
Influence of Cellular Trafficking Pathway on Bluetongue Virus Infection in Ovine Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Virus and Antibodies
2.2. Plasmids and Site-Directed Mutagenesis
2.3. Virus Growth Kinetics and Virus Release
2.4. BTV Release Assay
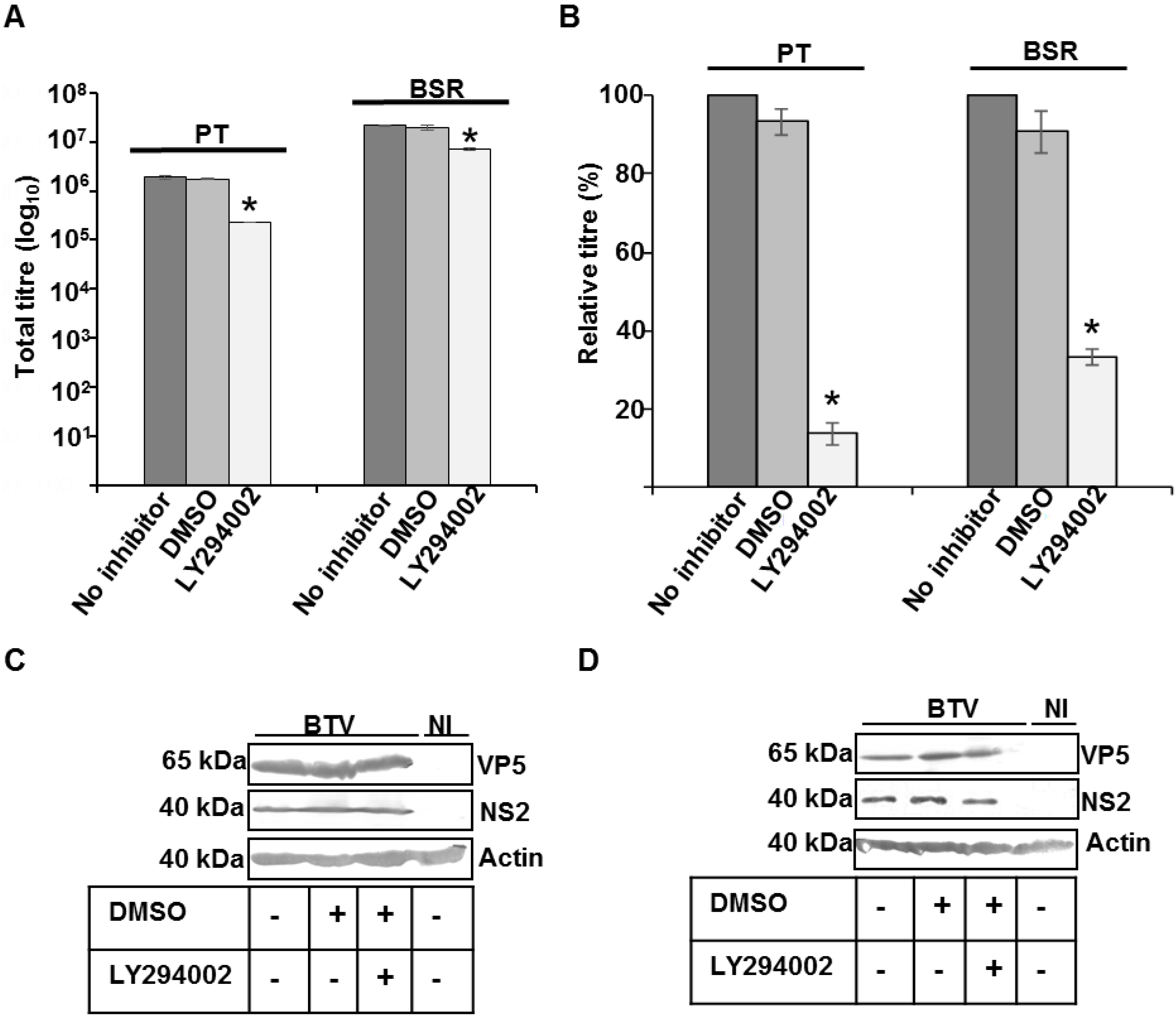
2.5. Ubiquitin and PI 3-Kinase Inhibition Assay
2.6. Confocal Microscopy
2.7. Electron Microscopy
3. Results
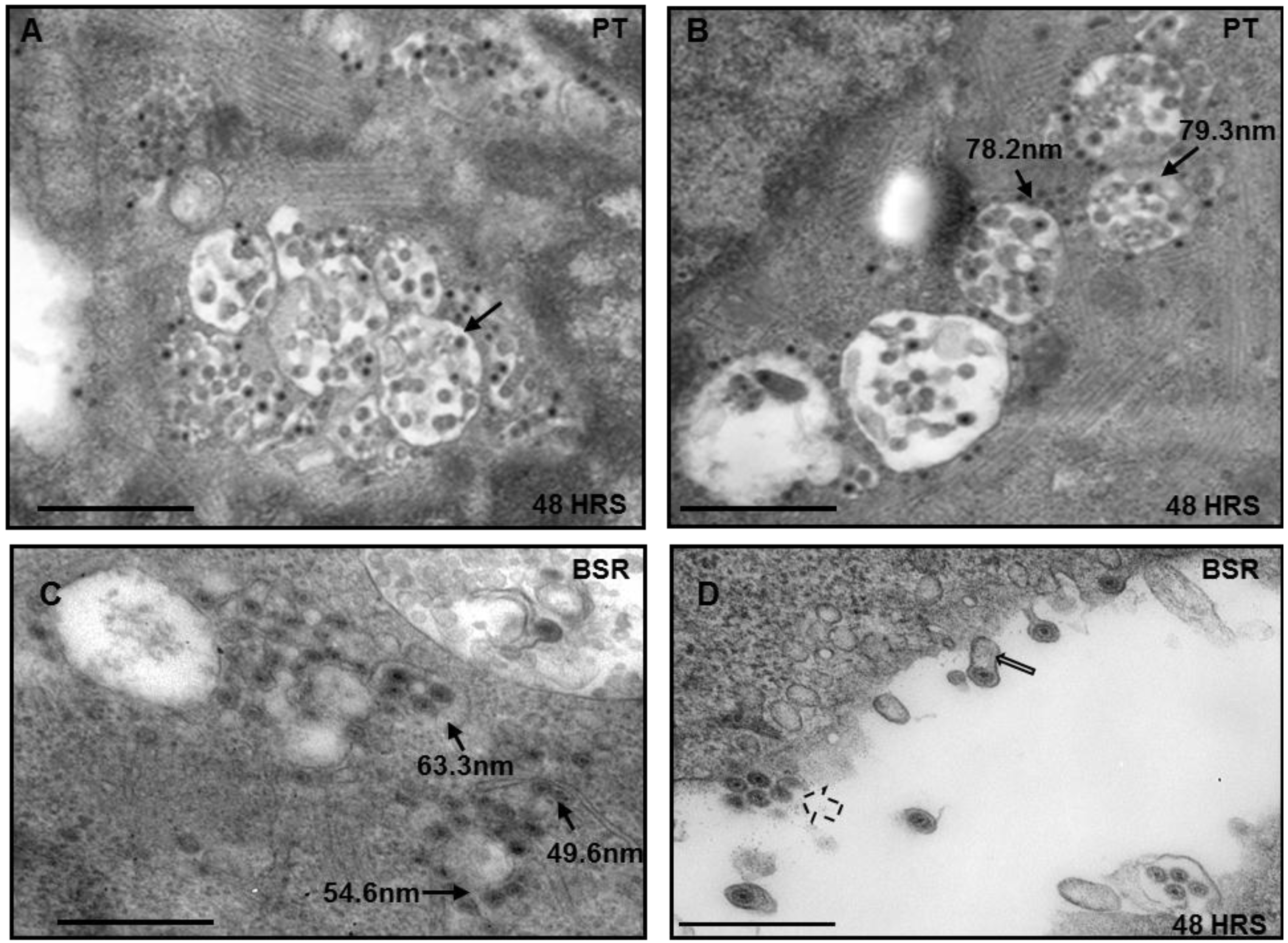
3.1. Vesicular Distribution of BTV Particles in Sheep Cells
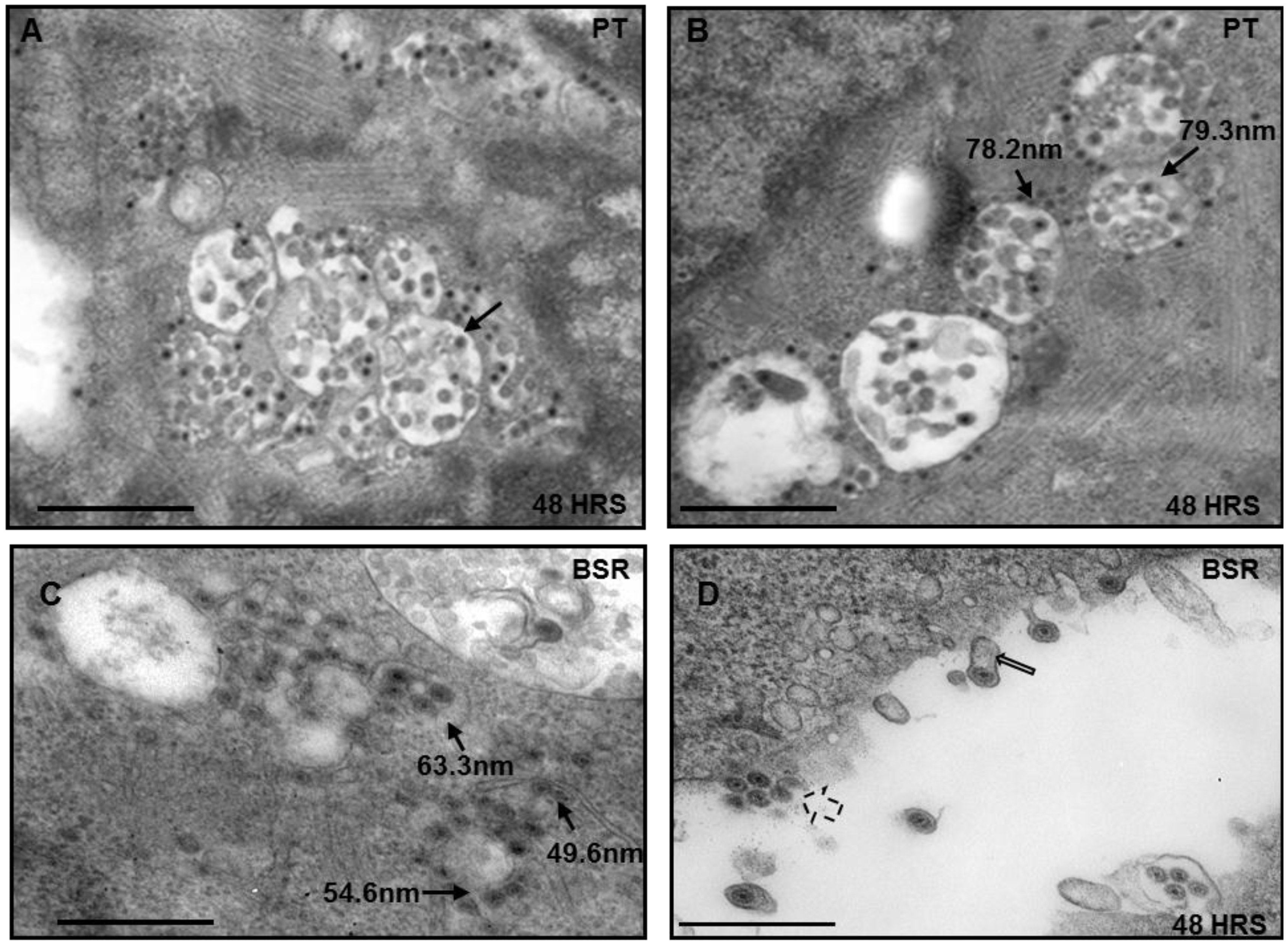
3.2. Disruption of MVB and BTV Growth
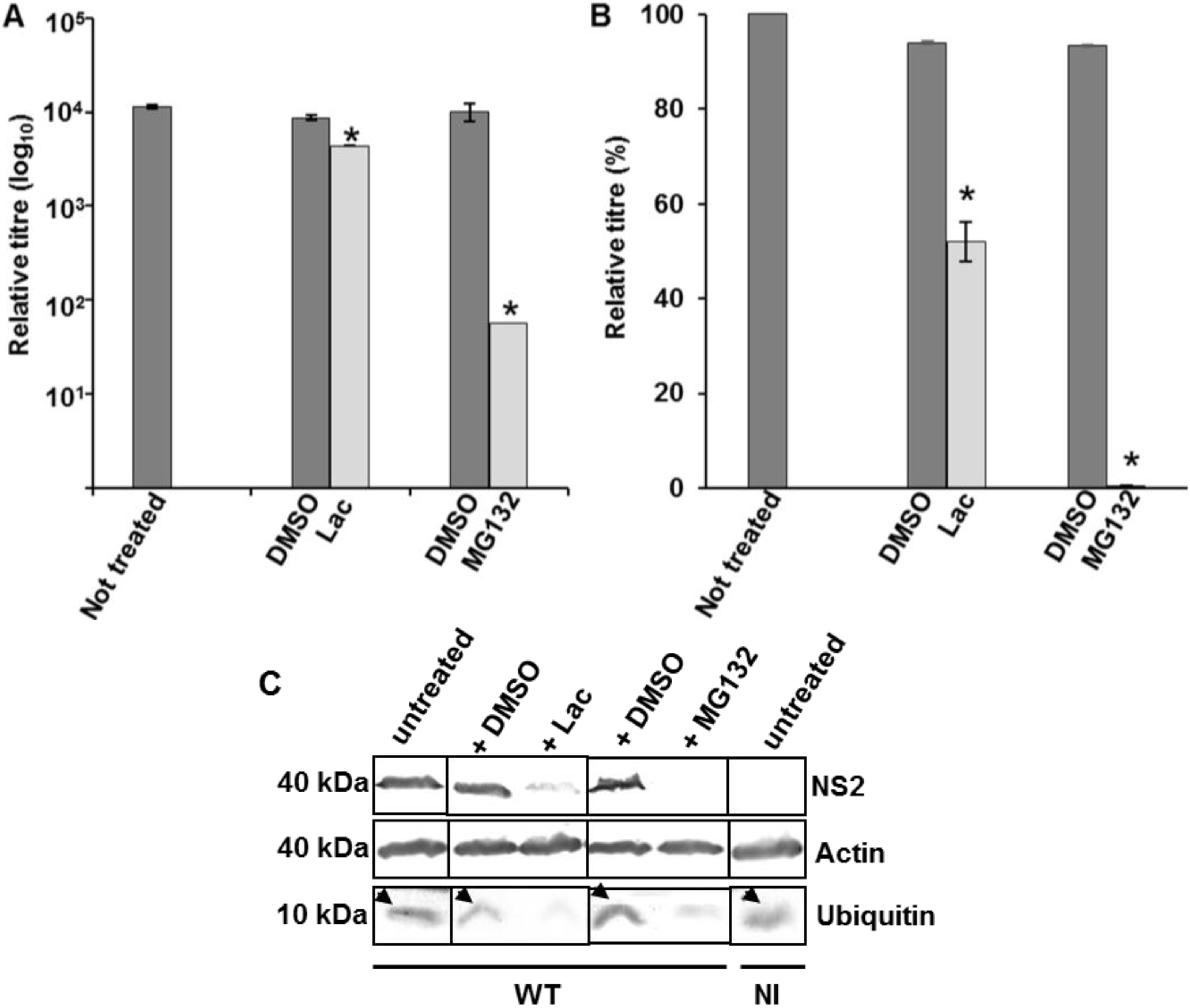
3.3. Ubiquitin, the Cellular Partner for Trafficking of BTV to Vesicles
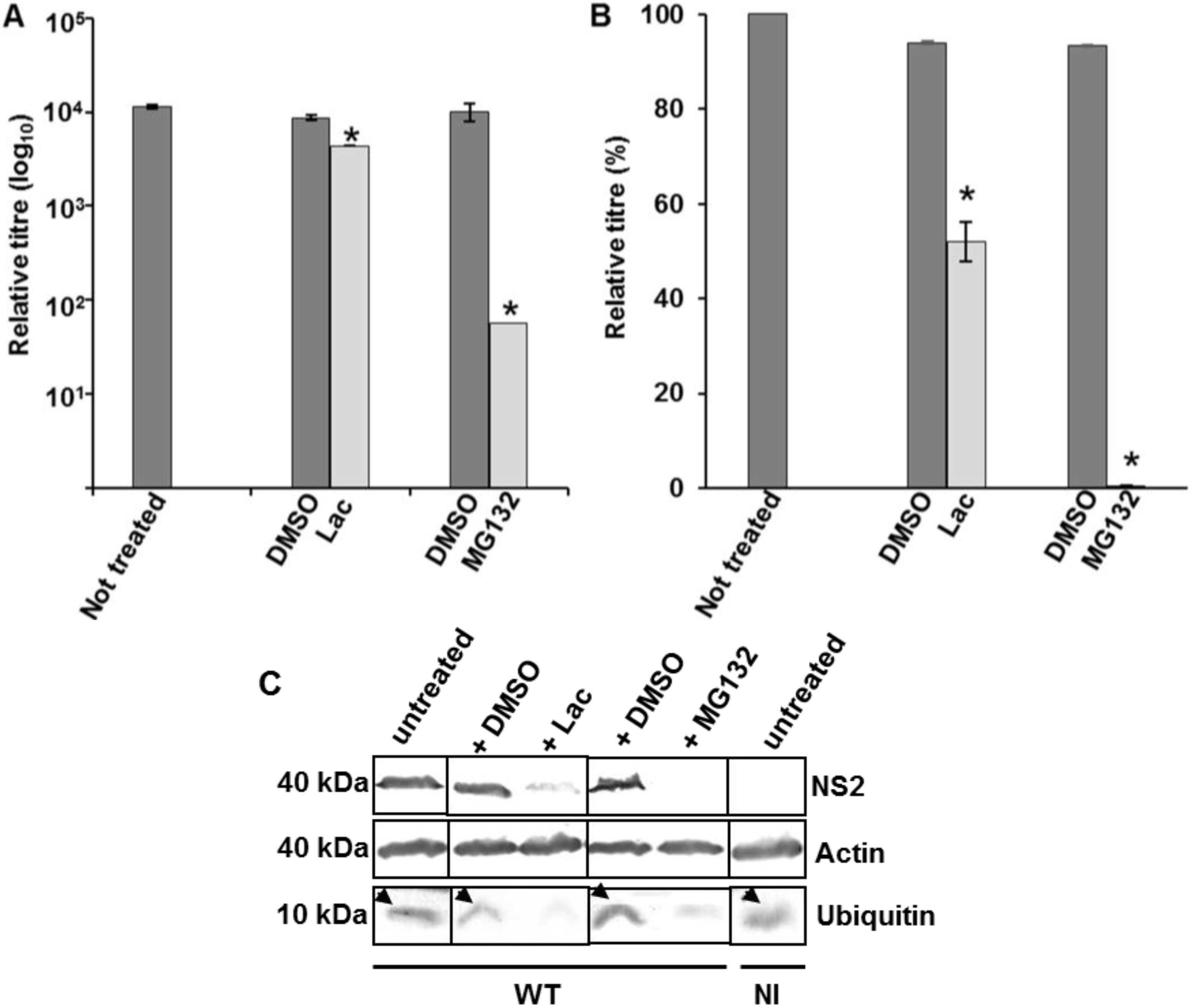
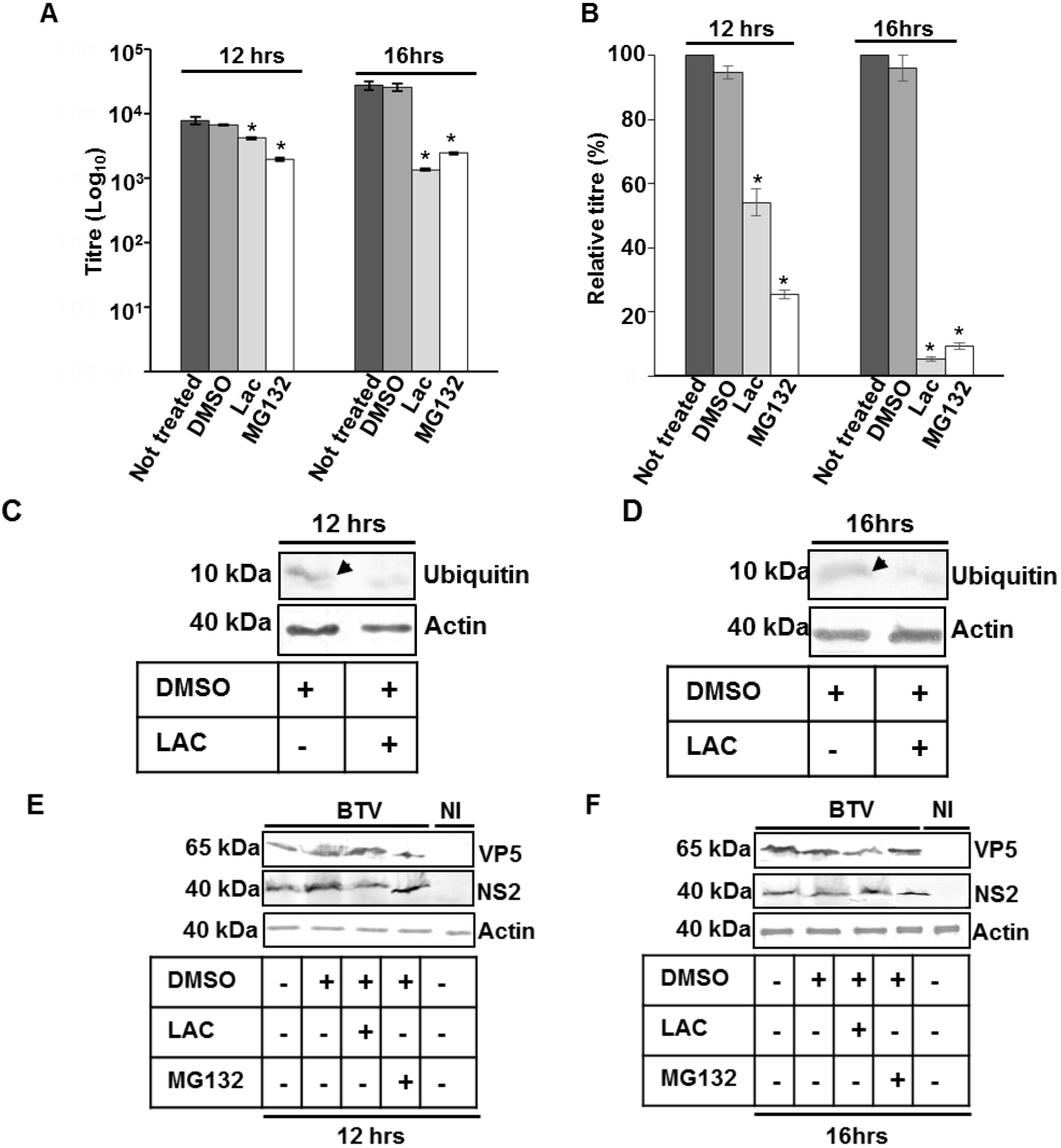
3.4. Influence of PPRY Late Domain on BTV Distribution
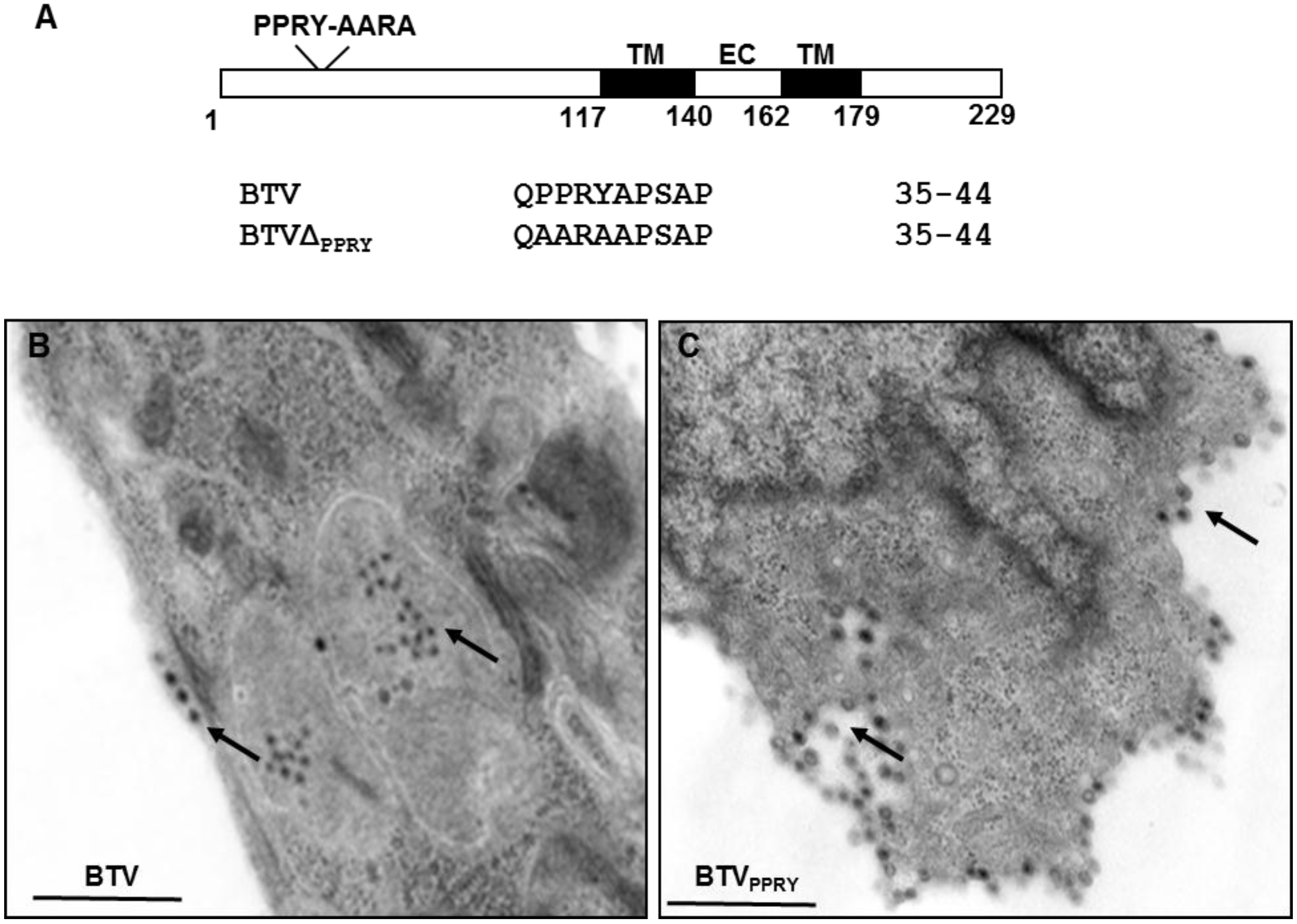
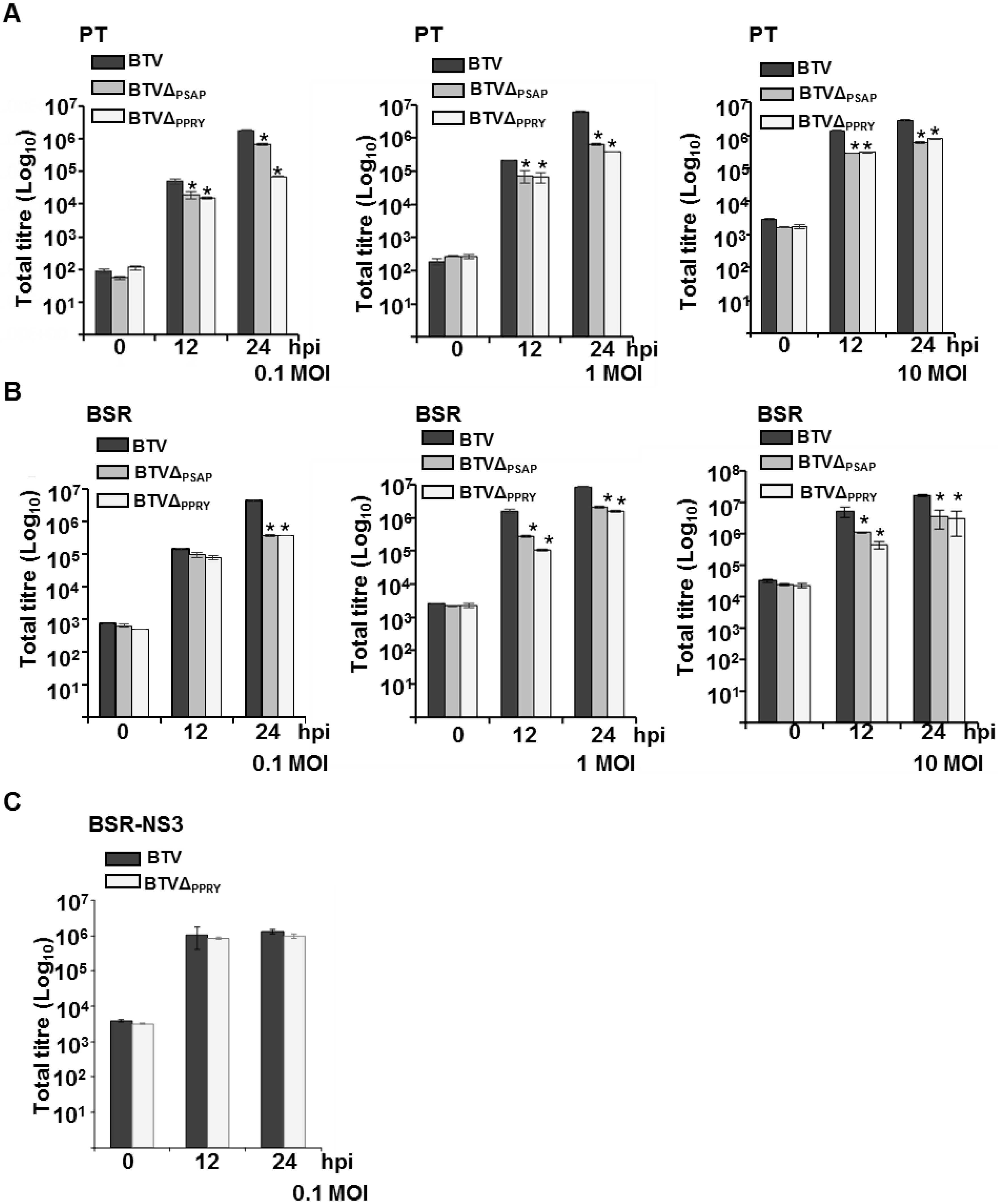
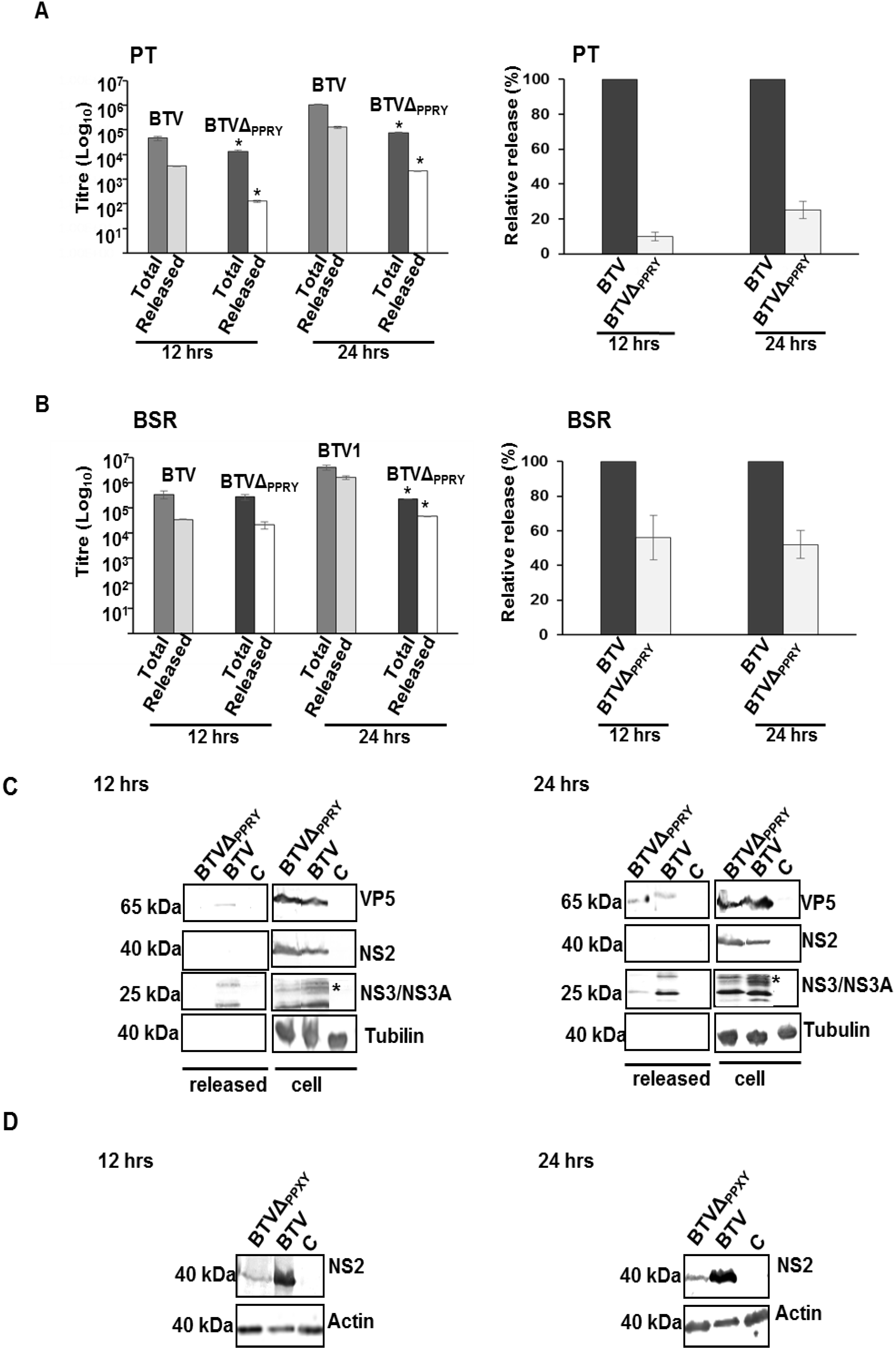
3.5. The Conserved PPRY Late Domain in NS3 Influences BTV Growth
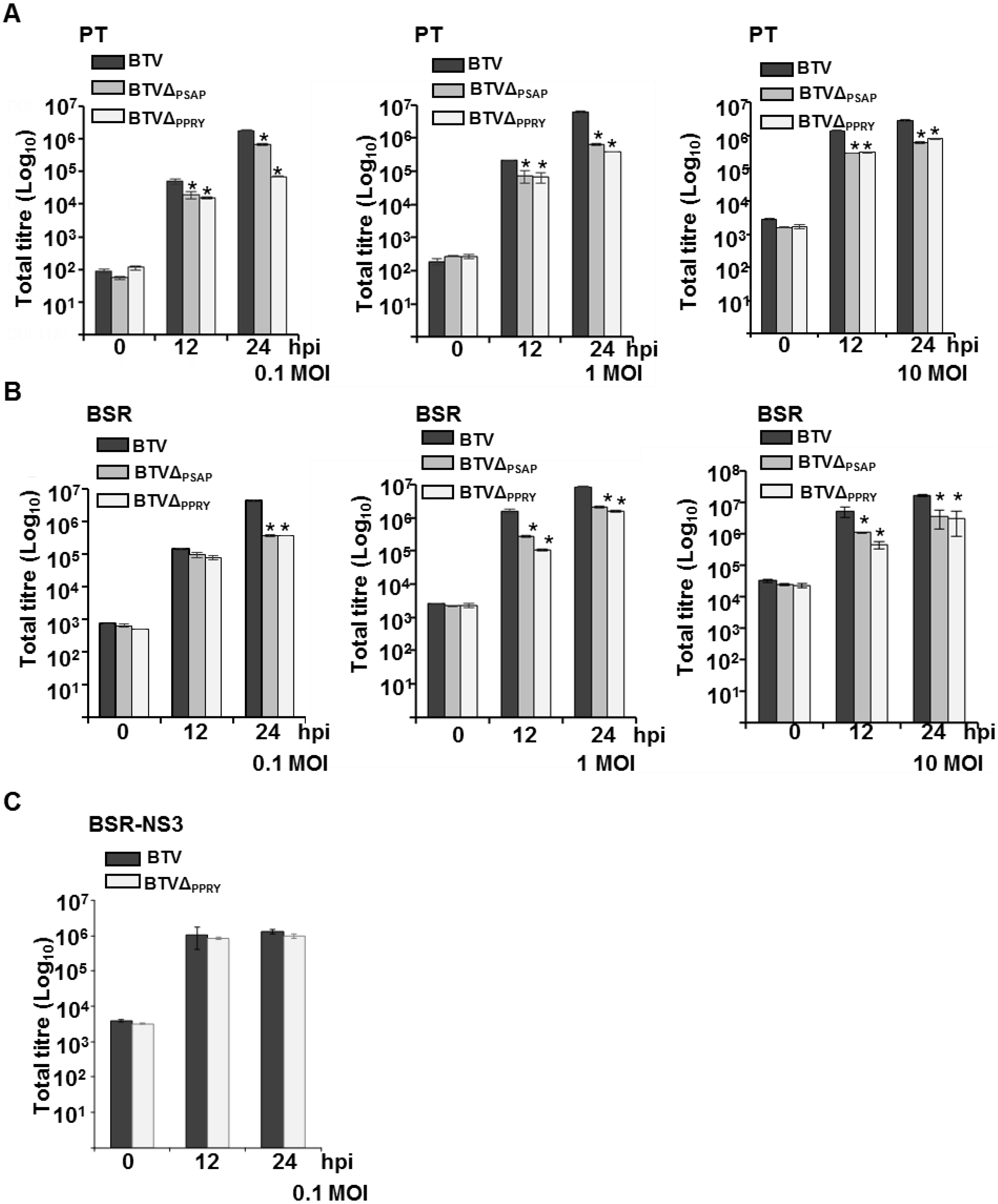
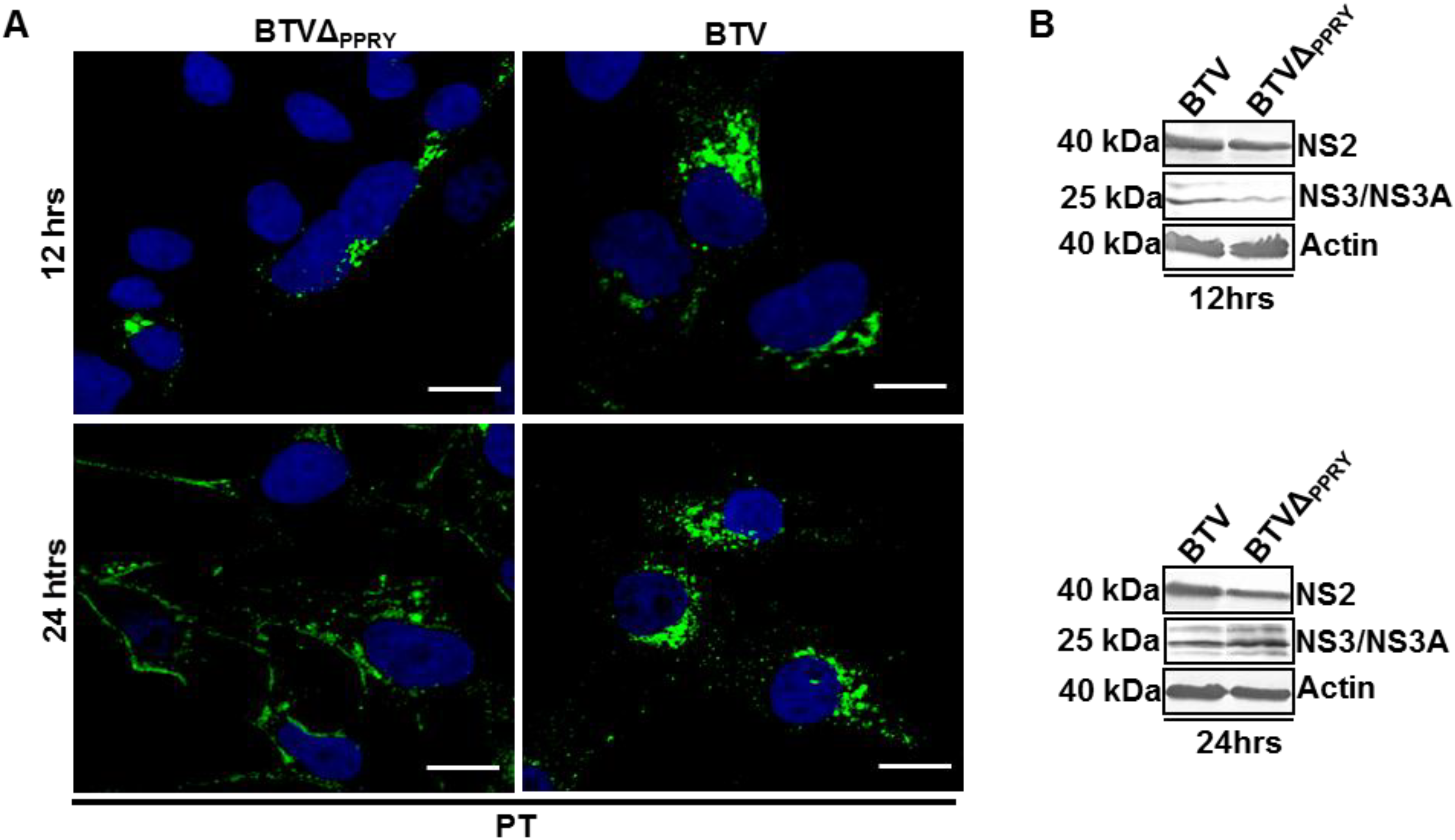
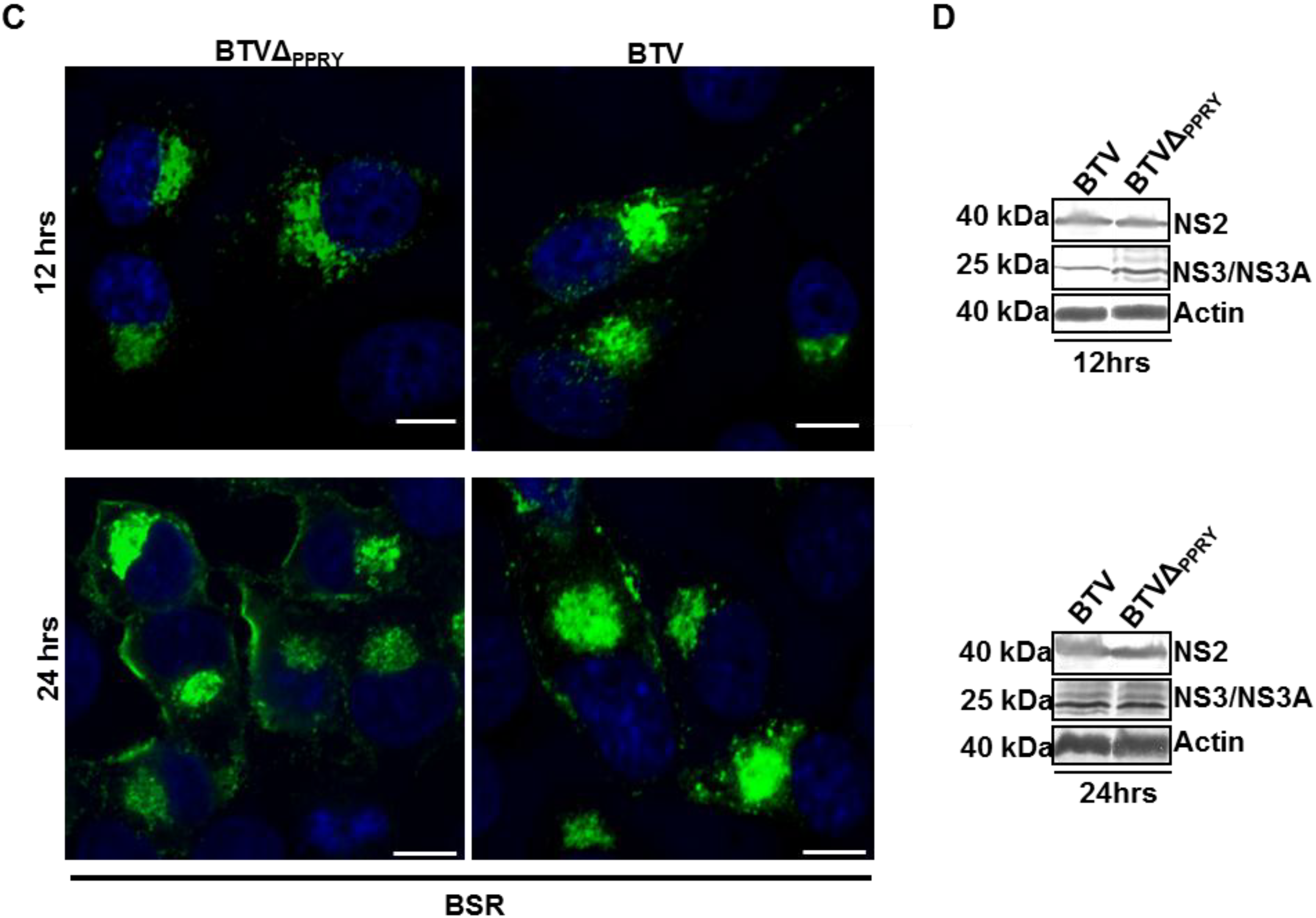
3.6. Mutation of PPRY Domain Alters the Distribution of NS3 in Sheep Cells


3.7. Effect of Mutations in the PPRY Domain of NS3 on Virus Release
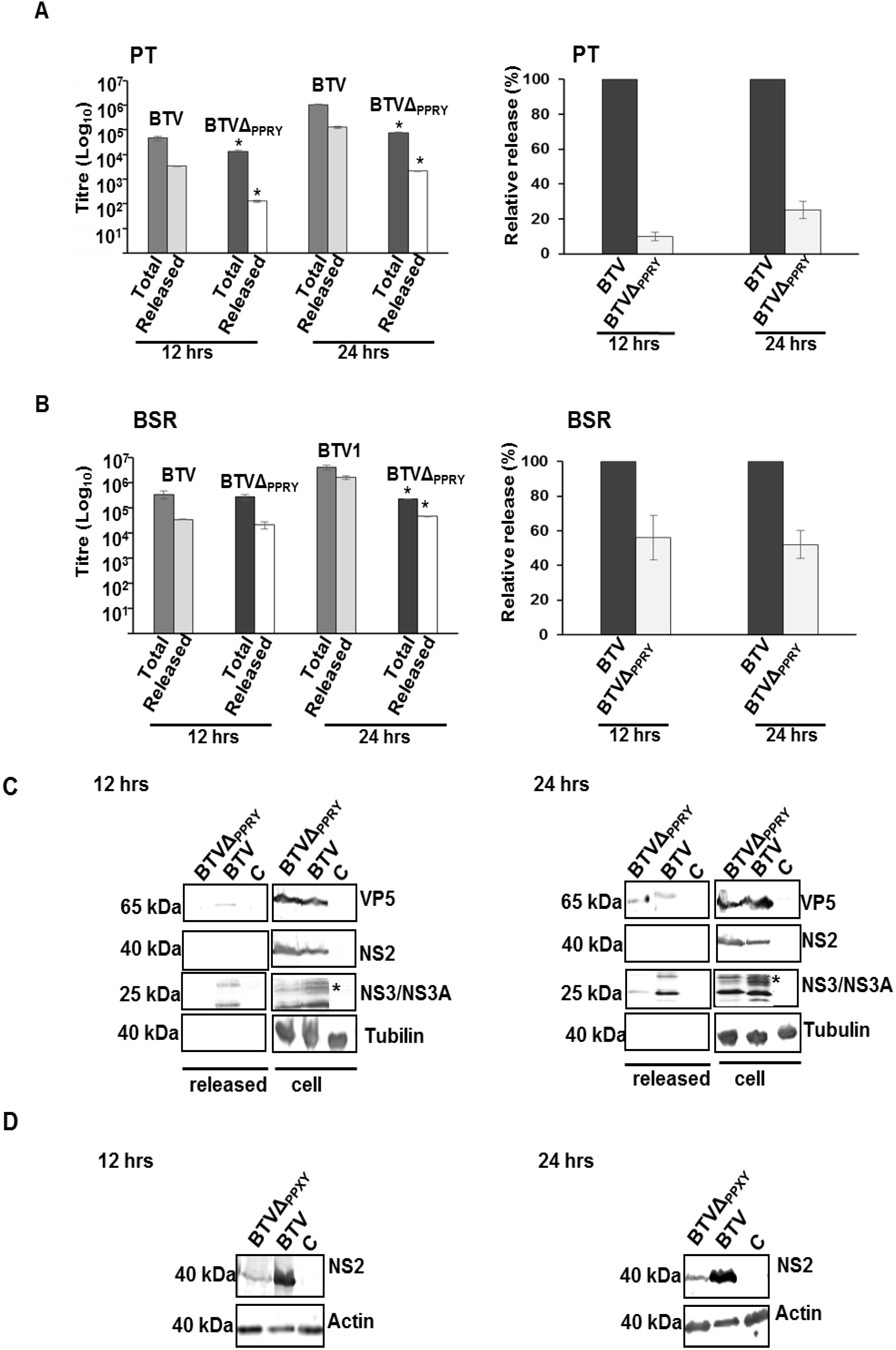
4. Discussion
Acknowledgement
Author Contributions
Conflicts of Interest
References
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef]
- Taylor, M.P.; Burgon, T.B.; Kirkegaard, K.; Jackson, W.T. Role of microtubules in extracellular release of poliovirus. J. Virol. 2009, 83, 6599–6609. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, A.D.; Eaton, B.T.; Brookes, S.M. The release of bluetongue virus from infected cells and their superinfection by progeny virus. Virology 1989, 173, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Celma, C.C.; Roy, P. A viral nonstructural protein regulates bluetongue virus trafficking and release. J. Virol. 2009, 83, 6806–6816. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, D.P.; Russell, M.R.G.; Odorizzi, G. A concentric circle model of multivesicular body cargo sorting. EMBO Rep. 2007, 8, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Gottlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef] [PubMed]
- Puffer, B.A.; Parent, L.J.; Wills, J.W.; Montelaro, R.C. Equine infectious anemia virus utilizes a YXXL motif within the late assembly domain of the Gag p9 protein. J. Virol. 1997, 71, 6541–6546. [Google Scholar] [PubMed]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Gottlinger, H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Wiesner, S.; Sudol, M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002, 513, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, S.B.; Losko, S.; Kaiser, C.A. Components of a ubiquitin ligase complex specify polyubiquitination and intracellular trafficking of the general amino acid permease. J. Cell Biol. 2001, 153, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Pelham, H.R. Sorting of proteins into multivesicular bodies: Ubiquitin-dependent and -independent targeting. EMBO J. 2001, 20, 5176–5186. [Google Scholar] [CrossRef] [PubMed]
- Urbanowski, J.L.; Piper, R.C. Ubiquitin sorts proteins into the intralumenal degradative compartment of the late-endosome/vacuole. Traffic 2001, 2, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Strack, B.; Calistri, A.; Accola, M.A.; Palu, G.; Gottlinger, H.G. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 2000, 97, 13063–13068. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Ott, D.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Krausslich, H.G.; Yewdell, J.W. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Shehu-Xhilaga, M.; Ablan, S.; Demirov, D.G.; Chen, C.; Montelaro, R.C.; Freed, E.O. Late domain-dependent inhibition of equine infectious anemia virus budding. J. Virol. 2004, 78, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Borja, M.; Wubbolts, R.; Calafat, J.; Janssen, H.; Divecha, N.; Dusseljee, S.; Neefjes, J. Multivesicular body morphogenesis requires phosphatidyl-inositol 3-kinase activity. Curr. Biol. 1999, 9, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Futter, C.E.; Collinson, L.M.; Backer, J.M.; Hopkins, C.R. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J. Cell Biol. 2001, 155, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, D.J.; Odorizzi, G.; Emr, S.D. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 2002, 3, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Piper, R.C.; Katzmann, D.J. Biogenesis and Function of Multivesicular Bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef] [PubMed]
- Gillooly, D.J.; Morrow, I.C.; Lindsay, M.; Gould, R.; Bryant, N.J.; Gaullier, J.M.; Parton, R.G.; Stenmark, H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000, 19, 4577–4588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Boyce, M.; Bhattacharya, B.; Schein, S.; Roy, P.; Zhou, Z.H. Bluetongue virus coat protein VP2 contains sialic acid-binding domains, and VP5 resembles enveloped virus fusion proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 6292–6297. [Google Scholar] [CrossRef] [PubMed]
- Roy, P. Orbiviruses and their replication. In Fields’ Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA; New York, NY, USA, 2007; Volume 2, pp. 1975–1997. [Google Scholar]
- Ratinier, M.; Caporale, M.; Golder, M.; Franzoni, G.; Allan, K.; Nunes, S.F.; Armezzani, A.; Bayoumy, A.; Rixon, F.; Shaw, A.; et al. Identification and characterization of a novel non-structural protein of bluetongue virus. PLoS Pathog. 2011, 7, e1002477. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, A.D.; Gould, A.R.; Coupar, B.; Eaton, B.T. Localization of the non-structural protein NS3 in bluetongue virus-infected cells. J. Gen. Virol. 1991, 72, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, S.Y.; Iwata, H.; Compans, R.W.; Roy, P. Multiple glycoproteins synthesized by the smallest RNA segment (S10) of bluetongue virus. J. Virol. 1992, 66, 7104–7112. [Google Scholar] [PubMed]
- Celma, C.C.; Roy, P. Interaction of calpactin light chain (S100A10/p11) and a viral NS protein is essential for intracellular trafficking of nonenveloped bluetongue virus. J. Virol. 2011, 85, 4783–4791. [Google Scholar] [CrossRef] [PubMed]
- Bansal, O.B.; Stokes, A.; Bansal, A.; Bishop, D.H.L.; Roy, P. Membrane organization of bluetongue virus non-structural glycoprotein NS3. J. Virol. 1998, 72, 3362–3369. [Google Scholar] [PubMed]
- Beaton, A.R.; Rodriguez, J.; Reddy, Y.K.; Roy, P. The membrane trafficking protein calpactin forms a complex with bluetongue virus protein NS3 and mediates virus release. Proc. Natl. Acad. Sci. USA 2002, 99, 13154–13159. [Google Scholar] [CrossRef] [PubMed]
- Wirblich, C.; Bhattacharya, B.; Roy, P. Nonstructural protein 3 of bluetongue virus assists virus release by recruiting ESCRT-I protein Tsg101. J. Virol. 2006, 80, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Celma, C.C.; Bhattacharya, B.; Eschbaumer, M.; Wernike, K.; Beer, M.; Roy, P. Pathogenicity study in sheep using reverse-genetics-based reassortant bluetongue viruses. Vet. Microbiol. 2014, 174, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Modrof, J.; Lymperopoulos, K.; Roy, P. Phosphorylation of bluetongue virus nonstructural Protein 2 Is Essential for Formation of Viral Inclusion Bodies. J. Virol. 2005, 79, 10023–10031. [Google Scholar] [CrossRef] [PubMed]
- Forzan, M.; Marsh, M.; Roy, P. Bluetongue virus entry into the cells. J. Virol. 2007, 81, 4819–4827. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.P.; Costa, G.L.; Schoelttin, W.; Cline, J.; Mathur, E.; Bauer, J.C. Site directed mutagenesis of double stranded DNA by the polymerase chain reaction. Gene 1994, 151, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Celma, C.C.; Roy, P. Development of reverse genetics systems for bluetongue virus: Recovery of infectious virus from synthetic RNA transcripts. J. Virol. 2008, 82, 8339–8348. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, E.; Roy, P. Minimum requirements for bluetongue virus primary replication in vivo. J. Virol. 2013, 87, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Roy, P. Bluetongue virus outer capsid protein VP5 interacts with membrane lipid rafts via a SNARE domain. J. Virol. 2008, 82, 10600–10612. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Koike, M.; Moriishi, E.; Kawabata, A.; Tang, H.; Oyaizu, H.; Uchiyama, Y.; Yamanishi, K. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 2008, 9, 1728–1742. [Google Scholar] [CrossRef] [PubMed]
- Calistri, A.; Salata, C.; Parolin, C.; Palù, G. Role of multivesicular bodies and their components in the egress of enveloped RNA viruses. Rev. Med. Virol. 2009, 19, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Bhattacharya, B.; Ward, T.H.; Roy, P. Trafficking of bluetongue virus visualized by recovery of tetracysteine-tagged virion particles. J. Virol. 2014, 88, 12656–12668. [Google Scholar] [CrossRef] [PubMed]
- Huismans, H.; van Dijk, A.A.; Els, H.J. Uncoating of parental bluetongue virus to core and subcore particles in infected L cells. Virology 1987, 157, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S. Ubiquitin: Same molecule, different degradation pathways. Cell 2010, 143, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Mimnaugh, E.G.; Chen, H.Y.; Davie, J.R.; Celis, J.E.; Neckers, L. Rapid deubiquitination of nucleosomal histones in human tumor cells caused by proteasome inhibitors and stress response inducers: Effects on replication, transcription, translation, and the cellular stress response. Biochemistry 1997, 36, 14418–14429. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Fujimoto, T.; Otoguro, K.; Matsuzaki, K.; Moriguchi, R.; Tanaka, H.; Sasaki, Y. Lactacystin, a novel microbial metabolite, induces neuritogenesis of neuroblastoma cells. J. Antibiot. (Tokyo) 1991, 44, 113–116. [Google Scholar] [CrossRef]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.K.; Bhattacharya, B.; Roy, P. Bluetongue virus RNA binding protein NS2 is a modulator of viral replication and assembly. BMC Mol. Biol. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- López, T.; Silva-Ayala, D.; López, S.; Arias, C.F. Replication of the rotavirus genome requires an active ubiquitin-proteasome system. J. Virol. 2011, 85, 11964–11971. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [PubMed]
- Pelchen-Matthews, A.; Raposo, G.; Marsh, M. Endosomes, exosomes and Trojan viruses. Trends Microbiol. 2004, 12, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Estes, M.K. Viroporin-mediated calcium-activated autophagy. Autophagy 2013, 9, 797–798. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harty, R.N.; Brown, M.E.; McGettigan, J.P.; Wang, G.; Jayakar, H.R.; Huibregtse, J.M.; Whitt, M.A.; Schnell, M.J. Rhabdoviruses and the cellular ubiquitin-proteasome system: A budding interaction. J. Virol. 2001, 75, 10623–10629. [Google Scholar] [CrossRef] [PubMed]
- Segura-Morales, C.; Pescia, C.; Chatellard-Causse, C.; Sadoul, R.; Bertrand, E.; Basyuk, E. Tsg101 and Alix interact with murine leukemia virus Gag and cooperate with Nedd4 ubiquitin ligases during budding. J. Biol. Chem. 2005, 280, 27004–27012. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Lu, J.; Liu, Y.; Davis, B.; Lee, M.S.; Olson, M.A.; Ruthel, G.; Freedman, B.D.; Schnell, M.J.; Wrobel, J.E.; et al. Small-molecule probes targeting the viral PPxY-host Nedd4 interface block egress of a broad range of RNA viruses. J. Virol. 2014, 88, 7294–7306. [Google Scholar] [CrossRef] [PubMed]
- Stoorvogel, W.; Kleijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.L.; Urbe, S. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 2007, 8, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteomics 2010, 73, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Staubach, S.; Razawi, H.; Hanisch, F.G. Proteomics of MUC1-containing lipid rafts from plasma membranes and exosomes of human breast carcinoma cells MCF-7. Proteomics 2009, 9, 2820–2835. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; de Milito, A.; Lugini, L.; Borghi, M.; Calabrò, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE 2009, 4, e5219. [Google Scholar] [CrossRef]
- Barreto, A.; Rodríguez, L.S.; Rojas, O.L.; Wolf, M.; Greenberg, H.B.; Franco, M.A.; Angel, J. Membrane vesicles released by intestinal epithelial cells infected with rotavirus inhibit T-cell function. Viral Immunol. 2010, 23, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Spehner, D.; Drillien, R. Extracellular vesicles containing virus-encoded membrane proteins are a byproduct of infection with modified vaccinia virus Ankara. Virus Res. 2008, 137, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.D.; Maier, C.L.; Pober, J.S. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J. Immunol. 2009, 182, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Masciopinto, F.; Giovani, C.; Campagnoli, S.; Galli-Stampino, L.; Colombatto, P.; Brunetto, M.; Yen, T.S.; Houghton, M.; Pileri, P.; Abrignani, S. Association of hepatitis C virus envelope proteins with exosomes. Eur. J. Immunol. 2004, 34, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, G.; Babst, M.; Emr, S.D. Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem. Sci. 2000, 25, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.K.; Overduin, M.; Emr, S.D. Location, location, location: Membrane targeting directed by PX domains. Science 2001, 294, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, G.; Babst, M.; Emr, S.D. Fab1p PtdIns3P 5-kinase function essential for protein sorting in the multivesicular body. Cell 1998, 95, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Schubert, H.L.; Kelly, B.N.; Hil, G.C.; Holton, J.M.; Hill, C.P. Ubiquitin recognition by the human TSG101 protein. Mol. Cell 2004, 13, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Perez-Caballero, D.; Bieniasz, P.D. Context-dependent effects of L domains and ubiquitination on viral budding. J. Virol. 2004, 78, 5554–5563. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Eastman, S.W.; Chung, W.; Bieniasz, P.D. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J. Cell Biol. 2005, 168, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Weissman, A.M. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol. 1998, 14, 19–57. [Google Scholar] [CrossRef] [PubMed]
- Rocca, A.; Lamaze, C.; Subtil, A.; Dautry-Varsat, A. Involvement of the ubiquitin/proteasome system in sorting of the interleukin 2 receptor beta chain to late endocytic compartments. Mol. Biol. Cell 2001, 12, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Van Kerkhof, P.; Alves dos Santos, C.M.; Sachse, M.; Klumperman, J.; Bu, G.; Strous, G.J. Proteasome inhibitors block a late step in lysosomal transport of selected membrane but not soluble proteins. Mol. Biol. Cell 2001, 12, 2556–2566. [Google Scholar] [CrossRef] [PubMed]
- Longva, K.E.; Blystad, F.D.; Stang, E.; Larsen, A.M.; Johannessen, L.E.; Madshus, I.H. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol. 2002, 156, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Contin, R.; Arnoldi, F.; Mano, M.; Burrone, O.R. Rotavirus replication requires a functional proteasome for effective assembly of viroplasms. J. Virol. 2011, 85, 2781–2792. [Google Scholar] [CrossRef] [PubMed]
- Zhadina, M.; McClure, M.O.; Johnson, M.C.; Bieniasz, P.D. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. USA 2007, 104, 20031–20036. [Google Scholar] [CrossRef] [PubMed]
- Watson, H.; Bonifacino, J.S. Direct binding to Rsp5p regulates ubiquitination-independent vacuolar transport of Sna3p. Mol. Biol. Cell 2007, 18, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, A.J.; Aboian, M.; Lee, J.; Azmi, I.; Payne, J.; Issaka, R.; Davies, B.A.; Katzmann, D.J. Characterization of multiple multivesicular body sorting determinants within Sna3: A role for the ubiquitin ligase Rsp5. Mol. Biol. Cell 2007, 18, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Blot, V.; Perugi, F.; Gay, B.; Prevost, M.C.; Briant, L.; Tangy, F.; Abriel, H.; Staub, O.; Dokhelar, M.C.; Pique, C. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J. Cell Sci. 2004, 117, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; McKittrick, I.; West, M.; Odorizzi, G. Direct binding to Rsp5 mediates ubiquitin-independent sorting of Sna3 via the multivesicular body pathway. Mol. Biol. Cell 2007, 18, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Harty, R.N.; Brown, M.E.; Wang, G.; Huibregtse, J.; Hayes, F.P. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: Implications for filovirus budding. Proc. Natl. Acad. Sci. USA 2000, 97, 13871–13876. [Google Scholar] [CrossRef] [PubMed]
- Kikonyogo, A.; Bouamr, F.; Vana, M.L.; Xiang, Y.; Aiyar, A.; Carter, C.; Leis, J. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. USA 2001, 98, 11199–11204. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, J.; Hunter, E.; Nakao, M.; Shida, H. Functional involvement of a novel Nedd4-like ubiquitin ligase on retrovirus budding. EMBO. Rep. 2002, 3, 636–640. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, B.; Celma, C.C.; Roy, P. Influence of Cellular Trafficking Pathway on Bluetongue Virus Infection in Ovine Cells. Viruses 2015, 7, 2378-2403. https://doi.org/10.3390/v7052378
Bhattacharya B, Celma CC, Roy P. Influence of Cellular Trafficking Pathway on Bluetongue Virus Infection in Ovine Cells. Viruses. 2015; 7(5):2378-2403. https://doi.org/10.3390/v7052378
Chicago/Turabian StyleBhattacharya, Bishnupriya, Cristina C. Celma, and Polly Roy. 2015. "Influence of Cellular Trafficking Pathway on Bluetongue Virus Infection in Ovine Cells" Viruses 7, no. 5: 2378-2403. https://doi.org/10.3390/v7052378
APA StyleBhattacharya, B., Celma, C. C., & Roy, P. (2015). Influence of Cellular Trafficking Pathway on Bluetongue Virus Infection in Ovine Cells. Viruses, 7(5), 2378-2403. https://doi.org/10.3390/v7052378