
RNA Viruses and RNAi: Quasispecies Implications for Viral Escape

Abstract
:1. RNA Virus Replication and Evolution
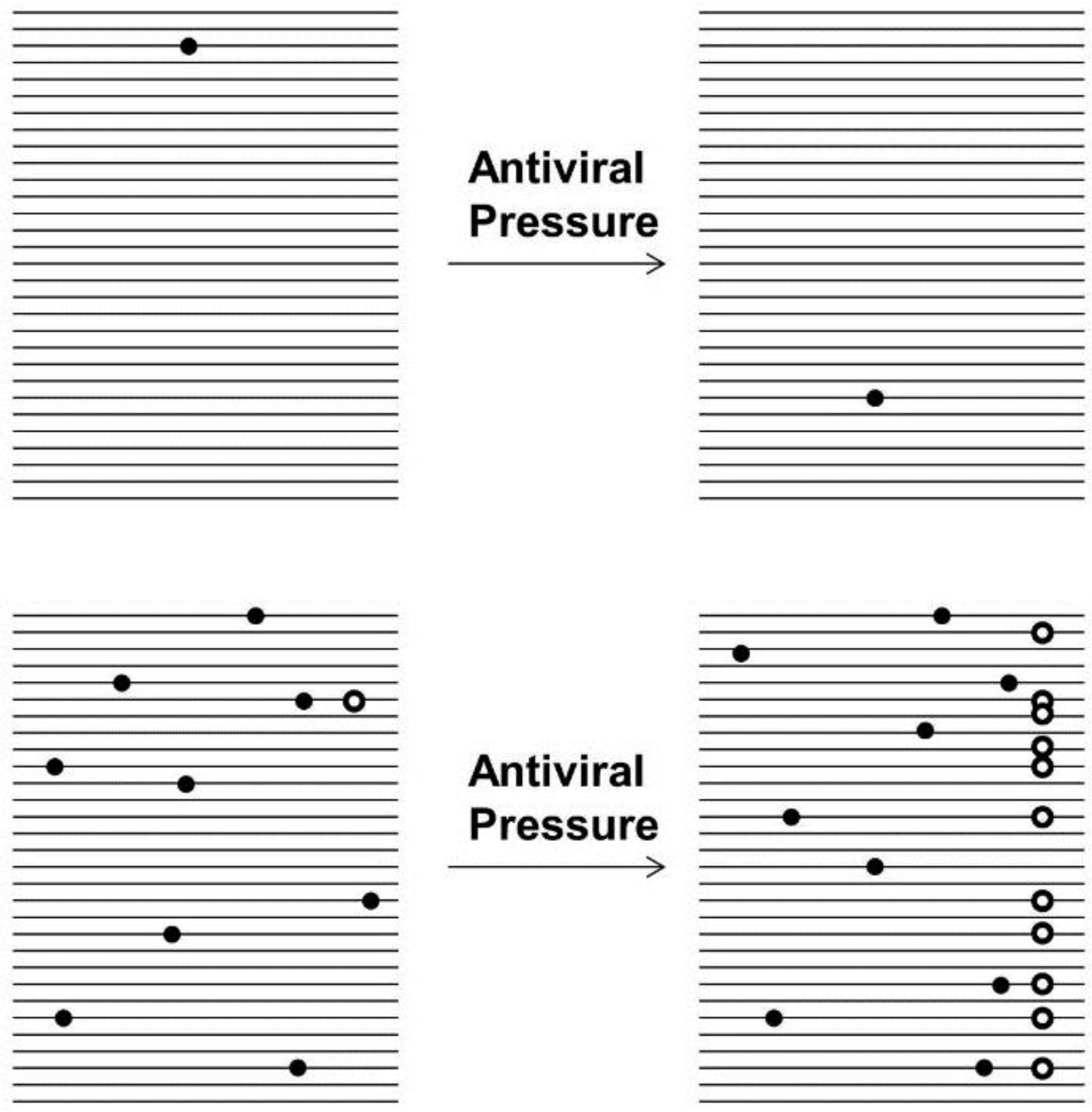
4. Beyond siRNA: The Problem with Resistance

{kind=link}
| Virus | siRNA target(s) | References |
|---|---|---|
| HIV | nef | [75,77,85] |
| tat | [74] | |
| int | [76] | |
| att | [76] | |
| Multiple siRNA, mutations outside targeted sequence | [79,81] | |
| Poliovirus | Multiple siRNAs | [69] |
| Hepatitis C | Multiple siRNA | [71] |
| JEV | Multiple siRNAs | [70,76] |
| FMDV | siRNA-resistant, no mutations in targeted sequence | [78] |
| Turnip Mosaic Virus | HC-Pro | [72] |
| PPRV | Nucleoprotein | [73] |
5. Combinational Therapy with siRNA
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Drake, J.W. The distribution of rates of spontaneous mutation over viruses, prokaryotes, and eukaryotes. Ann. N. Y. Acad. Sci. 1999, 870, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Steps toward Life; Oxford University Press: Oxford, UK, 1992; p. 173. [Google Scholar]
- Eigen, M. On the nature of virus quasispecies. Trends Microbiol. 1996, 4, 216–218. [Google Scholar] [CrossRef]
- Schuster, P.; Swetina, J. Stationary mutant distributions and evolutionary optimization. Bull. Math. Biol. 1988, 50, 635–660. [Google Scholar] [CrossRef] [PubMed]
- Swetina, J.; Schuster, P. Self-replication with errors. A model for polynucleotide replication. Biophys. Chem. 1982, 16, 329–345. [Google Scholar] [CrossRef]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Wilke, C.O.; Forster, R.; Novella, I.S. Quasispecies in time-dependent environments. Curr. Top. Microbiol. Immunol. 2006, 299, 33–50. [Google Scholar] [PubMed]
- Bull, J.J.; Meyers, L.A.; Lachmann, M. Quasispecies made simple. PLoS Comput. Biol. 2005, 1, e61. [Google Scholar] [CrossRef] [PubMed]
- Najera, I.; Holguin, A.; Quinones-Mateu, M.E.; Munoz-Fernandez, M.A.; Najera, R.; Lopez-Galindez, C.; Domingo, E. Pol gene quasispecies of human immunodeficiency virus: Mutations associated with drug resistance in virus from patients undergoing no drug therapy. J. Virol. 1995, 69, 23–31. [Google Scholar] [PubMed]
- Van Valen, L. A new evolutionary law. Evol. Theory 1973, 1, 1–30. [Google Scholar]
- Woo, H.J.; Reifman, J. A quantitative quasispecies theory-based model of virus escape mutation under immune selection. Proc. Natl. Acad. Sci. USA 2012, 109, 12980–12985. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Menendez-Arias, L.; Quinones-Mateu, M.E.; Holguin, A.; Gutierrez-Rivas, M.; Martinez, M.A.; Quer, J.; Novella, I.S.; Holland, J.J. Viral quasispecies and the problem of vaccine-escape and drug-resistant mutants. Prog. Drug Res. 1997, 48, 99–128. [Google Scholar] [PubMed]
- Muraille, E. Redefining the immune system as a social interface for cooperative processes. PLoS Pathog. 2013, 9, e1003203. [Google Scholar] [CrossRef] [PubMed]
- Bedford, T.; Suchard, M.A.; Lemey, P.; Dudas, G.; Gregory, V.; Hay, A.J.; McCauley, J.W.; Russell, C.A.; Smith, D.J.; Rambaut, A. Integrating influenza antigenic dynamics with molecular evolution. eLife 2014, 3, e01914. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Holmes, E.C. The evolution of epidemic influenza. Nat. Rev. Genet. 2007, 8, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Bloom, J.D. The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Wasilewski, L.N.; Snider, A.E.; El-Diwany, R.; Osburn, W.O.; Keck, Z.; Foung, S.K.; Ray, S.C. Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J. Clin. Investig. 2015, 125, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Rolls, J.K.; Szabo, G. The genetics of hepatitis C virus underlie its ability to escape humoral immunity. J. Clin. Investig. 2015, 125, 97–98. [Google Scholar]
- Batorsky, R.; Sergeev, R.A.; Rouzine, I.M. The route of HIV escape from immune response targeting multiple sites is determined by the cost-benefit tradeoff of escape mutations. PLoS Comput. Biol. 2014, 10, e1003878. [Google Scholar] [CrossRef] [PubMed]
- Leslie, A.J.; Pfafferott, K.J.; Chetty, P.; Draenert, R.; Addo, M.M.; Feeney, M.; Tang, Y.; Holmes, E.C.; Allen, T.; Prado, J.G.; et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 2004, 10, 282–289. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The immune response during acute HIV-1 infection: Clues for vaccine development. Nat. Rev. Immunol. 2010, 10, 11–23. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.; Picker, L.J.; Moore, J.P.; Burton, D.R. Another HIV vaccine failure: Where to next? Nat. Med. 2013, 19, 1576–1577. [Google Scholar] [PubMed]
- Esparza, J. A brief history of the global effort to develop a preventive HIV vaccine. Vaccine 2013, 31, 3502–3518. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.N.; Ogawa, H.; Trinh, D.Q.; Nguyen, T.H.T.; Pham, N.T.; Truong, D.A.; Bui, A.N.; Runstadler, J.; Imai, K.; Nguyen, K.V. Genetic characterization of an H5N1 avian influenza virus from a vaccinated duck flock in Vietnam. Virus Genes 2014, 49, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Schotsaert, M.; Garcia-Sastre, A. Influenza vaccines: A moving interdisciplinary field. Viruses 2014, 6, 3809–3826. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G. Antivirals for influenza: Historical perspectives and lessons learned. Antiviral Res. 2006, 71, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Moscona, A. Global transmission of oseltamivir-resistant influenza. N. Engl. J. Med. 2009, 360, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res. 2013, 98, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Takashita, E.; Meijer, A.; Lackenby, A.; Gubareva, L.; Rebelo-de-Andrade, H.; Besselaar, T.; Fry, A.; Gregory, V.; Leang, S.K.; Huang, W.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2013–2014. Antiviral Res. 2015, 117, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Behera, A.K.; Basu, S.; Cherian, S.S. Molecular mechanism of the enhanced viral fitness contributed by secondary mutations in the hemagglutinin protein of oseltamivir resistant H1N1 influenza viruses: Modeling studies of antibody and receptor binding. Gene 2015, 557, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Gillman, A.; Muradrasoli, S.; Soderstrom, H.; Holmberg, F.; Latorre-Margalef, N.; Tolf, C.; Waldenstrom, J.; Gunnarsson, G.; Olsen, B.; Jarhult, J.D. Oseltamivir-resistant influenza a (H1N1) virus strain with an H274Y mutation in neuraminidase persists without drug pressure in infected mallards. Appl. Environ. Microbiol. 2015, 81, 2378–2383. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.A.; Brun-Vezinet, F.; Clotet, B.; Gunthard, H.F.; Kuritzkes, D.R.; Pillay, D.; Schapiro, J.M.; Richman, D.D. Update of the drug resistance mutations in HIV-1: 2007. Top. HIV Med. 2007, 15, 119–125. [Google Scholar] [PubMed]
- Shi, J.; Zhou, J.; Shah, V.B.; Aiken, C.; Whitby, K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 2011, 85, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L. Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antiviral Res. 2013, 98, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Cameron, C.; Andino, R. Ribavirin’s antiviral mechanism of action: Lethal mutagenesis? J. Mol. Med. 2002, 80, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Cameron, C.E.; Andino, R. RNA virus error catastrophe: Direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 2001, 98, 6895–6900. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Andino, R. Ribavirin and lethal mutagenesis of poliovirus: Molecular mechanisms, resistance and biological implications. Virus Res. 2005, 107, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Beaucourt, S.; Vignuzzi, M. Ribavirin: A drug active against many viruses with multiple effects on virus replication and propagation. Molecular basis of ribavirin resistance. Curr. Opin. Virol. 2014, 8, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Emerson, S.U.; Wang, Y.; Pan, Q.; Balzarini, J.; Dallmeier, K.; Neyts, J. Ribavirin inhibits in vitro hepatitis E virus replication through depletion of cellular GTP pools and is moderately synergistic with alpha interferon. Antimicrob. Agents Chemother. 2014, 58, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Bitko, V.; Barik, S. Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild type negative-strand RNA viruses. BMC Microbiol. 2001, 1, e34. [Google Scholar] [CrossRef]
- Lee, N.S.; Dohjima, T.; Bauer, G.; Li, H.; Li, M.J.; Ehsani, A.; Salvaterra, P.; Rossi, J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol. 2002, 20, 500–505. [Google Scholar] [PubMed]
- Xie, P.W.; Xie, Y.; Zhang, X.J.; Huang, H.; He, L.N.; Wang, X.J.; Wang, S.Q. Inhibition of Dengue virus 2 replication by artificial microRNAs targeting the conserved regions. Nucleic Acid Ther. 2013, 23, 244–252. [Google Scholar] [PubMed]
- Kapadia, S.B.; Brideau-Andersen, A.; Chisari, F.V. Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl. Acad Sci. USA 2003, 100, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Kittler, R.; Buchholz, F.; Windisch, M.P.; Pietschmann, T.; Bartenschlager, R.; Frese, M. Alternative approaches for efficient inhibition of hepatitis C virus RNA replication by small interfering RNAs. J. Virol. 2004, 78, 3436–3446. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Steele, R.; Ray, R.; Ray, R.B. Small interfering RNA targeted to hepatitis C virus 5′ nontranslated region exerts potent antiviral effect. J. Virol. 2007, 81, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yan, W.; Du, Q.; Fei, L.; Liu, M.; Ni, Z.; Sheng, Z.; Zheng, Z. RNA interference targeting VP1 inhibits foot-and-mouth disease virus replication in BHK-21 cells and suckling mice. J. Virol. 2004, 78, 6900–6907. [Google Scholar] [CrossRef] [PubMed]
- Kahana, R.; Kuznetzova, L.; Rogel, A.; Shemesh, M.; Hai, D.; Yadin, H.; Stram, Y. Inhibition of foot-and-mouth disease virus replication by small interfering RNA. J. Gen. Virol. 2004, 85, 3213–3217. [Google Scholar] [CrossRef] [PubMed]
- Achazi, K.; Patel, P.; Paliwal, R.; Radonic, A.; Niedrig, M.; Donoso-Mantke, O. RNA interference inhibits replication of tick-borne encephalitis virus in vitro. Antiviral Res. 2012, 93, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; McManus, M.T.; Nguyen, T.; Shen, C.H.; Sharp, P.A.; Eisen, H.N.; Chen, J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 2718–2723. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.K.; Yap, E.M.; An, D.S.; Chen, I.S.; Nayak, D.P. Inhibition of influenza virus matrix (M1) protein expression and virus replication by U6 promoter-driven and lentivirus-mediated delivery of siRNA. J. Gen. Virol. 2004, 85, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Coburn, G.A.; Cullen, B.R. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J. Virol. 2002, 76, 9225–9231. [Google Scholar] [CrossRef] [PubMed]
- Jacque, J.M.; Triques, K.; Stevenson, M. Modulation of HIV-1 replication by RNA interference. Nature 2002, 418, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cheng, T.; Wei, L.; Cai, Y.; Yeo, A.E.; Han, J.; Yuan, Y.A.; Zhang, J.; Xia, N. Efficient inhibition of HIV-1 replication by an artificial polycistronic miRNA construct. Virol. J. 2012, 9, e118. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Huang, W.; Wang, Y.; Guan, W.; Li, R.; Yang, Z.; Zhong, N. Gene silencing of beta-galactosamide alpha-2,6-sialyltransferase 1 inhibits human influenza virus infection of airway epithelial cells. BMC Microbiol. 2014, 14, e78. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Lee, S.K.; Dykxhoorn, D.M.; Novina, C.; Zhang, D.; Crawford, K.; Cerny, J.; Sharp, P.A.; Lieberman, J.; Manjunath, N.; et al. Sustained small interfering RNA-mediated human immunodeficiency virus type 1 inhibition in primary macrophages. J. Virol. 2003, 77, 7174–7181. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Geerts, D.; Jeeninga, R.E.; Berkhout, B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antiviral Res. 2011, 89, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, S.M.; Lo, C.Y.; Tumpey, T.M.; Epstein, S.L. Protection against lethal influenza virus challenge by RNA interference in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 8682–8686. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Hensley, L.E.; Kagan, E.; Yu, E.Z.; Geisbert, J.B.; Daddario-DiCaprio, K.; Fritz, E.A.; Jahrling, P.B.; McClintock, K.; Phelps, J.R.; et al. Postexposure protection of guinea pigs against a lethal Ebola virus challenge is conferred by RNA interference. J. Infect. Dis. 2006, 193, 1650–1657. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Lee, A.C.; Robbins, M.; Geisbert, J.B.; Honko, A.N.; Sood, V.; Johnson, J.C.; de Jong, S.; Tavakoli, I.; Judge, A.; et al. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: A proof-of-concept study. Lancet 2010, 375, 1896–1905. [Google Scholar] [CrossRef]
- DeVincenzo, J.; Lambkin-Williams, R.; Wilkinson, T.; Cehelsky, J.; Nochur, S.; Walsh, E.; Meyers, R.; Gollob, J.; Vaishnaw, A. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2010, 107, 8800–8805. [Google Scholar] [CrossRef] [PubMed]
- DeVincenzo, J.P.; Whitley, R.J.; Mackman, R.L.; Scaglioni-Weinlich, C.; Harrison, L.; Farrell, E.; McBride, S.; Lambkin-Williams, R.; Jordan, R.; Xin, Y.; et al. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N. Engl. J. Med. 2014, 371, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. siRNA for influenza therapy. Viruses 2010, 2, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Haasnoot, J.; Westerhout, E.M.; Berkhout, B. RNA interference against viruses: Strike and counterstrike. Nat. Biotechnol. 2007, 25, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, C.; Liang, W. Development of RNAi technology for targeted therapy—A track of siRNA based agents to RNAi therapeutics. J. Control. Release 2014, 193, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Lares, M.R.; Rossi, J.J.; Ouellet, D.L. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010, 28, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lee, S.K.; Shankar, P.; Manjunath, N. A single siRNA suppresses fatal encephalitis induced by two different flaviviruses. PLoS Med. 2006, 3, e96. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Antiretroviral drugs. Curr. Opin. Pharmacol. 2010, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Ko, T.M.; Ma, H.I.; Wu, H.L.; Xiao, X.; Li, J.; Chang, C.M.; Wu, P.Y.; Chen, C.H.; Han, J.M.; et al. Long-term inhibition of hepatitis B virus in transgenic mice by double-stranded adeno-associated virus 8-delivered short hairpin RNA. Gene Ther. 2007, 14, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jiang, P.; Li, Y.; Wang, X.; Huang, J.; Bai, J.; Cao, J.; Wu, B.; Chen, N.; Zeshan, B. Inhibition of porcine reproductive and respiratory syndrome virus replication by adenovirus-mediated RNA interference both in porcine alveolar macrophages and swine. Antiviral Res. 2009, 82, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Stone, J.K.; Andino, R. Poliovirus escape from RNA interference: Short interfering RNA-target recognition and implications for therapeutic approaches. J. Virol. 2005, 79, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xue, Y.; Wang, B.; Du, J.; Jin, Q. Broad-spectrum antiviral activity of RNA interference against four genotypes of Japanese encephalitis virus based on single microRNA polycistrons. PLoS ONE 2011, 6, e26304. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.A.; Richardson, C.D. Hepatitis C virus replicons escape RNA interference induced by a short interfering RNA directed against the NS5B coding region. J. Virol. 2005, 79, 7050–7058. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.; Lafforgue, G.; Morelli, M.J.; Gonzalez-Candelas, F.; Chua, N.H.; Daros, J.A.; Elena, S.F. Ultradeep sequencing analysis of population dynamics of virus escape mutants in RNAi-mediated resistant plants. Mol. Biol. Evol. 2012, 29, 3297–3307. [Google Scholar] [CrossRef] [PubMed]
- Holz, C.L.; Albina, E.; Minet, C.; Lancelot, R.; Kwiatek, O.; Libeau, G.; Servan de Almeida, R. RNA interference against animal viruses: How morbilliviruses generate extended diversity to escape small interfering RNA control. J. Virol. 2012, 86, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, H.; Kohara, M.; Kannagi, M.; Masuda, T. Effective suppression of human immunodeficiency virus type 1 through a combination of short- or long-hairpin RNAs targeting essential sequences for retroviral integration. J. Virol. 2006, 80, 7658–7666. [Google Scholar] [CrossRef] [PubMed]
- Sabariegos, R.; Gimenez-Barcons, M.; Tapia, N.; Clotet, B.; Martinez, M.A. Sequence homology required by human immunodeficiency virus type 1 to escape from short interfering RNAs. J. Virol. 2006, 80, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Gismondi, M.I.; Ortiz, X.P.; Curra, A.P.; Asurmendi, S.; Taboga, O. Artificial microRNAs as antiviral strategy to FMDV: Structural implications of target selection. J. Virol. Methods 2014, 199, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.S.; Pham, N.P.; Schaffer, D.V. HIV develops indirect cross-resistance to combinatorial RNAi targeting two distinct and spatially distant sites. Mol. Ther. 2012, 20, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; Das, A.T. HIV-1 escape from RNAi antivirals: Yet another houdini action? Mol. Ther. Nucleic Acids 2012, 1, e26. [Google Scholar] [CrossRef] [PubMed]
- Westerhout, E.M.; Ooms, M.; Vink, M.; Das, A.T.; Berkhout, B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005, 33, 796–804. [Google Scholar] [CrossRef] [PubMed]
- DeVincenzo, J.P. The promise, pitfalls and progress of RNA-interference-based antiviral therapy for respiratory viruses. Antivir. Ther. 2012, 17, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D. Current issues of RNAi therapeutics delivery and development. J. Control. Release 2014, 195, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Von Eije, K.J.; ter Brake, O.; Berkhout, B. Human immunodeficiency virus type 1 escape is restricted when conserved genome sequences are targeted by RNA interference. J. Virol. 2008, 82, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; von Eije, K.J.; Schopman, N.C.; Westerink, J.T.; ter Brake, O.; Haasnoot, J.; Berkhout, B. Combinatorial RNAi against HIV-1 using extended short hairpin RNAs. Mol. Ther. 2009, 17, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.S.; Freed, E.O. Novel approaches to inhibiting HIV-1 replication. Antiviral Res. 2010, 85, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.N.; Shah, P.S.; Burnett, J.C.; Schaffer, D.V. HIV evades RNA interference directed at TAR by an indirect compensatory mechanism. Cell Host Microbe 2008, 4, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Boutimah, F.; Eekels, J.J.; Liu, Y.P.; Berkhout, B. Antiviral strategies combining antiretroviral drugs with RNAi-mediated attack on HIV-1 and cellular co-factors. Antiviral Res. 2013, 98, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Tiemann, K.; Rossi, J.J. RNAi-based therapeutics-current status, challenges and prospects. EMBO Mol. Med. 2009, 1, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Uprichard, S.L. The therapeutic potential of RNA interference. FEBS Lett. 2005, 579, 5996–6007. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Presloid, J.B.; Novella, I.S. RNA Viruses and RNAi: Quasispecies Implications for Viral Escape. Viruses 2015, 7, 3226-3240. https://doi.org/10.3390/v7062768
Presloid JB, Novella IS. RNA Viruses and RNAi: Quasispecies Implications for Viral Escape. Viruses. 2015; 7(6):3226-3240. https://doi.org/10.3390/v7062768
Chicago/Turabian StylePresloid, John B., and Isabel S. Novella. 2015. "RNA Viruses and RNAi: Quasispecies Implications for Viral Escape" Viruses 7, no. 6: 3226-3240. https://doi.org/10.3390/v7062768
APA StylePresloid, J. B., & Novella, I. S. (2015). RNA Viruses and RNAi: Quasispecies Implications for Viral Escape. Viruses, 7(6), 3226-3240. https://doi.org/10.3390/v7062768