062771