
Modulation of DNA Damage and Repair Pathways by Human Tumour Viruses

Abstract
:1. Introduction
2. Human Tumour Viruses
3. The DNA Damage Response (DDR)
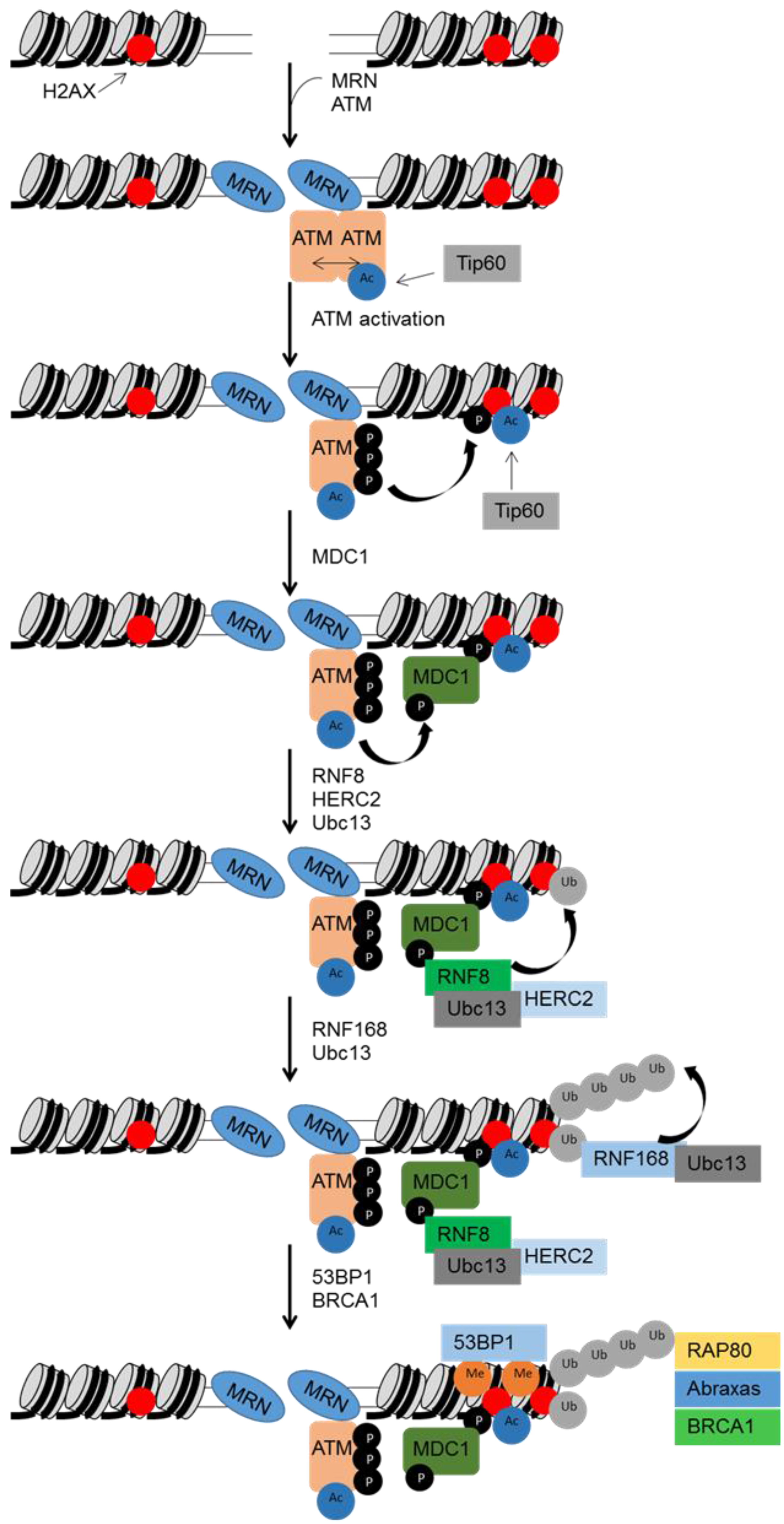
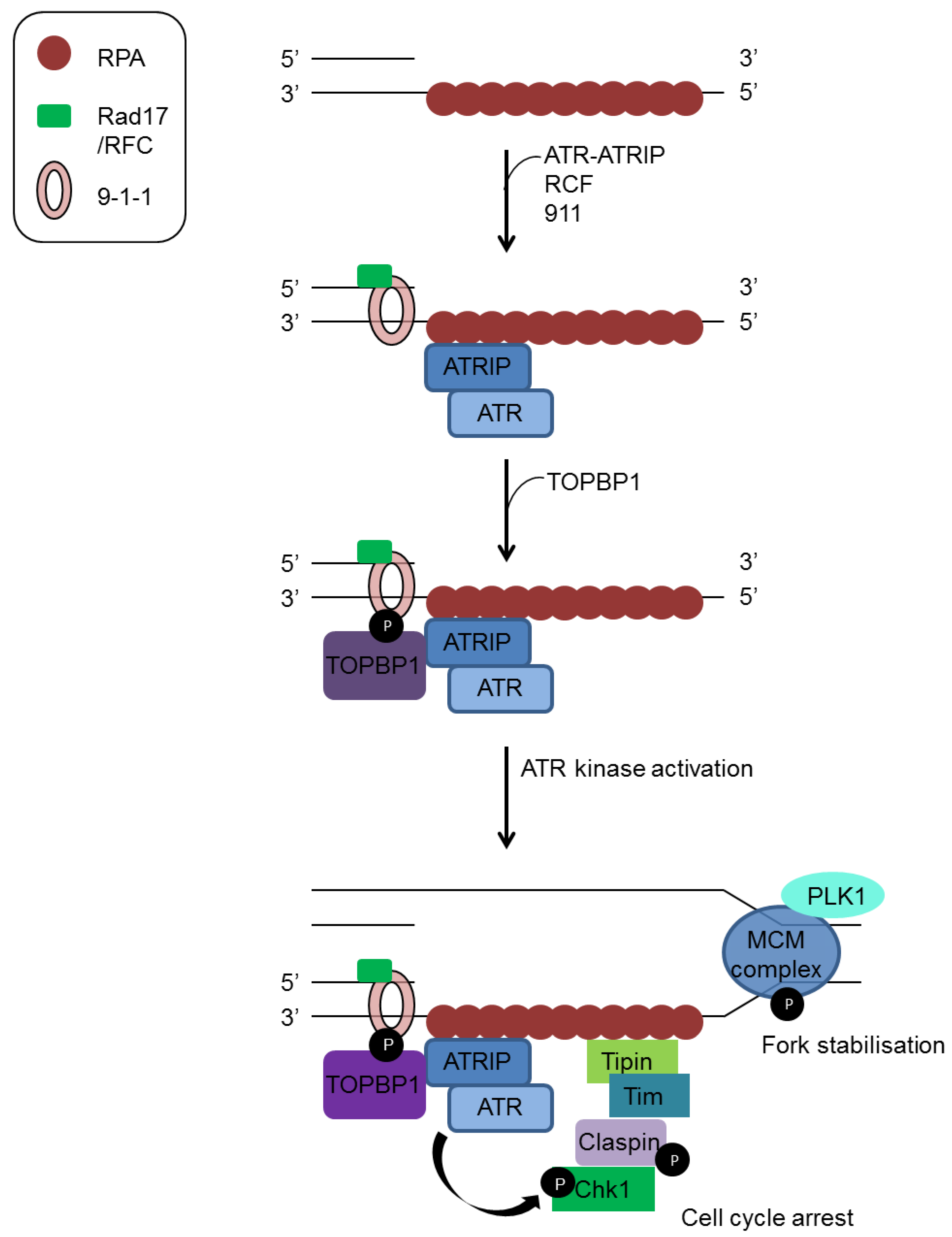
3.1. Sensors and Transducers
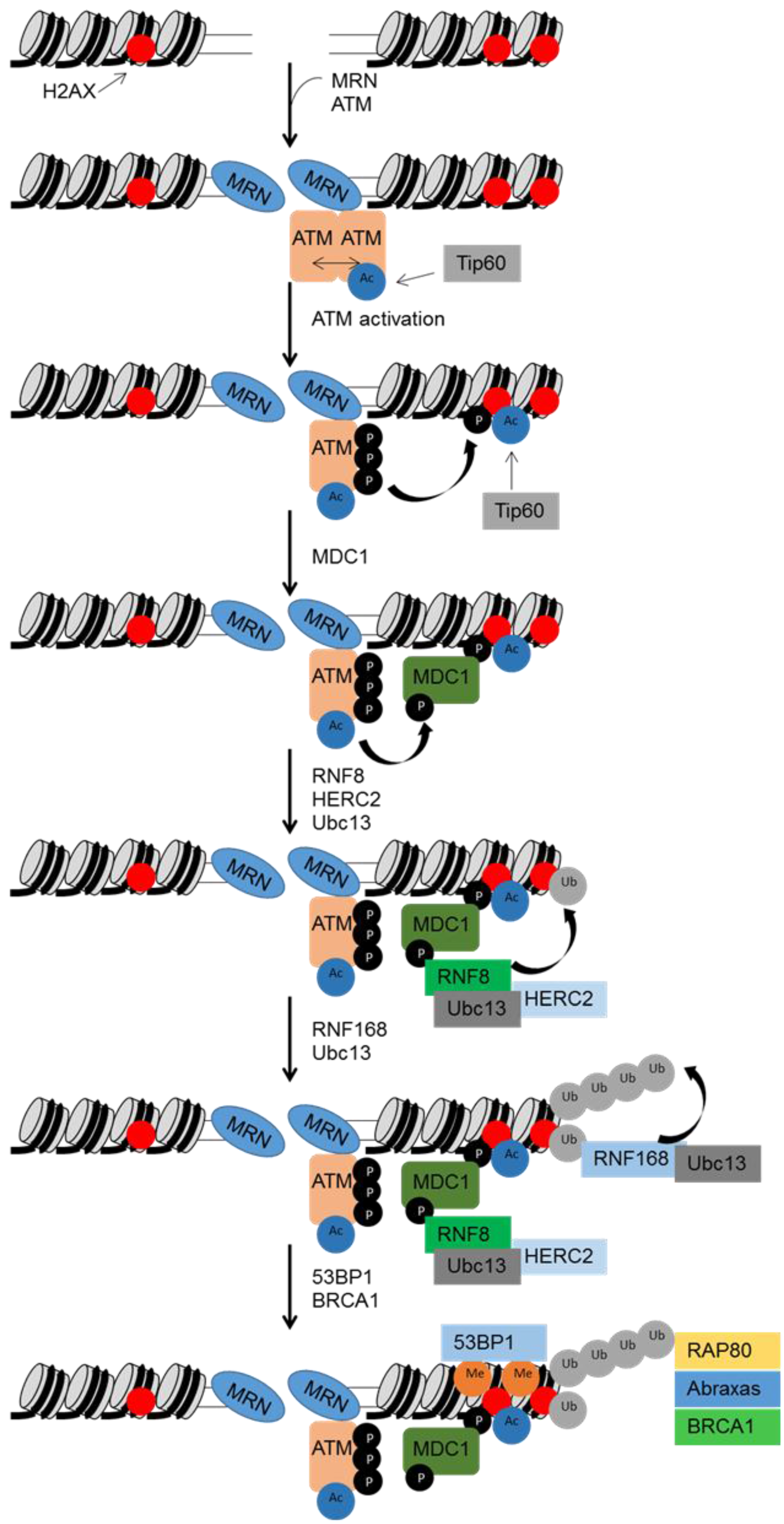
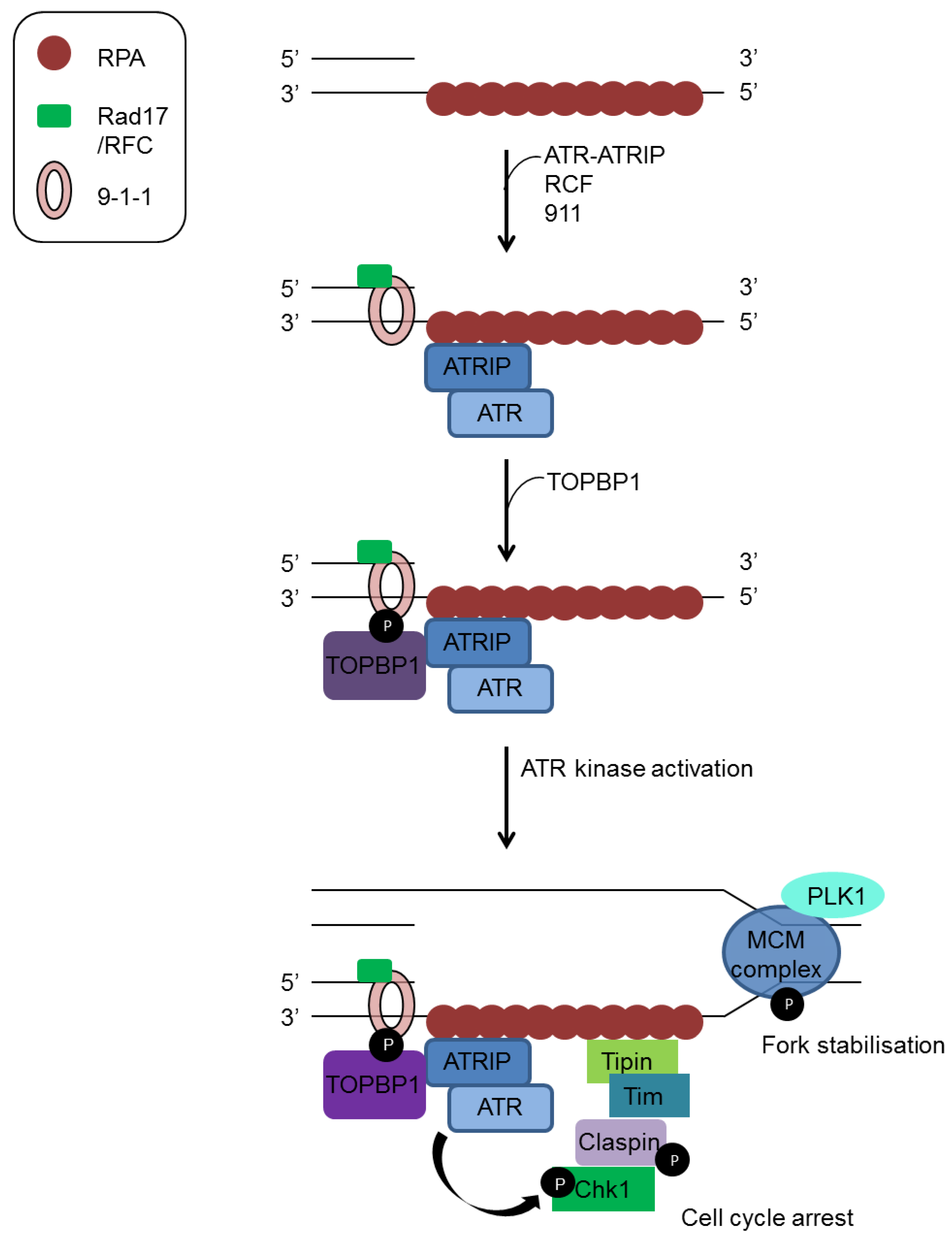
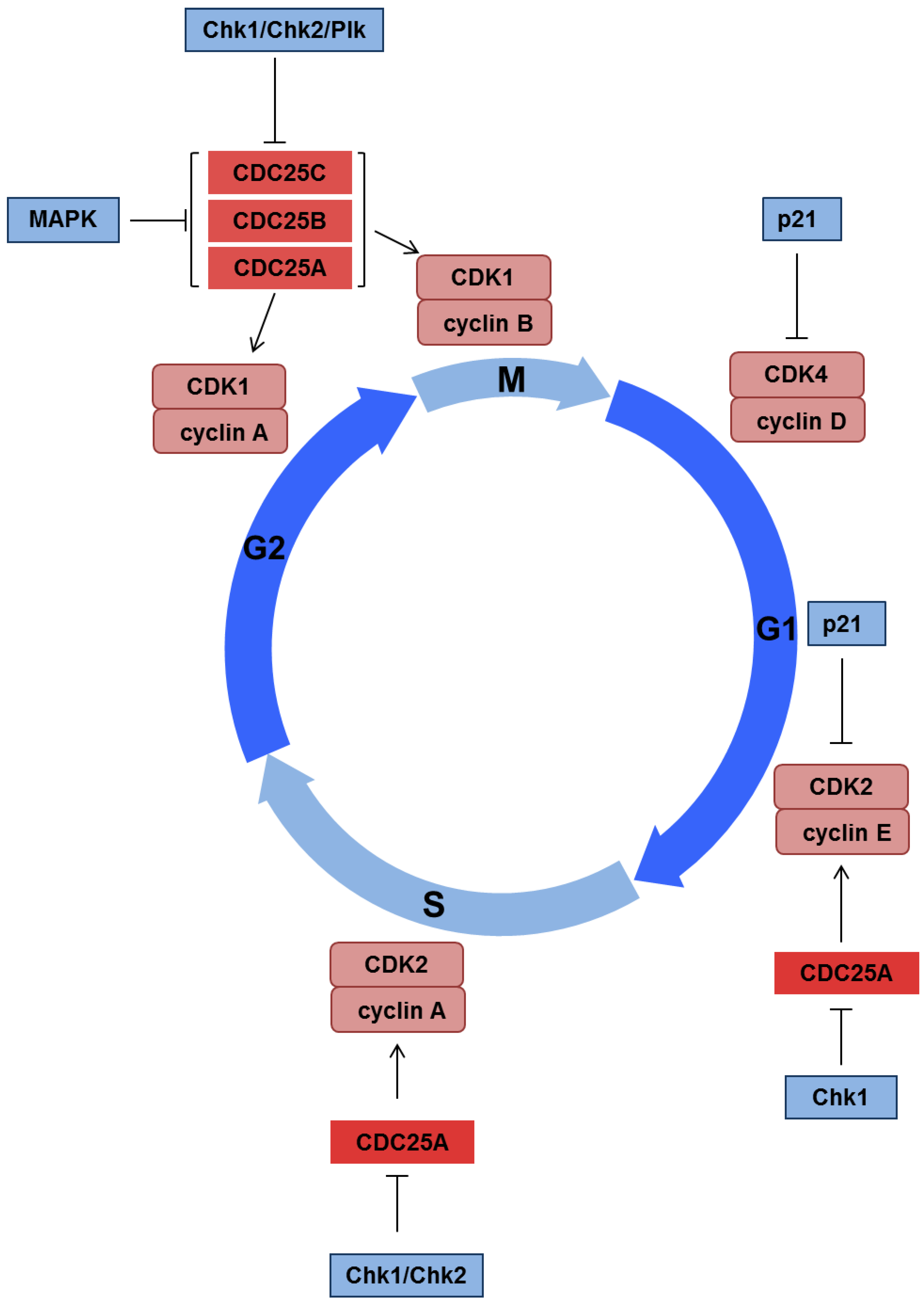
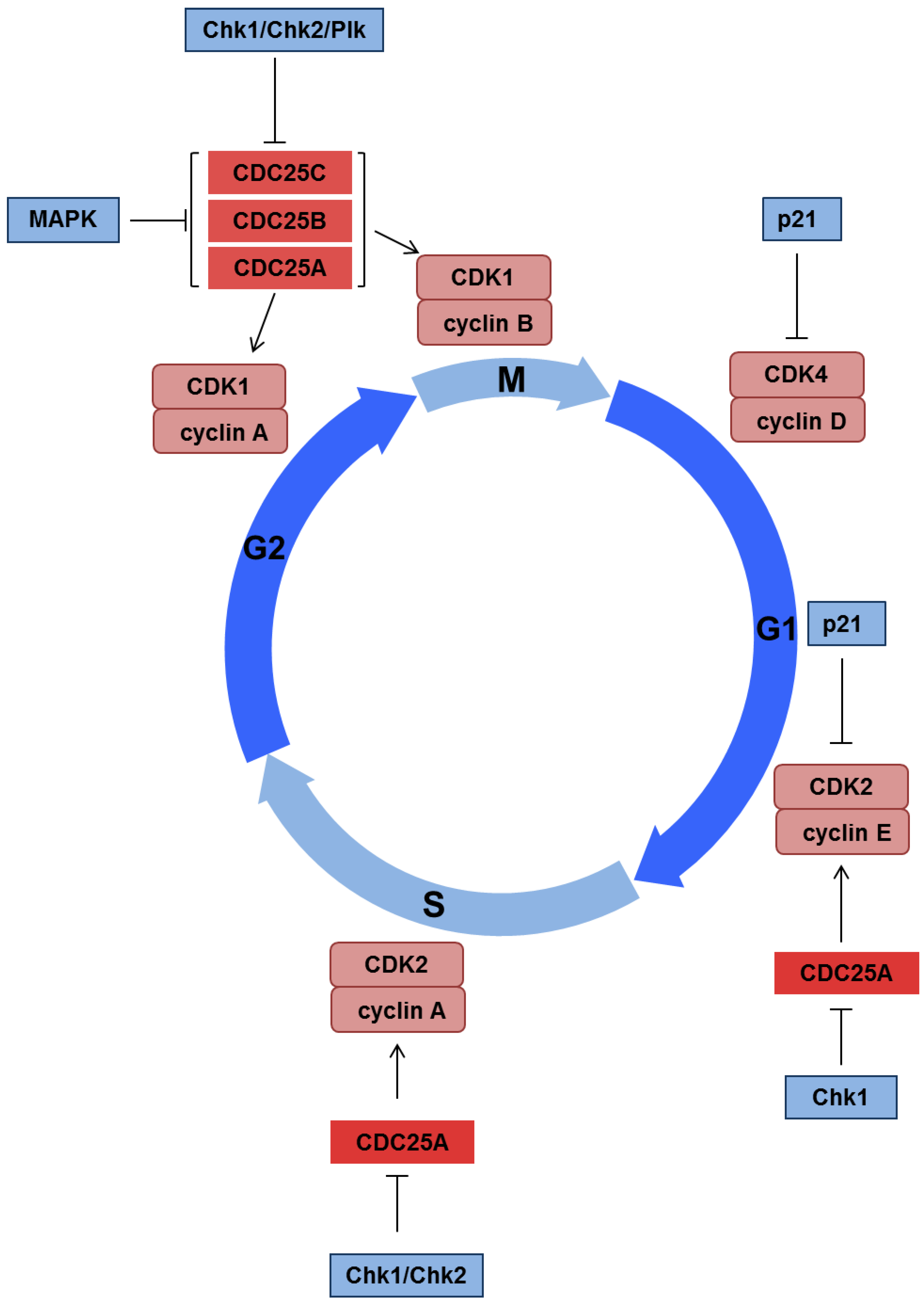
3.2. Activation of Cell Cycle Checkpoints
3.3. Apoptosis and Senescence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Genome | Viral Oncoproteins * | Associated Cancer |
|---|---|---|---|
| Human papilloma virus (HPV) | dsDNA | E6 and E7 | Cervical cancer, penile cancer, anogenital carcinoma, head and neck cancer |
| Merkel cell polyoma virus (MCPyV) | dsDNA | Large T antigen | Merkel cell carcinoma (MCC) |
| Human T cell leukaemia virus-1 (HTLV-1) | ssRNA | Tax | Adult T cell leukaemia (ATL) |
| Epstein Barr virus (EBV) | dsDNA | EBNA2, EBNA3C, LMP-1, LMP-2 | Nasopharyngeal carcinoma (NPC), Burkitt’s lymphoma, Hodgkin’s lymphoma, post-transplant lymphoproliferative disease (PTLD), T cell lymphoma, gastric cancer |
| Kaposi’s sarcoma-associated herpesvirus (KSHV) | dsDNA | LANA, v-cyclin, vGPCR, vIL-6, vBcl-2, vFLIP, Kaposin B | Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), multicentric Castleman’s disease (MCD) |
| Hepatitis B virus (HBV) | ssDNA + ssRNA | HBx | Hepatocellular carcinoma (HCC) |
| Hepatitis C virus (HCV) | ssRNA | Core, NS3, NS5A | Hepatocellular carcinoma (HCC), B-cell lymphoma |
4. DNA Damage Repair Pathways
| Repair Pathway | DDR Target Protein | Virus/Viral Protein | REFERENCE |
|---|---|---|---|
| Direct repair | MGMT | HPV E6 | [50] |
| Base excision repair (BER) | XRCC1 Β polymerase | HPV E6 HTLV-1 Tax | [51] [52] |
| Nucleotide excision repair (GG-NER) | XPC PCNA DDB1 TFIIH subunits (XPB, XPD) | MCPy LT HTLV-1 Tax EBV BPLF1 HBx HBx | [53] [54] [55] [56,57] [58,59] |
| Mismatch repair (MMR) | MSH2, MSH6 MSH2, MSH6, MLH1, hPSM2 | KSHV VRCs HTLV-1 Tax EBV VRCs | [60] [61] [62] |
| Single strand break repair | PARP-1 XRCC1 | KSHV VRCs HPV E6 | [60] [51] |
| Homologous recombination (HR) | Rad51 BRCA1 Rad52 | HPV16 E7 EBV VRCs HPV31 VRCs HPV31 VRCs HTLV-1 VRCs EBV VRCs | [63] [64] [65] [65] [66] [64] |
| Non-homologous end joining (NHEJ) | DNA-PKcs Ku70/Ku80 | KSHV VRCs HTLV-1 VRCs KSHVorf59 KSHV VRCs | [60] [66] [67] [60] |
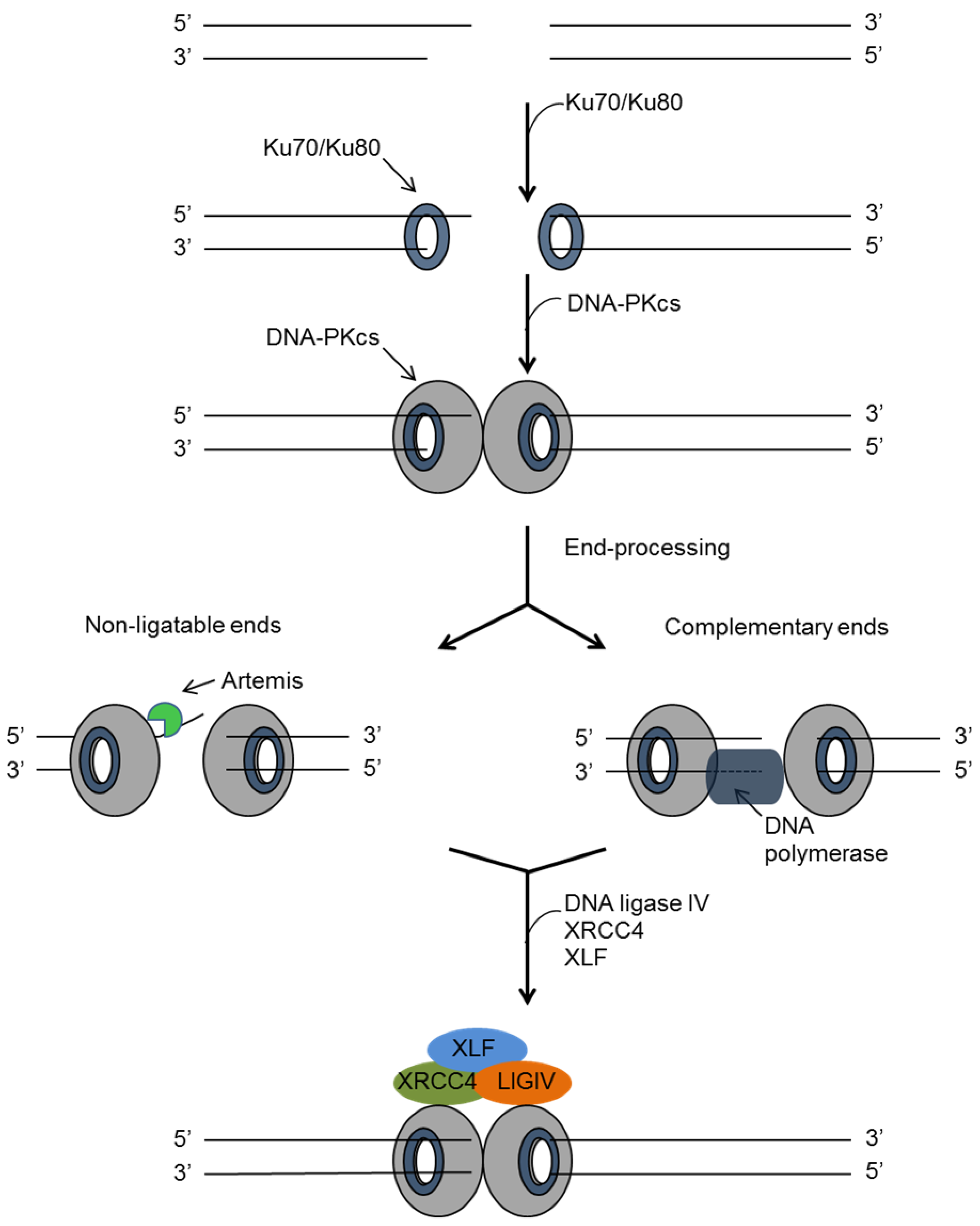
4.1. Non-Homologues End Joining (NHEJ)
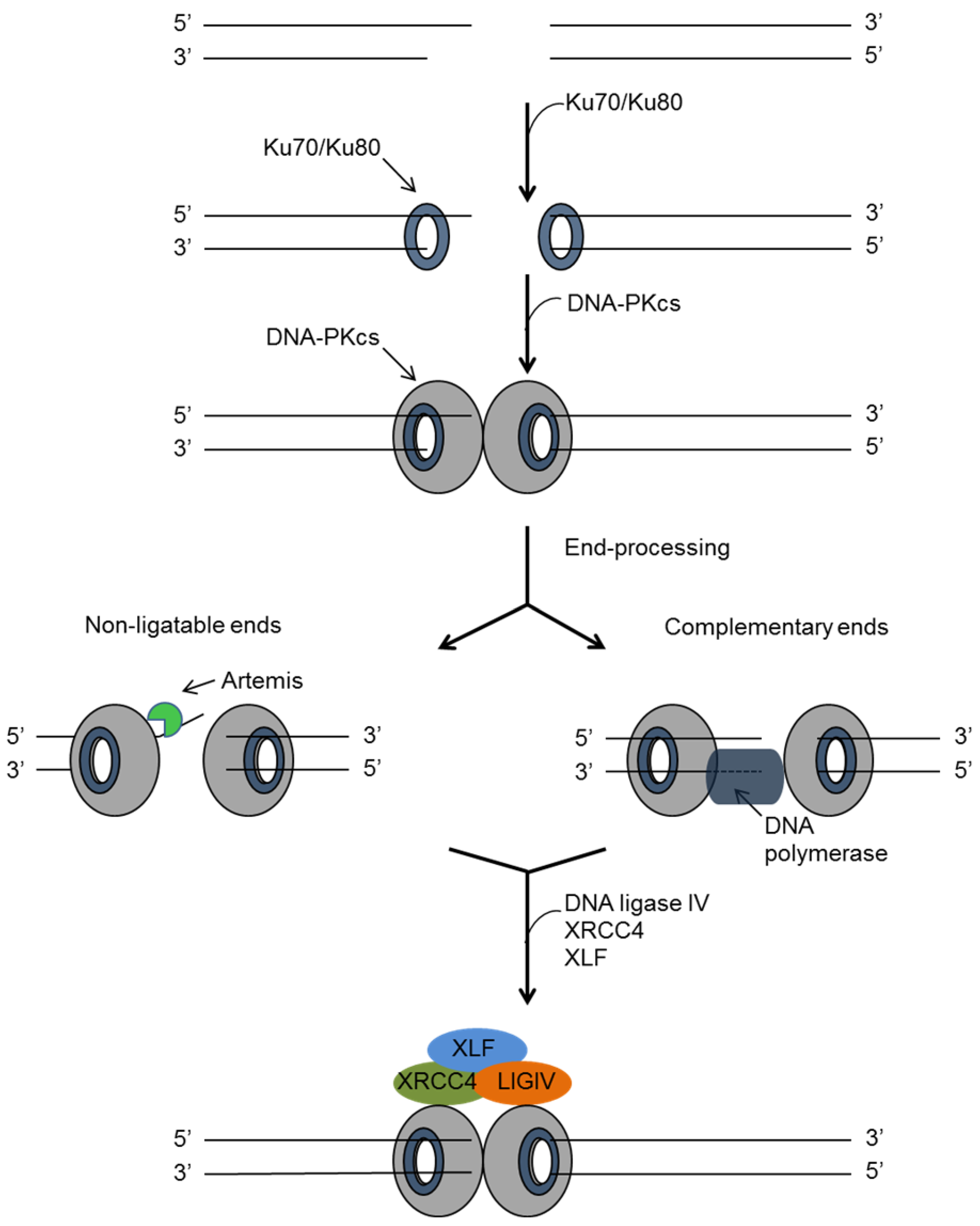
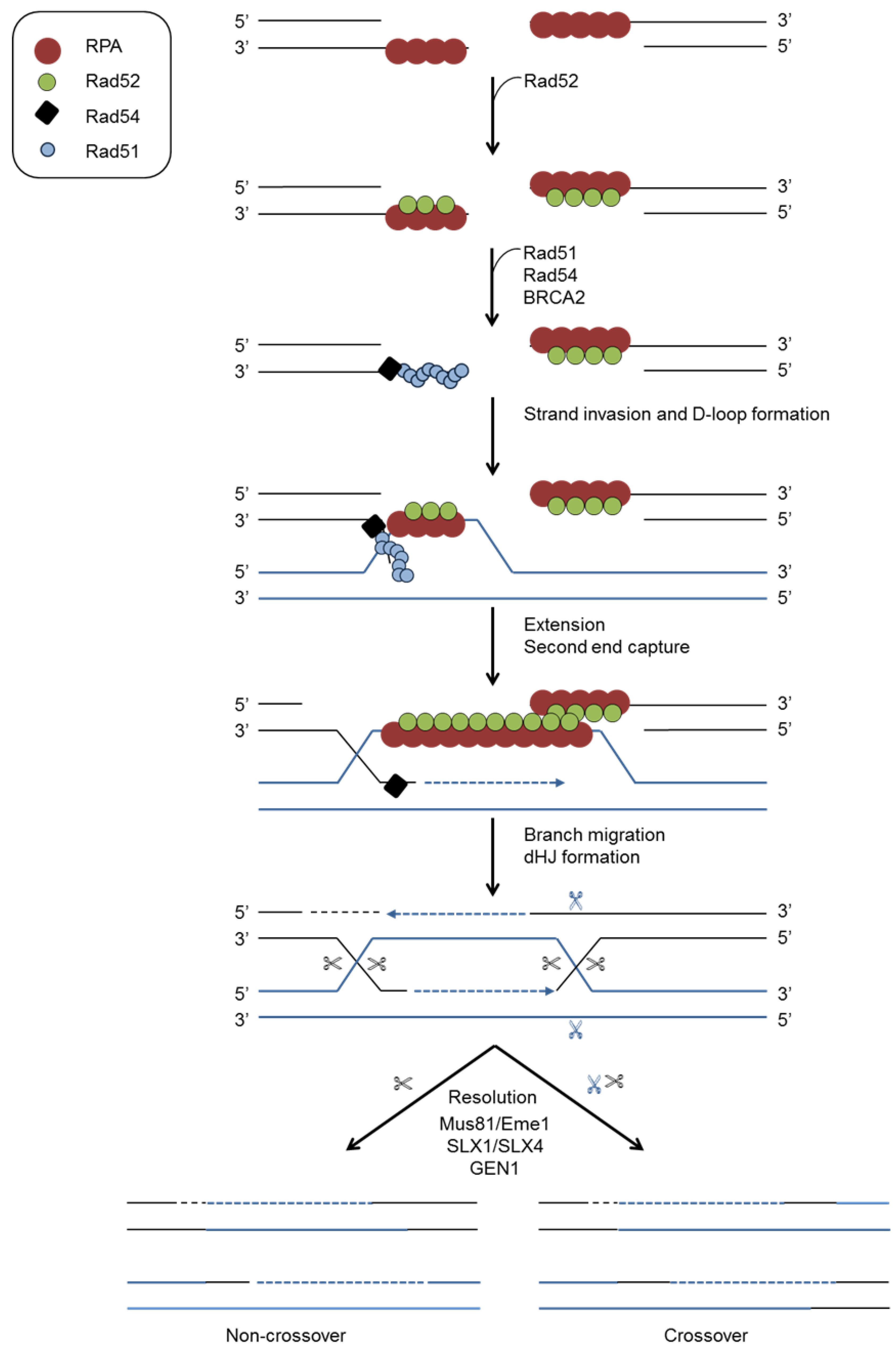
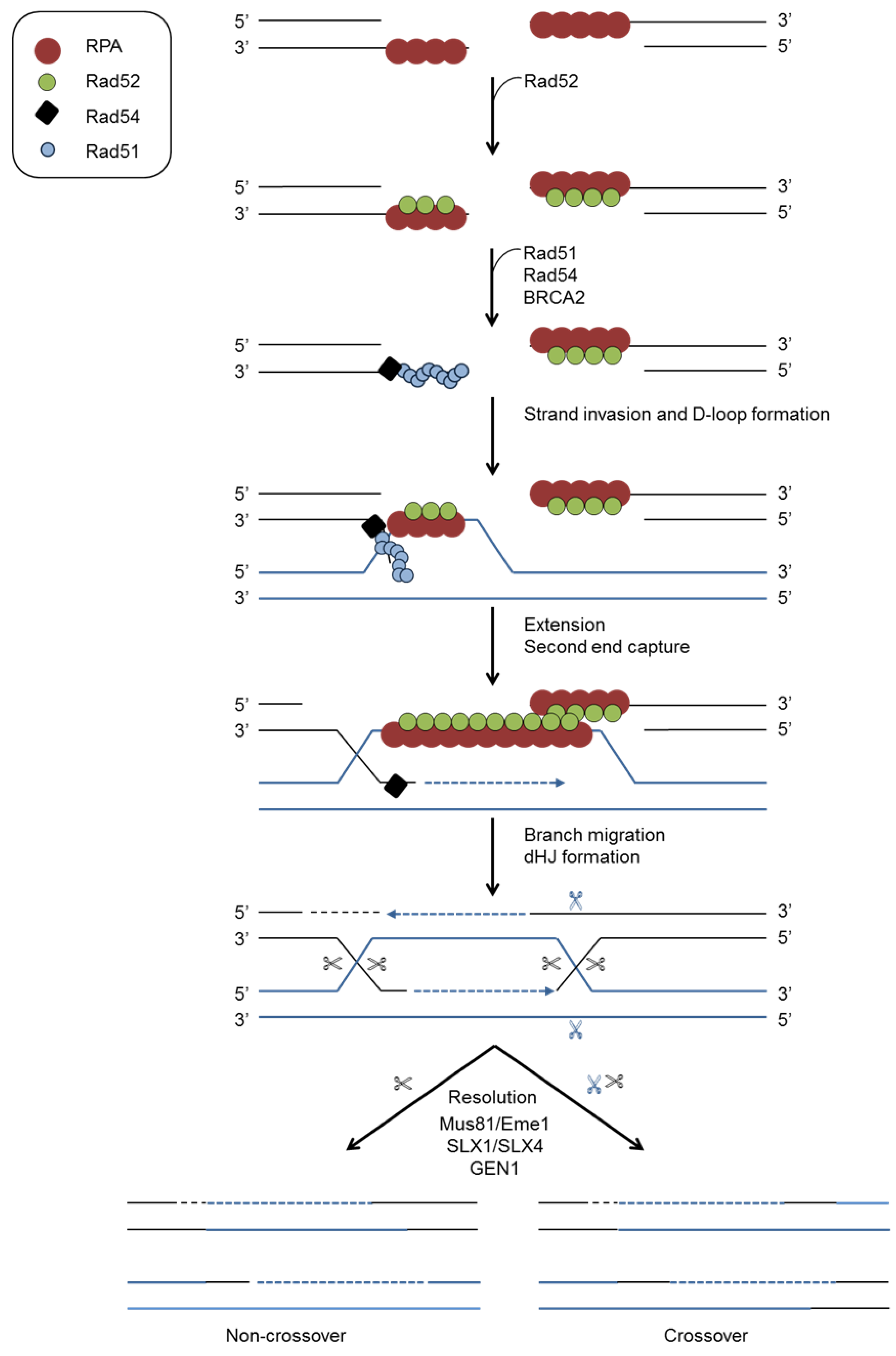
4.2. Homologous Recombination (HR)
4.3. Single Strand Break Repair
4.4. Base Excision Repair (BER)
4.5. Nucleotide Excision Repair (NER)
4.6. Mismatch Repair (MMR)
4.7. Fanconi Anaemia (FA) Pathway
4.8. DNA Repair Pathways and the Cell Cycle
5. Human Papillomaviruses (HPV)
5.1. Activation of the DDR by HPV
5.2. Involvement of DDR Factors in HPV Replication
5.3. Deregulation of DDR Signalling by E6 and E7
5.4. Summary
6. Merkel Cell Polyomavirus (MCPyV)
6.1. Activation of the DDR by MCPyV
6.2. Involvement of DDR Factors in MCPyV Replication
6.3. Interference with DNA Repair by LT
6.4. Summary
7. Epstein-Barr Virus (EBV)
7.1. Genetic Instability Induced by EBV Proteins
7.2. Involvement of DDR Factors in EBV Replication
7.3. Dysregulation of Cell Cycle Checkpoints during EBV Infection
7.4. Summary
8. Kaposi’s Sarcoma Related Virus (KSHV)
8.1. DDR Activation during Latent KSHV Infection
8.2. Involvement of DDR Factors in KSHV Replication
8.3. DDR Activation during Lytic Replication of KSHV
8.4. Summary
9. Human T-cell Leukemia Virus Type 1 (HTLV-1)
9.1. Genetic Instability in Tax-Expressing Cells
9.2. Interference with DNA Repair by HTLV-1 Proteins
9.3. Interaction between Tax and Checkpoint Kinases
9.4. Summary
10. Hepatitis B virus (HBV)
10.1. Introduction of Oxidative DNA Damage by HBV
10.2. Interference with DNA Repair by HBV
10.3. Activation of the ATR Pathway by HBx
10.4. Summary
11. Hepatitis C virus (HCV)
11.1. Introduction of Oxidative DNA Damage by HCV
11.2. Interference with DNA Repair by HCV
11.3. Interaction between HCV Proteins and the ATM Pathway
11.4. Summary
12. Conclusions
Acknowledgments
Conflicts of Interest
References
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef] [PubMed]
- Meerbrey, K.L.; Hu, G.; Kessler, J.D.; Roarty, K.; Li, M.Z.; Fang, J.E.; Herschkowitz, J.I.; Burrows, A.E.; Ciccia, A.; Sun, T.; et al. The pinducer lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 3665–3670. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Chouteau, P.; Lerat, H. 'Liver let die': Oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 2014, 95, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Marriott, S.J.; Semmes, O.J. Impact of HTLV-I tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 2005, 24, 5986–5995. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.W.; Levan, A.; Aula, P.; Norrby, E. Chromosome damage associated with the measles virus in vitro. Hereditas 1965, 54, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, E.A.; Spector, D.H. Viral induction of site-specific chromosome damage. Rev. Med. Virol. 2003, 13, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-pk phosphorylation of rpa32 ser4/ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Changing the ubiquitin landscape during viral manipulation of the DNA damage response. FEBS Lett. 2011, 585, 2897–2906. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Weitzman, M.D. Chromatin at the intersection of viral infection and DNA damage. Biochim. Biophys. Acta 2010, 1799, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bekker-Jensen, S.; Mailand, N.; Lukas, C.; Bartek, J.; Lukas, J. Claspin operates downstream of topbp1 to direct atr signaling towards chk1 activation. Mol. Cell. Biol. 2006, 26, 6056–6064. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.J.; Stewart, G.S. A single strand that links multiple neuropathologies in human disease. Brain 2013, 136, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Lobrich, M. DNA double-strand breaks: Their cellular and clinical impact? Oncogene 2007, 26, 7717–7719. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-pk: A dynamic enzyme in a versatile dsb repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. Atr: An essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of atm, atr and DNA-pkcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Petrini, J.H.; Stracker, T.H. The cellular response to DNA double-strand breaks: Defining the sensors and mediators. Trends Cell Biol. 2003, 13, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Lukas, C.; Kitagawa, R.; Melander, F.; Kastan, M.B.; Bartek, J.; Lukas, J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006, 173, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates atm through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; FeRNAndes, N.; Price, B.D. A role for the tip60 histone acetyltransferase in the acetylation and activation of atm. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. Mdc1 maintains genomic stability by participating in the amplification of atm-dependent DNA damage signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The riddle syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Rendtlew Danielsen, J.; Fugger, K.; Gromova, I.; Nerstedt, A.; Lukas, C.; Bartek, J.; Lukas, J.; Mailand, N. Herc2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 2010, 12, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Ellison, V.; Stillman, B. Biochemical characterization of DNA damage checkpoint complexes: Clamp loader and clamp complexes with specificity for 5' recessed DNA. PLoS Biol. 2003, 1, e33. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Kim, S.M.; Dunphy, W.G. Claspin and the activated form of atr-atrip collaborate in the activation of chk1. J. Biol. Chem. 2004, 279, 49599–49608. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Akan, Z.; Yilmaz, S.; Grillo, M.; Smith-Roe, S.L.; Kang, T.H.; Cordeiro-Stone, M.; Kaufmann, W.K.; Abraham, R.T.; Sancar, A.; et al. Tipin-replication protein a interaction mediates chk1 phosphorylation by atr in response to genotoxic stress. J. Biol. Chem. 2010, 285, 16562–16571. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Cortez, D. Atr signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Lukas, C.; Bartek, J. Mammalian cell cycle checkpoints: Signalling pathways and their organization in space and time. DNA Repair 2004, 3, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A. How the rb tumor suppressor structure and function was revealed by the study of adenovirus and sv40. Virology 2009, 384, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. Atm- and cell cycle-dependent regulation of atr in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Gonzalez, A.M.; Contreras-Paredes, A.; Manzo-Merino, J.; Lizano, M. The modulation of apoptosis by oncogenic viruses. Virol. J. 2013, 10, e182. [Google Scholar] [CrossRef]
- Cheng, E.H.; Nicholas, J.; Bellows, D.S.; Hayward, G.S.; Guo, H.G.; Reitz, M.S.; Hardwick, J.M. A bcl-2 homolog encoded by kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with bax or bak. Proc. Natl. Acad. Sci. USA 1997, 94, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Rowe, M.; Gregory, C.; Croom-Carter, D.; Wang, F.; Longnecker, R.; Kieff, E.; Rickinson, A. Induction of bcl-2 expression by epstein-barr virus latent membrane protein 1 protects infected b cells from programmed cell death. Cell 1991, 65, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Gross-Mesilaty, S.; Reinstein, E.; Bercovich, B.; Tobias, K.E.; Schwartz, A.L.; Kahana, C.; Ciechanover, A. Basal and human papillomavirus e6 oncoprotein-induced degradation of myc proteins by the ubiquitin pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 8058–8063. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M. Warts, cancer and ubiquitylation: Lessons from the papillomaviruses. Trans. Am. Clin. Climatol. Assoc. 2006, 117, 113–126, discussion 126–117. [Google Scholar] [PubMed]
- Krueger, A.; Fas, S.C.; Giaisi, M.; Bleumink, M.; Merling, A.; Stumpf, C.; Baumann, S.; Holtkotte, D.; Bosch, V.; Krammer, P.H.; et al. HTLV-1 tax protects against cd95-mediated apoptosis by induction of the cellular flice-inhibitory protein (c-flip). Blood 2006, 107, 3933–3939. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Tumor viruses and replicative immortality—avoiding the telomere hurdle. Semin. Cancer Biol. 2014, 26, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, J.; Zheng, B.; Fang, X.; Angstrom, T.; Liu, C.; Li, X.; Erlandsson, F.; Bjorkholm, M.; Nordenskjord, M.; et al. Telomere attrition predominantly occurs in precursor lesions during in vivo carcinogenic process of the uterine cervix. Oncogene 2004, 23, 7441–7447. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Horikawa, I.; Nishimoto, A.; Ohmura, H.; Ito, H.; Hirohashi, S.; Shay, J.W.; Oshimura, M. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet. Cytogenet. 1997, 93, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase activation by human papillomavirus type 16 e6 protein: Induction of human telomerase reverse transcriptase expression through myc and gc-rich sp1 binding sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, N.; Zhang, H.; You, J.; Wang, H.; Ye, L. Effects of Hepatitis B virus x protein on human telomerase reverse transcriptase expression and activity in hepatoma cells. J. Lab. Clin. Med. 2005, 145, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Bais, C.; van Geelen, A.; Eroles, P.; Mutlu, A.; Chiozzini, C.; Dias, S.; Silverstein, R.L.; Rafii, S.; Mesri, E.A. Kaposi's sarcoma associated herpesvirus g protein-coupled receptor immortalizes human endothelial cells by activation of the vegf receptor-2/ kdr. Cancer Cell 2003, 3, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Kumakura, S.; Uchida, M.; Barrett, J.C.; Tsutsui, T. Immortalization of normal human embryonic fibroblasts by introduction of either the human papillomavirus type 16 e6 or e7 gene alone. Int. J. Cancer 2003, 106, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Srivenugopal, K.S.; Ali-Osman, F. The DNA repair protein, o(6)-methylguanine-DNA methyltransferase is a proteolytic target for the e6 human papillomavirus oncoprotein. Oncogene 2002, 21, 5940–5945. [Google Scholar] [CrossRef] [PubMed]
- Iftner, T.; Elbel, M.; Schopp, B.; Hiller, T.; Loizou, J.I.; Caldecott, K.W.; Stubenrauch, F. Interference of papillomavirus e6 protein with single-strand break repair by interaction with xrcc1. EMBO J. 2002, 21, 4741–4748. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K.T.; Widen, S.G.; Semmes, O.J.t.; Wilson, S.H. HTLV-I trans-activator protein, tax, is a trans-repressor of the human beta-polymerase gene. Science 1990, 247, 1082–1084. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, S.K.; Ona-Vu, K.; Sullivan, E.M.; Dong, T.K.; Hsu, S.W.; Oh, D.H. Defective DNA repair and cell cycle arrest in cells expressing merkel cell polyomavirus T antigen. Int. J. Cancer 2012, 131, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.Y.; Marriott, S.J. Disruption of nucleotide excision repair by the human T-cell leukemia virus type 1 tax protein. J. Virol. 1999, 73, 4299–4304. [Google Scholar] [PubMed]
- Whitehurst, C.B.; Vaziri, C.; Shackelford, J.; Pagano, J.S. Epstein-barr virus bplf1 deubiquitinates pcna and attenuates polymerase eta recruitment to DNA damage sites. J. Virol. 2012, 86, 8097–8106. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Elledge, S.J.; Butel, J.S. Hepatitis B virus x protein interacts with a probable cellular DNA repair protein. J. Virol. 1995, 69, 1107–1114. [Google Scholar] [PubMed]
- Sitterlin, D.; Lee, T.H.; Prigent, S.; Tiollais, P.; Butel, J.S.; Transy, C. Interaction of the UV-damaged DNA-binding protein with Hepatitis B virus x protein is conserved among mammalian hepadnaviruses and restricted to transactivation-proficient x-insertion mutants. J. Virol. 1997, 71, 6194–6199. [Google Scholar] [PubMed]
- Qadri, I.; Conaway, J.W.; Conaway, R.C.; Schaack, J.; Siddiqui, A. Hepatitis B virus transactivator protein, hbx, associates with the components of tfiih and stimulates the DNA helicase activity of tfiih. Proc. Natl. Acad. Sci. USA 1996, 93, 10578–10583. [Google Scholar] [CrossRef] [PubMed]
- Qadri, I.; Fatima, K.; Abde, L.H.H. Hepatitis B virus x protein impedes the DNA repair via its association with transcription factor, tfiih. BMC Microbiol. 2011, 11, e48. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Tang, Q.; Maul, G.G.; Yuan, Y. Kaposi's sarcoma-associated herpesvirus ori-lyt-dependent DNA replication: Involvement of host cellular factors. J. Virol. 2008, 82, 2867–2882. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Tsukada, J.; Kominato, Y.; Tanaka, Y. Reduced expression of human mismatch repair genes in adult T-cell leukemia. Am. J. Hematol. 2005, 78, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Kudoh, A.; Sugaya, Y.; Iwahori, S.; Shirata, N.; Isomura, H.; Tsurumi, T. Postreplicative mismatch repair factors are recruited to epstein-barr virus replication compartments. J. Biol. Chem. 2006, 281, 11422–11430. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Nickel, K.P.; Torres, A.D.; Lee, D.; Lambert, P.F.; Kimple, R.J. Human papillomavirus type 16 e7 oncoprotein causes a delay in repair of DNA damage. Radiother Oncol. 2014, 113, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, A.; Iwahori, S.; Sato, Y.; Nakayama, S.; Isomura, H.; Murata, T.; Tsurumi, T. Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in epstein-barr virus replication compartments. J. Virol. 2009, 83, 6641–6651. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Belgnaoui, S.M.; Fryrear, K.A.; Nyalwidhe, J.O.; Guo, X.; Semmes, O.J. The viral oncoprotein tax sequesters DNA damage response factors by tethering mdc1 to chromatin. J. Biol. Chem. 2010, 285, 32897–32905. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, J.; Liao, Q.; Wu, Y.; Peng, C.; Chen, X. Lytic infection of kaposi's sarcoma-associated herpesvirus induces DNA double-strand breaks and impairs non-homologous end joining. J. Gen. Virol. 2013, 94, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Jackson, S.P. The DNA-dependent protein kinase: Requirement for DNA ends and association with ku antigen. Cell 1993, 72, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Nick McElhinny, S.A.; Snowden, C.M.; McCarville, J.; Ramsden, D.A. Ku recruits the xrcc4-ligase iv complex to DNA ends. Mol. Cell. Biol. 2000, 20, 2996–3003. [Google Scholar] [CrossRef] [PubMed]
- Hartlerode, A.J.; Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009, 423, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Grawunder, U.; Wilm, M.; Wu, X.; Kulesza, P.; Wilson, T.E.; Mann, M.; Lieber, M.R. Activity of DNA ligase iv stimulated by complex formation with xrcc4 protein in mammalian cells. Nature 1997, 388, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. Xlf interacts with the xrcc4-DNA ligase iv complex to promote DNA nonhomologous end-joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Sica, R.A.; Kowalczykowski, S.C. Rad52 promotes second-end DNA capture in double-stranded break repair to form complement-stabilized joint molecules. Proc. Natl. Acad. Sci. USA 2009, 106, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. Blm-dna2-rpa-mrn and exo1-blm-rpa-mrn constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, M.J.; West, S.C. DNA repair synthesis facilitates rad52-mediated second-end capture during dsb repair. Mol. Cell 2008, 29, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Kowalczykowski, S.C. Second-end DNA capture in double-strand break repair: How to catch a DNA by its tail. Cell Cycle 2009, 8, 1816–1817. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8. [Google Scholar] [CrossRef] [PubMed]
- D'Amours, D.; Desnoyers, S.; D'Silva, I.; Poirier, G.G. Poly(adp-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [PubMed]
- Caldecott, K.W.; Aoufouchi, S.; Johnson, P.; Shall, S. Xrcc1 polypeptide interacts with DNA polymerase beta and possibly poly(adp-ribose) polymerase, and DNA ligase iii is a novel molecular 'nick-sensor' in vitro. Nucl. Acids Res. 1996, 24, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. Xrcc1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair 2007, 6, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B. Repairing DNA-methylation damage. Nat Rev Mol. Cell. Biol. 2004, 5, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B.; Bates, P.A.; Paik, J.; Jacobs, S.C.; Lindahl, T. Repair of alkylated DNA: Recent advances. DNA Repair 2007, 6, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Dianov, G.L. Co-ordination of base excision repair and genome stability. DNA Repair 2013, 12, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Yang, C.; Sengupta, S.; Mitra, S.; Hegde, M.L. New paradigms in the repair of oxidative damage in human genome: Mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins. Cell. Mol. Life Sci. 2015, 72, 1679–1698. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, S.; Coin, F. Orchestral maneuvers at the damaged sites in nucleotide excision repair. Cell. Mol. Life Sci. 2015, 72, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, W.; Fousteri, M. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- de Laat, W.L.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell. Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. New insights and challenges in mismatch repair: Getting over the chromatin hurdle. DNA Repair 2014, 19, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat Rev Mol. Cell. Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Genschel, J.; Modrich, P. Mechanism of 5'-directed excision in human mismatch repair. Mol. Cell 2003, 12, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.S.; Kunkel, T.A. Ribonucleotides in DNA: Origins, repair and consequences. DNA Repair 2014, 19, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Deans, A.J. The fanconi anemia DNA repair pathway: Structural and functional insights into a complex disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D'Andrea, A.D. Regulation of DNA cross-link repair by the fanconi anemia/brca pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell. Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Germani, A.; Prabel, A.; Mourah, S.; Podgorniak, M.P.; Di Carlo, A.; Ehrlich, R.; Gisselbrecht, S.; Varin-Blank, N.; Calvo, F.; Bruzzoni-Giovanelli, H. Siah-1 interacts with CtIP and promotes its degradation by the proteasome pathway. Oncogene 2003, 22, 8845–8851. [Google Scholar] [CrossRef] [PubMed]
- Schroering, A.G.; Edelbrock, M.A.; Richards, T.J.; Williams, K.J. The cell cycle and DNA mismatch repair. Exp. Cell Res. 2007, 313, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, M.A. Base excision repair of ionizing radiation-induced DNA damage in g1 and g2 cell cycle phases. Cancer Cell Int. 2007, 7, e15. [Google Scholar] [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Clifford, G.M.; Rana, R.K.; Franceschi, S.; Smith, J.S.; Gough, G.; Pimenta, J.M. Human papillomavirus genotype distribution in low-grade cervical lesions: Comparison by geographic region and with cervical cancer. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.; Herrington, C.S.; Stickland, J.E.; Evans, M.F.; McGee, J.O. Episomal and integrated human papillomavirus in cervical neoplasia shown by non-isotopic in situ hybridisation. J. Clin. Pathol. 1991, 44, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.M.; Baker, C.C. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [PubMed]
- Crook, T.; Tidy, J.A.; Vousden, K.H. Degradation of p53 can be targeted by HPV e6 sequences distinct from those required for p53 binding and trans-activation. Cell 1991, 67, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Munger, K. Interactions of the human papillomavirus e7 protein with cell cycle regulators. Semin. Cancer Biol. 1996, 7, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the atm DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus e1 helicase blocks s-phase progression and triggers an atm-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus e1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the atr-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic instability of the host cell induced by the human papillomavirus replication machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Spardy, N.; Duensing, A.; Charles, D.; Haines, N.; Nakahara, T.; Lambert, P.F.; Duensing, S. The human papillomavirus type 16 e7 oncoprotein activates the fanconi anemia (fa) pathway and causes accelerated chromosomal instability in fa cells. J. Virol. 2007, 81, 13265–13270. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Pitot, H.C.; Strati, K.; Spardy, N.; Duensing, S.; Grompe, M.; Lambert, P.F. Deficiencies in the fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer Res. 2010, 70, 9959–9968. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Mackintosh, L.J.; Bodily, J.M.; DoRNAn, E.S.; Laimins, L.A.; Morgan, I.M. An interaction between human papillomavirus 16 e2 and topbp1 is required for optimum viral DNA replication and episomal genome establishment. J. Virol. 2012, 86, 12806–12815. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Simpson, D.A.; Zhou, Y.; Mitra, A.; Mitchell, D.L.; Cordeiro-Stone, M.; Kaufmann, W.K. Human papilloma virus type16 e6 deregulates chk1 and sensitizes human fibroblasts to environmental carcinogens independently of its effect on p53. Cell Cycle 2009, 8, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 e6 abrogate atr activity resulting in increased persistence of uvb induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef] [PubMed]
- Winder, D.M.; Pett, M.R.; Foster, N.; Shivji, M.K.; Herdman, M.T.; Stanley, M.A.; Venkitaraman, A.R.; Coleman, N. An increase in DNA double-strand breaks, induced by ku70 depletion, is associated with human papillomavirus 16 episome loss and de novo viral integration events. J. Pathol. 2007, 213, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.M.; Filippova, M.; Filippov, V.; Payne, K.J.; Duerksen-Hughes, P. Human papillomavirus type 16 e6* induces oxidative stress and DNA damage. J. Virol. 2014, 88, 6751–6761. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Garau, X.; Peter, M.; Avril, M.F.; Laude, H.; Couturier, J.; Rozenberg, F.; Almeida, A.; Boitier, F.; Carlotti, A.; Couturaud, B.; et al. Merkel cell carcinoma of the skin: Pathological and molecular evidence for a causative role of mcv in oncogenesis. J. Pathol. 2009, 218, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4e-bp1 translation regulator. J. Clin. Invest. 2011, 121, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.A.; Abdul-Sada, H.; Knight, L.M.; Jackson, B.R.; Richards, K.; Prescott, E.L.; Peach, A.H.; Blair, G.E.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus small T antigen targets the nemo adaptor protein to disrupt inflammatory signaling. J. Virol. 2013, 87, 13853–13867. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Wang, X.; Li, J.; Buck, C.B.; You, J. Host DNA damage response factors localize to merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. J. Virol. 2014, 88, 3285–3297. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Kuppermann, B.D.; Wolitz, R.A.; Palestine, A.G.; Li, H.; Robinson, C.A. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche ganciclovir study group. N. Engl. J. Med. 1999, 340, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Diaz, J.; Wang, X.; Tsang, S.H.; You, J. Phosphorylation of merkel cell polyomavirus large tumor antigen at serine 816 by atm kinase induces apoptosis in host cells. J. Biol. Chem. 2015, 290, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.M.; Yu, K.J.; Mbulaiteye, S.M.; Hildesheim, A.; Bhatia, K. The extent of genetic diversity of epstein-barr virus and its geographic and disease patterns: A need for reappraisal. Virus Res. 2009, 143, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Ganem, D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in kaposi sarcoma pathogenesis. J. Clin. Invest. 2004, 113, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Gruhne, B.; Szeles, A.; Masucci, M.G. Epstein-barr virus promotes genomic instability in burkitt's lymphoma. Oncogene 2007, 26, 5115–5123. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Kamranvar, S.A.; Masucci, M.G.; Sompallae, R. Ebv and genomic instability—a new look at the role of the virus in the pathogenesis of burkitt's lymphoma. Semin. Cancer Biol. 2009, 19, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Masucci, M.G. The epstein-barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 2011, 25, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.T.; Chen, Y.R.; Chen, S.C.; Hu, C.Y.; Lin, C.S.; Chang, Y.T.; Wang, W.B.; Chen, J.Y. Epstein-barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene 2004, 23, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Liu, M.T.; Chang, Y.T.; Wu, C.C.; Hu, C.Y.; Chen, J.Y. Epstein-barr virus latent membrane protein 1 represses DNA repair through the pi3k/akt/foxo3a pathway in human epithelial cells. J. Virol. 2008, 82, 8124–8137. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.H.; Wu, C.C.; Fang, C.Y.; Yu, S.L.; Hsu, H.Y.; Chow, Y.H.; Chen, J.Y. Epstein-barr virus balf3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 2014, 5, 8583–8601. [Google Scholar] [PubMed]
- Chang, Y.H.; Lee, C.P.; Su, M.T.; Wang, J.T.; Chen, J.Y.; Lin, S.F.; Tsai, C.H.; Hsieh, M.J.; Takada, K.; Chen, M.R. Epstein-barr virus bglf4 kinase retards cellular s-phase progression and induces chromosomal abnormality. PLoS ONE 2012, 7, e39217. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Liu, M.T.; Chang, Y.T.; Fang, C.Y.; Chou, S.P.; Liao, H.W.; Kuo, K.L.; Hsu, S.L.; Chen, Y.R.; Wang, P.W.; et al. Epstein-barr virus dnase (bglf5) induces genomic instability in human epithelial cells. Nucl. Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, A.; Fujita, M.; Zhang, L.; Shirata, N.; Daikoku, T.; Sugaya, Y.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Epstein-barr virus lytic replication elicits atm checkpoint signal transduction while providing an s-phase-like cellular environment. J. Biol. Chem. 2005, 280, 8156–8163. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.G.; Verrall, E.; Schelcher, C.; Rhie, A.; Doherty, A.J.; Sinclair, A.J. Functional interaction between epstein-barr virus replication protein zta and host DNA damage response protein 53bp1. J. Virol. 2009, 83, 11116–11122. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.R.; Hu, S.; Woodard, C.; Lin, J.; TaveRNA, S.D.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and tip60 histone acetyltransferase to promote virus replication. Cell Host Microbe 2011, 10, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Hagemeier, S.R.; Barlow, E.A.; Meng, Q.; Kenney, S.C. The cellular ataxia telangiectasia-mutated kinase promotes epstein-barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J. Virol. 2012, 86, 13360–13370. [Google Scholar] [CrossRef] [PubMed]
- Hau, P.M.; Deng, W.; Jia, L.; Yang, J.; Tsurumi, T.; Chiang, A.K.; Huen, M.S.; Tsao, S.W. Role of atm in the formation of the replication compartment during lytic replication of epstein-barr virus in nasopharyngeal epithelial cells. J. Virol. 2015, 89, 652–668. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An atm/chk2-mediated DNA damage-responsive signaling pathway suppresses epstein-barr virus transformation of primary human b cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Koganti, S.; Hui-Yuen, J.; McAllister, S.; Gardner, B.; Grasser, F.; Palendira, U.; Tangye, S.G.; Freeman, A.F.; Bhaduri-McIntosh, S. Stat3 interrupts atr-chk1 signaling to allow oncovirus-mediated cell proliferation. Proc. Natl. Acad. Sci. USA 2014, 111, 4946–4951. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, T.; Verma, S.C.; Lan, K.; Murakami, M.; Robertson, E.S. The atm/atr signaling effector chk2 is targeted by epstein-barr virus nuclear antigen 3c to release the g2/m cell cycle block. J. Virol. 2007, 81, 6718–6730. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; A, J.M.; Saha, A.; Banerjee, S.; Lu, J.; Robertson, E.S. Epstein-barr virus essential antigen ebna3c attenuates h2ax expression. J. Virol. 2014, 88, 3776–3788. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Pang, P.S.; Tsang, C.M.; Hau, P.M.; Yip, Y.L.; Cheung, A.L.; Tsao, S.W. Epstein-barr virus-encoded latent membrane protein 1 impairs g2 checkpoint in human nasopharyngeal epithelial cells through defective chk1 activation. PLoS ONE 2012, 7, e39095. [Google Scholar] [CrossRef] [PubMed]
- Dheekollu, J.; Deng, Z.; Wiedmer, A.; Weitzman, M.D.; Lieberman, P.M. A role for mre11, nbs1, and recombination junctions in replication and stable maintenance of ebv episomes. PLoS ONE 2007, 2, e1257. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in aids-associated kaposi's sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in aids-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d'Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric castleman's disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Dittmer, D.P. Transcription profile of kaposi's sarcoma-associated herpesvirus in primary kaposi's sarcoma lesions as determined by real-time pcr arrays. Cancer Res. 2003, 63, 2010–2015. [Google Scholar] [PubMed]
- Guito, J.; Lukac, D.M. KSHV reactivation and novel implications of protein isomerization on lytic switch control. Viruses 2015, 7, 72–109. [Google Scholar] [CrossRef] [PubMed]
- Uppal, T.; Banerjee, S.; Sun, Z.; Verma, S.C.; Robertson, E.S. KSHV lana—the master regulator of KSHV latency. Viruses 2014, 6, 4961–4998. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Upadhyay, S.K.; M, A.J.P.; Lu, J.; Cai, Q.; Saha, A.; Robertson, E.S. H2ax phosphorylation is important for lana-mediated kaposi's sarcoma-associated herpesvirus episome persistence. J. Virol. 2013, 87, 5255–5269. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.V.; Dutta, D.; Ansari, M.A.; Dutta, S.; Chandran, B. Kaposi's sarcoma-associated herpesvirus induces the atm and h2ax DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J. Virol. 2014, 88, 2821–2834. [Google Scholar] [CrossRef] [PubMed]
- Koopal, S.; Furuhjelm, J.H.; Jarviluoma, A.; Jaamaa, S.; Pyakurel, P.; Pussinen, C.; Wirzenius, M.; Biberfeld, P.; Alitalo, K.; Laiho, M.; et al. Viral oncogene-induced DNA damage response is activated in kaposi sarcoma tumorigenesis. PLoS Pathog. 2007, 3, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, E.W.; Klefstrom, J.; Evan, G.I.; Jones, N. The oncogenic potential of kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2002, 2, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Zhou, F.; Gao, S.J. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 2004, 64, 4064–4068. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sahu, S.K.; Mohanty, S.; Chakrabarti, S.; Maji, S.; Reddy, R.R.; Jha, A.K.; Goswami, C.; Kundu, C.N.; Rajasubramaniam, S.; et al. Kaposi sarcoma herpes virus latency associated nuclear antigen protein release the g2/m cell cycle blocks by modulating atm/atr mediated checkpoint pathway. PLoS ONE 2014, 9, e100228. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Nakamura, H.; Lee, S.H.; Souvlis, J.; Yustein, J.T.; Gygi, S.; Kung, H.J.; Jung, J.U. Poly(adp-ribose) polymerase 1 and ste20-like kinase hkfc act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell. Biol. 2003, 23, 8282–8294. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.R.; Noerenberg, M.; Whitehouse, A. A novel mechanism inducing genome instability in kaposi's sarcoma-associated herpesvirus infected cells. PLoS Pathog. 2014, 10, e1004098. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Nakamura, H.; Liang, X.; Feng, P.; Chang, H.; Kowalik, T.F.; Jung, J.U. Inhibition of the atm/p53 signal transduction pathway by kaposi's sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006, 80, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of epstein-barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Franchini, G.; Nicot, C.; Johnson, J.M. Seizing of t cells by human T-cell leukemia/lymphoma virus type 1. Adv. Cancer Res. 2003, 89, 69–132. [Google Scholar] [PubMed]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Okayama, A.; Donnelly, R.; Tachibana, N.; Essex, M. Polymerase chain reaction analysis of defective human T-cell leukemia virus type i proviral genomes in leukemic cells of patients with adult T-cell leukemia. J. Virol. 1991, 65, 5471–5476. [Google Scholar] [PubMed]
- Grassmann, R.; Dengler, C.; Muller-Fleckenstein, I.; Fleckenstein, B.; McGuire, K.; Dokhelar, M.C.; Sodroski, J.G.; Haseltine, W.A. Transformation to continuous growth of primary human T lymphocytes by human T-cell leukemia virus type i x-region genes transduced by a herpesvirus saimiri vector. Proc. Natl. Acad. Sci. USA 1989, 86, 3351–3355. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, R.; Berchtold, S.; Radant, I.; Alt, M.; Fleckenstein, B.; Sodroski, J.G.; Haseltine, W.A.; Ramstedt, U. Role of human T-cell leukemia virus type 1 x region proteins in immortalization of primary human lymphocytes in culture. J. Virol. 1992, 66, 4570–4575. [Google Scholar] [PubMed]
- Hara, T.; Matsumura-Arioka, Y.; Ohtani, K.; Nakamura, M. Role of human T-cell leukemia virus type i tax in expression of the human telomerase reverse transcriptase (htert) gene in human T-cells. Cancer Sci 2008, 99, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Pise-Masison, C.A.; Mahieux, R.; Jiang, H.; Ashcroft, M.; Radonovich, M.; Duvall, J.; Guillerm, C.; Brady, J.N. Inactivation of p53 by human T-cell lymphotropic virus type 1 tax requires activation of the nf-kappab pathway and is dependent on p53 phosphorylation. Mol. Cell. Biol. 2000, 20, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
- Kehn, K.; Fuente Cde, L.; Strouss, K.; Berro, R.; Jiang, H.; Brady, J.; Mahieux, R.; Pumfery, A.; Bottazzi, M.E.; Kashanchi, F. The HTLV-I tax oncoprotein targets the retinoblastoma protein for proteasomal degradation. Oncogene 2005, 24, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Semmes, O.J.; Jeang, K.T. Localization of human T-cell leukemia virus type 1 tax to subnuclear compartments that overlap with interchromatin speckles. J. Virol. 1996, 70, 6347–6357. [Google Scholar] [PubMed]
- Bex, F.; McDowall, A.; Burny, A.; Gaynor, R. The human T-cell leukemia virus type 1 transactivator protein tax colocalizes in unique nuclear structures with nf-kappab proteins. J. Virol. 1997, 71, 3484–3497. [Google Scholar] [PubMed]
- Majone, F.; Semmes, O.J.; Jeang, K.T. Induction of micronuclei by HTLV-I tax: A cellular assay for function. Virology 1993, 193, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Philpott, S.M.; Buehring, G.C. Defective DNA repair in cells with human T-cell leukemia/bovine leukemia viruses: Role of tax gene. J. Natl. Cancer Inst. 1999, 91, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, H.H.; Bai, X.T.; Shelton, S.; Nicot, C. HTLV-I tax increases genetic instability by inducing DNA double strand breaks during DNA replication and switching repair to nhej. PLoS ONE 2012, 7, e42226. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, V.T.; Green, A.M.; Khurgel, M.; Semmes, O.J.; Kupfer, G.M. Human T-cell leukemia virus i tax protein sensitizes p53-mutant cells to DNA damage. Cancer Res. 2008, 68, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, T.; Ham-Terhune, J.; Peloponese, J.M., Jr.; Jeang, K.T. Induction of reactive oxygen species by human T-cell leukemia virus type 1 tax correlates with DNA damage and expression of cellular senescence marker. J. Virol. 2010, 84, 5431–5437. [Google Scholar] [CrossRef] [PubMed]
- Chaib-Mezrag, H.; Lemacon, D.; Fontaine, H.; Bellon, M.; Bai, X.T.; Drac, M.; Coquelle, A.; Nicot, C. Tax impairs DNA replication forks and increases DNA breaks in specific oncogenic genome regions. Mol. Cancer 2014, 13, e205. [Google Scholar] [CrossRef]
- Miyake, H.; Suzuki, T.; Hirai, H.; Yoshida, M. Trans-activator tax of human T-cell leukemia virus type 1 enhances mutation frequency of the cellular genome. Virology 1999, 253, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.S.; Hamzeh, F.; Moore, S.; Nicholas, J.; Ambinder, R.F. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J. Virol. 1999, 73, 4786–4793. [Google Scholar] [PubMed]
- Schavinsky-Khrapunsky, Y.; Priel, E.; Aboud, M. Dose-dependent dual effect of HTLV-1 tax oncoprotein on p53-dependent nucleotide excision repair in human T-cells. Int. J. Cancer 2008, 122, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Chandhasin, C.; Ducu, R.I.; Berkovich, E.; Kastan, M.B.; Marriott, S.J. Human T-cell leukemia virus type 1 tax attenuates the atm-mediated cellular DNA damage response. J. Virol. 2008, 82, 6952–6961. [Google Scholar] [CrossRef] [PubMed]
- Dayaram, T.; Lemoine, F.J.; Donehower, L.A.; Marriott, S.J. Activation of wip1 phosphatase by HTLV-1 tax mitigates the cellular response to DNA damage. PLoS ONE 2013, 8, e55989. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Majone, F.; Luisetto, R.; Zamboni, D.; Iwanaga, Y.; Jeang, K.T. Ku protein as a potential human T-cell leukemia virus type 1 (HTLV-1) tax target in clastogenic chromosomal instability of mammalian cells. Retrovirology 2005, 2, e45. [Google Scholar] [CrossRef]
- Ducu, R.I.; Dayaram, T.; Marriott, S.J. The HTLV-1 tax oncoprotein represses ku80 gene expression. Virology 2011, 416, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, H.H.; Pancewicz, J.; Nicot, C. Human T-lymphotropic type 1 virus p30 inhibits homologous recombination and favors unfaithful DNA repair. Blood 2011, 117, 5897–5906. [Google Scholar] [CrossRef] [PubMed]
- Haoudi, A.; Daniels, R.C.; Wong, E.; Kupfer, G.; Semmes, O.J. Human T-cell leukemia virus-i tax oncoprotein functionally targets a subnuclear complex involved in cellular DNA damage-response. J. Biol. Chem. 2003, 278, 37736–37744. [Google Scholar] [CrossRef] [PubMed]
- Park, H.U.; Jeong, J.H.; Chung, J.H.; Brady, J.N. Human T-cell leukemia virus type 1 tax interacts with chk1 and attenuates DNA-damage induced g2 arrest mediated by chk1. Oncogene 2004, 23, 4966–4974. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Guo, X.; Durkin, S.S.; Fryrear, K.F.; Ward, M.D.; Semmes, O.J. Human T-cell leukemia virus type 1 tax oncoprotein prevents DNA damage-induced chromatin egress of hyperphosphorylated chk2. J. Biol. Chem. 2007, 282, 29431–29440. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.S.; Guo, X.; Fryrear, K.A.; Mihaylova, V.T.; Gupta, S.K.; Belgnaoui, S.M.; Haoudi, A.; Kupfer, G.M.; Semmes, O.J. HTLV-1 tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J. Biol. Chem. 2008, 283, 36311–36320. [Google Scholar] [CrossRef] [PubMed]
- Chlichlia, K.; Khazaie, K. HTLV-1 tax: Linking transformation, DNA damage and apoptotic T-cell death. Chem Biol Interact 2010, 188, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Baydoun, H.H.; Yao, Y.; Nicot, C. HTLV-I tax-dependent and -independent events associated with immortalization of human primary t lymphocytes. Blood 2010, 115, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wood, C. Kaposi's sarcoma-associated herpesvirus transactivator rta induces cell cycle arrest in g0/g1 phase by stabilizing and promoting nuclear localization of p27kip. J. Virol. 2013, 87, 13226–13238. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.; Chen, J. Human claspin is required for replication checkpoint control. J. Biol. Chem. 2003, 278, 30057–30062. [Google Scholar] [CrossRef] [PubMed]
- Jarviluoma, A.; Koopal, S.; Rasanen, S.; Makela, T.P.; Ojala, P.M. KSHV viral cyclin binds to p27kip1 in primary effusion lymphomas. Blood 2004, 104, 3349–3354. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Schneider, R.J. The enigmatic x gene of hepatitis b virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Zhu, M.; Duan, L.X.; London, W.T. Hepatitis b x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene 1993, 8, 1109–1117. [Google Scholar] [PubMed]
- Wang, X.W.; Forrester, K.; Yeh, H.; Feitelson, M.A.; Gu, J.R.; Harris, C.C. Hepatitis B virus x protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ercc3. Proc. Natl. Acad. Sci. USA 1994, 91, 2230–2234. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Rho, H.M. Transcriptional repression of the human p53 gene by hepatitis B viral x protein. Oncogene 2000, 19, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Lu, M.; Gwack, Y.; Souvlis, J.; Zeichner, S.L.; Jung, J.U. Global changes in kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible rta transactivator. J. Virol. 2003, 77, 4205–4220. [Google Scholar] [CrossRef] [PubMed]
- Hagen, T.M.; Huang, S.; Curnutte, J.; Fowler, P.; Martinez, V.; Wehr, C.M.; Ames, B.N.; Chisari, F.V. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 12808–12812. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Chang, W.W.; Lei, H.Y.; Lai, M.D.; Chang, W.T.; Huang, W. Pre-s mutant surface antigens in chronic Hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004, 25, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.; Boehme, R. Antiviral activity and mechanism of action of ganciclovir. Rev. Infect. Dis. 1988, 10 Suppl. 3, S490–S494. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone h2ax is phosphorylated in an atr-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Burda, M.R.; Burkle, A.; Zuckerman, D.M.; Will, H.; Rogler, C.E.; Greten, H.; Petersen, J. Increase in de novo hbv DNA integrations in response to oxidative DNA damage or inhibition of poly(adp-ribosyl)ation. Hepatology 2002, 35, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus x protein stimulates viral genome replication via a ddb1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Bresnahan, W.A.; Boldogh, I.; Thompson, E.A.; Albrecht, T. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late g1. Virology 1996, 224, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.A.; Dyson, O.F.; Akula, S.M. Identifying cellular genes crucial for the reactivation of kaposi's sarcoma-associated herpesvirus latency. J. Gen. Virol. 2006, 87, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.A.; Lee, T.H.; Butel, J.S.; Slagle, B.L. Hepatitis B virus x protein interferes with cellular DNA repair. J. Virol. 1998, 72, 266–272. [Google Scholar] [PubMed]
- Martin-Lluesma, S.; Schaeffer, C.; Robert, E.I.; van Breugel, P.C.; Leupin, O.; Hantz, O.; Strubin, M. Hepatitis B virus x protein affects s phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 2008, 48, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Jaitovich-Groisman, I.; Benlimame, N.; Slagle, B.L.; Perez, M.H.; Alpert, L.; Song, D.J.; Fotouhi-Ardakani, N.; Galipeau, J.; Alaoui-Jamali, M.A. Transcriptional regulation of the tfiih transcription repair components xpb and xpd by the Hepatitis B virus x protein in liver cells and transgenic liver tissue. J. Biol. Chem. 2001, 276, 14124–14132. [Google Scholar] [PubMed]
- Prost, S.; Ford, J.M.; Taylor, C.; Doig, J.; Harrison, D.J. Hepatitis b x protein inhibits p53-dependent DNA repair in primary mouse hepatocytes. J. Biol. Chem. 1998, 273, 33327–33332. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.T.; Ren, J.; Wong, E.T.; Ban, K.H.; Lee, L.A.; Lee, C.G. The Hepatitis B virus x protein sensitizes hepg2 cells to uv light-induced DNA damage. J. Biol. Chem. 2005, 280, 33525–33535. [Google Scholar] [CrossRef] [PubMed]
- Van de Klundert, M.A.; van Hemert, F.J.; Zaaijer, H.L.; Kootstra, N.A. The Hepatitis B virus x protein inhibits thymine DNA glycosylase initiated base excision repair. PLoS ONE 2012, 7, e48940. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L. Defective DNA damage response and repair in liver cells expressing Hepatitis B virus surface antigen. FASEB J. 2013, 27, 2316–2327. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Hullinger, R.L.; Andrisani, O.M. Hepatitis B virus x protein via the p38mapk pathway induces e2f1 release and atr kinase activation mediating p53 apoptosis. J. Biol. Chem. 2008, 283, 25455–25467. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Hou, N.B.; Yang, X.L.; He, X.; Liu, Y.; Zhang, Y.H.; Wei, C.W.; Song, T.; Li, L.; Ma, Q.J.; et al. Ataxia telangiectasia-mutated-rad3-related DNA damage checkpoint signaling pathway triggered by Hepatitis B virus infection. World J. Gastroenterol. 2008, 14, 6163–6170. [Google Scholar] [CrossRef] [PubMed]
- Rakotomalala, L.; Studach, L.; Wang, W.H.; Gregori, G.; Hullinger, R.L.; Andrisani, O. Hepatitis B virus x protein increases the cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J. Biol. Chem. 2008, 283, 28729–28740. [Google Scholar] [CrossRef] [PubMed]
- Studach, L.; Wang, W.H.; Weber, G.; Tang, J.; Hullinger, R.L.; Malbrue, R.; Liu, X.; Andrisani, O. Polo-like kinase 1 activated by the Hepatitis B virus x protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. J. Biol. Chem. 2010, 285, 30282–30293. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, J.; Sun, J.; Song, T.; Wei, C.; Zhang, Y.; Rao, G.; Chen, G.; Li, D.; Yang, G.; et al. Inhibition of hbv replication by theophylline. Antiviral Res. 2011, 89, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Lin, S.F.; Ellison, T.J.; Levy, A.M.; Mayeur, G.L.; Izumiya, C.; Kung, H.J. Cell cycle regulation by kaposi's sarcoma-associated herpesvirus k-bzip: Direct interaction with cyclin-cdk2 and induction of g1 growth arrest. J. Virol. 2003, 77, 9652–9661. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Y.; Tang, Q.Q.; Chen, H.; ApRhys, C.; Farrell, C.; Chen, J.; Fujimuro, M.; Lane, M.D.; Hayward, G.S. Lytic replication-associated protein (rap) encoded by kaposi sarcoma-associated herpesvirus causes p21cip-1-mediated g1 cell cycle arrest through ccaat/enhancer-binding protein-alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 10683–10688. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Jung, E.J.; Flemington, E.K. Cell cycle analysis of epstein-barr virus-infected cells following treatment with lytic cycle-inducing agents. J. Virol. 2001, 75, 4482–4489. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D.; Weitzman, M.D. The intrinsic antiviral defense to incoming hsv-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein icp0. PLoS Pathog. 2011, 7, e1002084. [Google Scholar] [CrossRef] [PubMed]
- FeRNAndez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced g2-m checkpoint activation by histone h2ax and 53bp1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.; Sung, V.M.; Lee, K.J.; Levine, A.M.; Lai, M.M. Hepatitis C virus infection activates the immunologic (type ii) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J. Virol. 2004, 78, 8835–8843. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and stat3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kohara, M.; Izumi, K.; Kasama, Y.; Hirata, Y.; Huang, Y.; Shuda, M.; Mukaidani, C.; Takano, T.; Tokunaga, Y.; et al. Hepatitis C virus impairs p53 via persistent overexpression of 3beta-hydroxysterol delta24-reductase. J. Biol. Chem. 2009, 284, 36442–36452. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Hepatitis C virus-induced activation of beta-catenin promotes c-myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.R.; Sabry, G.M.; Bahnassy, A.A.; Shalaby, K.A.; Abdel-Wahabh, S.A.; Zakaria, S. Mismatch repair genes (hmlh1, hpms1, hpms2, gtbp/hmsh6, hmsh2) in the pathogenesis of hepatocellular carcinoma. World J. Gastroenterol. 2005, 11, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Polyak, S.J.; Bano, N.; Qiu, W.C.; Carithers, R.L.; Shuhart, M.; Gretch, D.R.; Das, A. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme neil1. J Gastroenterol Hepatol 2010, 25, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Downregulation of gadd45beta expression by Hepatitis C virus leads to defective cell cycle arrest. Cancer Res. 2010, 70, 4901–4911. [Google Scholar] [CrossRef] [PubMed]
- van Pelt, J.F.; Severi, T.; Crabbe, T.; Eetveldt, A.V.; Verslype, C.; Roskams, T.; Fevery, J. Expression of Hepatitis C virus core protein impairs DNA repair in human hepatoma cells. Cancer Lett 2004, 209, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; McNamara, G.; Cheng, K.T.; Huang, J.; Wang, C.H.; Comai, L.; Ou, J.H.; Lai, M.M. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the atm-nbs1/mre11/rad50 DNA repair pathway in monocytes and hepatocytes. J Immunol 2010, 185, 6985–6998. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kuroki, M.; Dansako, H.; Abe, K.; Ikeda, M.; Wakita, T.; Kato, N. The DNA damage sensors ataxia-telangiectasia mutated kinase and checkpoint kinase 2 are required for Hepatitis C virus RNA replication. J. Virol. 2008, 82, 9639–9646. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Cheng, Y.S.; Lai, M.M. Hepatitis C virus ns3/4a protein interacts with atm, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 2008, 370, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Bittar, C.; Shrivastava, S.; Bhanja Chowdhury, J.; Rahal, P.; Ray, R.B. Hepatitis C virus ns2 protein inhibits DNA damage pathway by sequestering p53 to the cytoplasm. PLoS ONE 2013, 8, e62581. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Dansako, H.; Kobayashi, N.; Ikeda, M.; Kato, N. Hepatitis C virus ns5b delays cell cycle progression by inducing interferon-beta via toll-like receptor 3 signaling pathway without replicating viral genomes. Virology 2006, 346, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Y.; Tong, W.; Pan, T.; Li, J.; Sun, S.; Shao, J.; Ding, H.; Toyoda, T.; Yuan, Z. Hepatitis C virus ns5b protein delays s phase progression in human hepatocyte-derived cells by relocalizing cyclin-dependent kinase 2-interacting protein (cinp). J. Biol. Chem. 2011, 286, 26603–26615. [Google Scholar] [CrossRef] [PubMed]
- Qadri, I.; Iwahashi, M.; Simon, F. Hepatitis C virus ns5a protein binds tbp and p53, inhibiting their DNA binding and p53 interactions with tbp and ercc3. Biochim. Biophys. Acta 2002, 1592, 193–204. [Google Scholar] [CrossRef]
- Luo, M.H.; Rosenke, K.; CzoRNAk, K.; Fortunato, E.A. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (atm)- and atm-rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 2007, 81, 1934–1950. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Neufeldt, C.J.; Pang, D.; Wilson, K.; Loewen-Dobler, D.; Joyce, M.A.; Wozniak, R.W.; Tyrrell, D.L. Functional characterization of nuclear localization and export signals in Hepatitis C virus proteins and their role in the membranous web. PLoS ONE 2014, 9, e114629. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Williams, V.; Filippova, M.; Filippov, V.; Duerksen-Hughes, P. Viral carcinogenesis: Factors inducing DNA damage and virus integration. Cancers 2014, 6, 2155–2186. [Google Scholar] [CrossRef] [PubMed]
- Mauser, A.; Holley-Guthrie, E.; Zanation, A.; Yarborough, W.; Kaufmann, W.; Klingelhutz, A.; Seaman, W.T.; Kenney, S. The epstein-barr virus immediate-early protein bzlf1 induces expression of e2f-1 and other proteins involved in cell cycle progression in primary keratinocytes and gastric carcinoma cells. J. Virol. 2002, 76, 12543–12552. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Dee, A.R.; Smith, S.; Schumacher, A.J.; Weller, S.K. Efficient herpes simplex virus 1 replication requires cellular atr pathway proteins. J. Virol. 2013, 87, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Barez, P.Y.; Boxus, M.; Willems, L. Checkpoints modulation by the human T-lymphotropic virus type 1 tax protein. Retrovirology 2014, 11, e90. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollingworth, R.; Grand, R.J. Modulation of DNA Damage and Repair Pathways by Human Tumour Viruses. Viruses 2015, 7, 2542-2591. https://doi.org/10.3390/v7052542
Hollingworth R, Grand RJ. Modulation of DNA Damage and Repair Pathways by Human Tumour Viruses. Viruses. 2015; 7(5):2542-2591. https://doi.org/10.3390/v7052542
Chicago/Turabian StyleHollingworth, Robert, and Roger J Grand. 2015. "Modulation of DNA Damage and Repair Pathways by Human Tumour Viruses" Viruses 7, no. 5: 2542-2591. https://doi.org/10.3390/v7052542
APA StyleHollingworth, R., & Grand, R. J. (2015). Modulation of DNA Damage and Repair Pathways by Human Tumour Viruses. Viruses, 7(5), 2542-2591. https://doi.org/10.3390/v7052542