
Roles of HTLV-1 basic Zip Factor (HBZ) in Viral Chronicity and Leukemic Transformation. Potential New Therapeutic Approaches to Prevent and Treat HTLV-1-Related Diseases

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HTLV-1 Infectivity and Spread in vivo
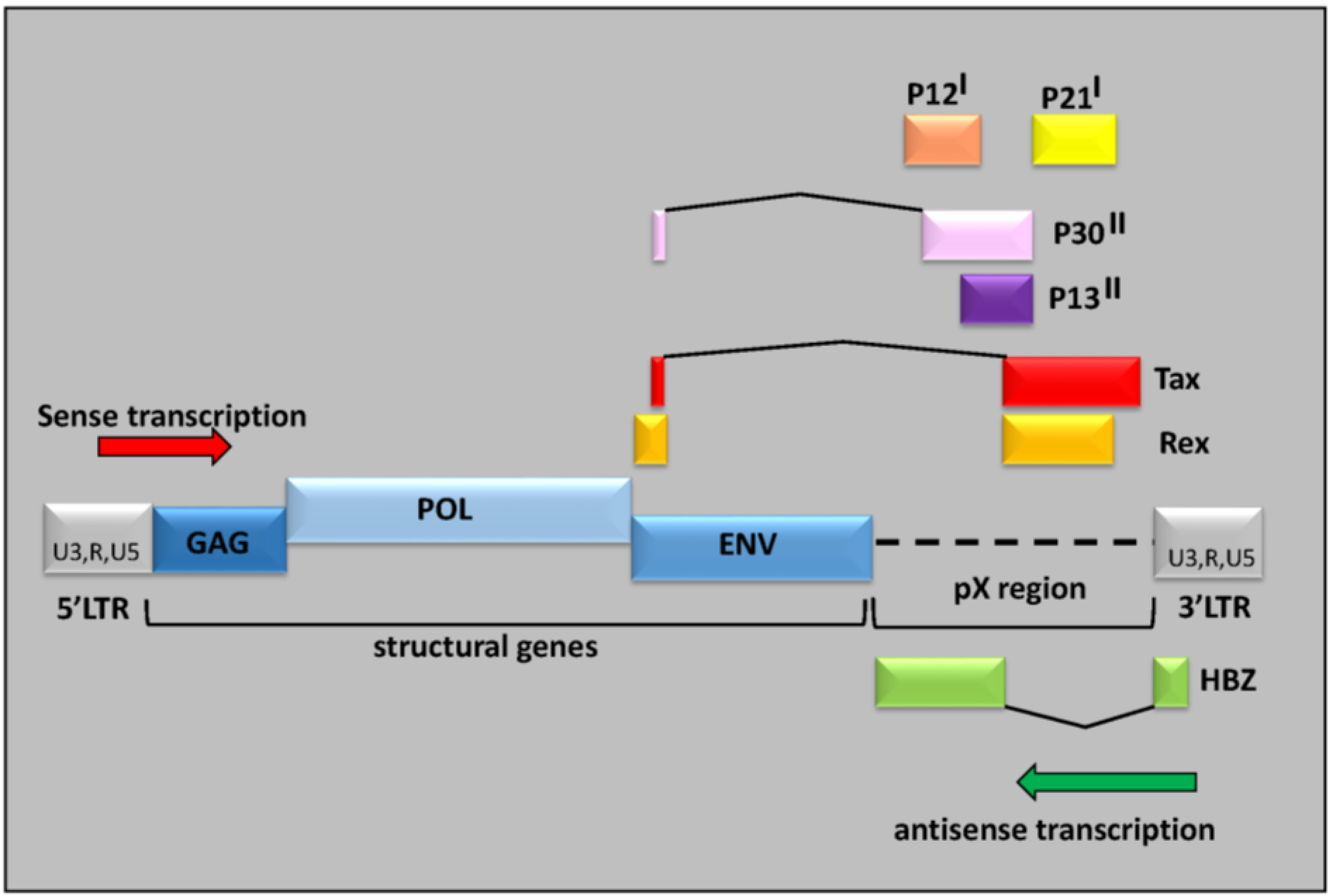
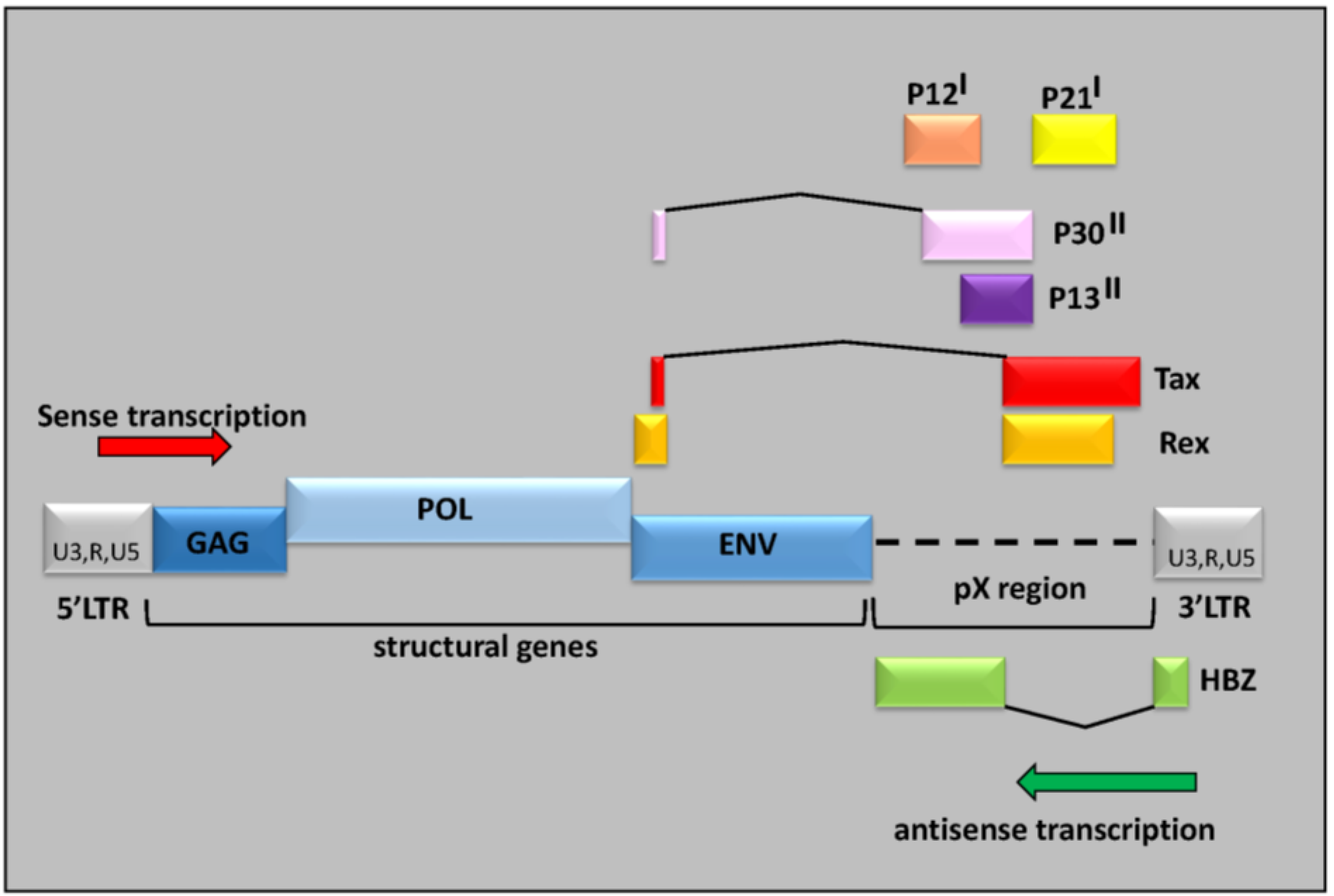
3. HTLV-1, Chronicity and Host Immune Response
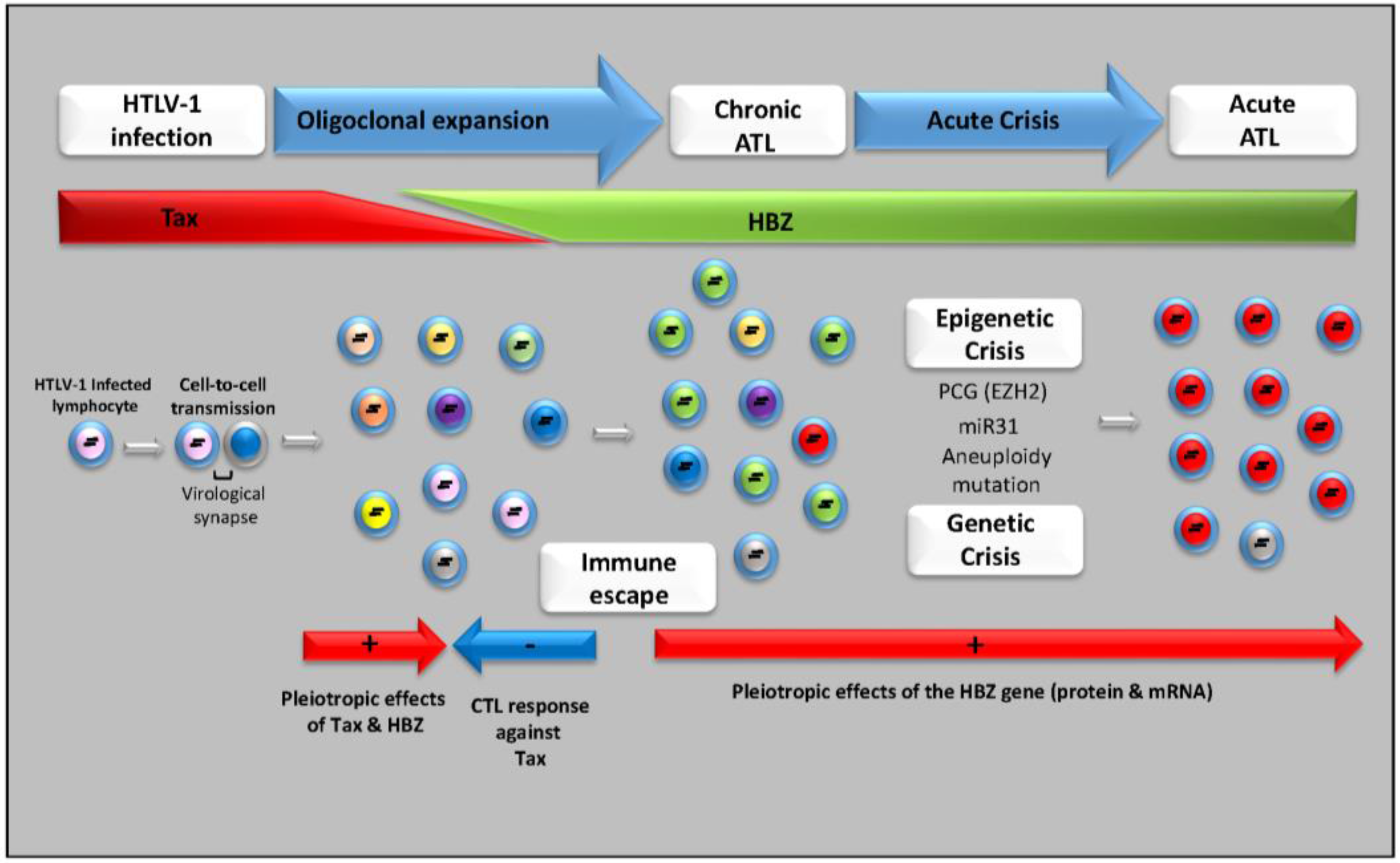
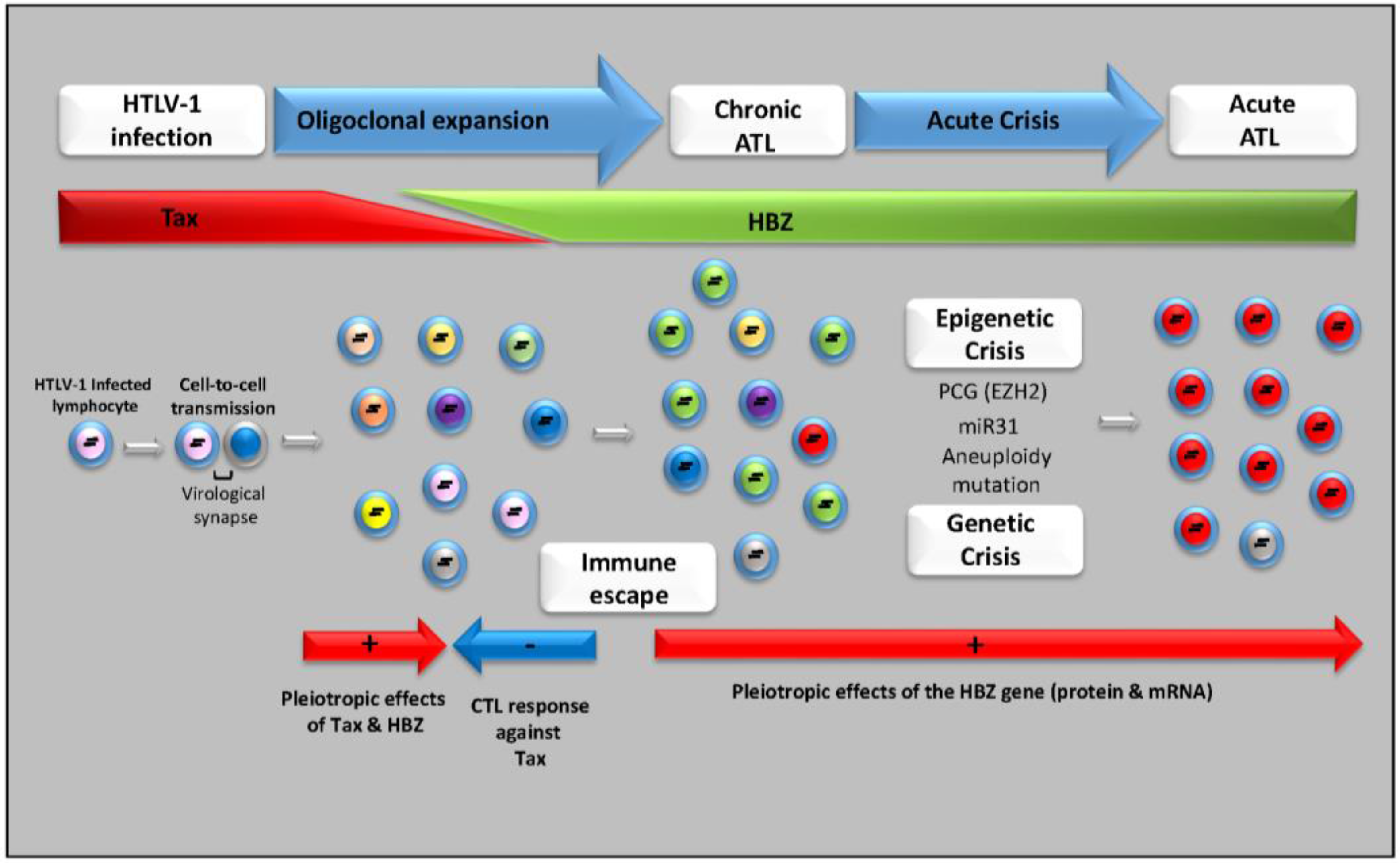
4. Multifaceted Processes in the Transformation of Infected Cells
5. HTLV-1 Antisense HBZ Transcripts and Viral Pathogenesis
6. Therapeutic Approaches for ATL
6.1. Chemotherapy
6.2. Molecular Targeted Therapy
6.3. Immunotherapy
6.4. Stem Cell Transplantation
7. Perspectives and Conclusions
Author Contributions
Conflicts of Interest
References
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef] [PubMed]
- Popovic, M.; Reitz, M.S., Jr.; Sarngadharan, M.G.; Robert-Guroff, M.; Kalyanaraman, V.S.; Nakao, Y.; Miyoshi, I.; Minowada, J.; Yoshida, M.; Ito, Y.; et al. The virus of Japanese adult T-cell leukaemia is a member of the human T-cell leukaemia virus group. Nature 1982, 300, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 1982, 79, 2031–2035. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de Thé, G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef]
- Osame, M.; Igata, A. The history of discovery and clinico-epidemiology of HTLV-I-associated myelopathy(HAM). Jpn. J. Med. 1989, 28, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, B.; Mesnard, J.M. Making Sense out of Antisense Transcription in Human T-Cell Lymphotropic Viruses (HTLVs). Viruses 2011, 3, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Gaudray, G.; Gachon, F.; Basbous, J.; Biard-Piechaczyk, M.; Devaux, C.; Mesnard, J.M. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J. Virol. 2002, 76, 12813–12822. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Yasunaga, J.; Yoshida, M.; Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, M.H.; Landry, S.; Audet, B.; Arpin-Andre, C.; Hivin, P.; Pare, M.E.; Thete, J.; Wattel, E.; Marriott, S.J.; Mesnard, J.M.; et al. HTLV-I antisense transcripts initiating in the 3′LTR are alternatively spliced and polyadenylated. Retrovirology 2006, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Green, P.L. The HBZ gene, a key player in HTLV-1 pathogenesis. Retrovirology 2009, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.S.; Petrow-Sadowski, C.; Huang, Y.K.; Bertolette, D.C.; Ruscetti, F.W. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat. Med. 2008, 14, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Kim, F.J.; Kinet, S.; Taylor, N.; Sitbon, M.; Battini, J.L. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 2003, 115, 449–459. [Google Scholar] [CrossRef]
- Wielgosz, M.M.; Rauch, D.A.; Jones, K.S.; Ruscetti, F.W.; Ratner, L. Cholesterol dependence of HTLV-I infection. AIDS Res. Hum. Retrovir. 2005, 21, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Bouttier, M.; Vassy, R.; Seigneuret, M.; Petrow-Sadowski, C.; Janvier, S.; Heveker, N.; Ruscetti, F.W.; Perret, G.; Jones, K.S.; et al. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood 2009, 113, 5176–5185. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, J.; Matsuoka, M. Molecular mechanisms of HTLV-1 infection and pathogenesis. Int. J. Hematol. 2011, 94, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.S.; Fugo, K.; Petrow-Sadowski, C.; Huang, Y.; Bertolette, D.C.; Lisinski, I.; Cushman, S.W.; Jacobson, S.; Ruscetti, F.W. Human T-cell leukemia virus type 1 (HTLV-1) and HTLV-2 use different receptor complexes to enter T cells. J. Virol. 2006, 80, 8291–8302. [Google Scholar] [CrossRef] [PubMed]
- Zane, L.; Sibon, D.; Legras, C.; Lachuer, J.; Wierinckx, A.; Mehlen, P.; Delfau-Larue, M.H.; Gessain, A.; Gout, O.; Pinatel, C.; et al. Clonal expansion of HTLV-1 positive CD8+ cells relies on cIAP-2 but not on c-FLIP expression. Virology 2010, 407, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Nejmeddine, M.; Negi, V.S.; Mukherjee, S.; Tanaka, Y.; Orth, K.; Taylor, G.P.; Bangham, C.R. HTLV-1-Tax and ICAM-1 act on T-cell signal pathways to polarize the microtubule-organizing center at the virological synapse. Blood 2009, 114, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Pais-Correia, A.M.; Sachse, M.; Guadagnini, S.; Robbiati, V.; Lasserre, R.; Gessain, A.; Gout, O.; Alcover, A.; Thoulouze, M.I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2010, 16, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Majorovits, E.; Nejmeddine, M.; Tanaka, Y.; Taylor, G.P.; Fuller, S.D.; Bangham, C.R. Human T-lymphotropic virus-1 visualized at the virological synapse by electron tomography. PLoS ONE 2008, 3, e2251. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Lloyd, P.; Derse, D. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 2010, 6, e1000788. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.P.; Goon, P.; Furukawa, Y.; Green, H.; Barfield, A.; Mosley, A.; Nose, H.; Babiker, A.; Rudge, P.; Usuku, K.; et al. Zidovudine plus lamivudine in Human T-Lymphotropic Virus type-I-associated myelopathy: A randomised trial. Retrovirology 2006, 3, 63. [Google Scholar] [CrossRef] [PubMed]
- Cavrois, M.; Leclercq, I.; Gout, O.; Gessain, A.; Wain-Hobson, S.; Wattel, E. Persistent oligoclonal expansion of human T-cell leukemia virus type 1-infected circulating cells in patients with Tropical spastic paraparesis/HTLV-1 associated myelopathy. Oncogene 1998, 17, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Etoh, K.; Tamiya, S.; Yamaguchi, K.; Okayama, A.; Tsubouchi, H.; Ideta, T.; Mueller, N.; Takatsuki, K.; Matsuoka, M. Persistent clonal proliferation of human T-lymphotropic virus type I-infected cells in vivo. Cancer Res. 1997, 57, 4862–4867. [Google Scholar] [PubMed]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.M.; Nicot, C.; Fullen, J.; Ciminale, V.; Casareto, L.; Mulloy, J.C.; Jacobson, S.; Franchini, G. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J. Virol. 2001, 75, 6086–6094. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.; Shida, H.; McFarlin, D.E.; Fauci, A.S.; Koenig, S. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV-I pX in patients with HTLV-I associated neurological disease. Nature 1990, 348, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kannagi, M.; Harada, S.; Maruyama, I.; Inoko, H.; Igarashi, H.; Kuwashima, G.; Sato, S.; Morita, M.; Kidokoro, M.; Sugimoto, M.; et al. Predominant recognition of human T cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-I-infected cells. Int. Immunol. 1991, 3, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Goon, P.K.; Biancardi, A.; Fast, N.; Igakura, T.; Hanon, E.; Mosley, A.J.; Asquith, B.; Gould, K.G.; Marshall, S.; Taylor, G.P.; et al. Human T cell lymphotropic virus (HTLV) type-1-specific CD8+ T cells: Frequency and immunodominance hierarchy. J. Infect. Dis. 2004, 189, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Nagasato, K.; Nakamura, T.; Shirabe, S.; Shibayama, K.; Ohishi, K.; Ichinose, K.; Tsujihata, M.; Nagataki, S. Presence of serum anti-human T-lymphotropic virus type I (HTLV-I) IgM antibodies means persistent active replication of HTLV-I in HTLV-I-associated myelopathy. J. Neurol. Sci. 1991, 103, 203–208. [Google Scholar] [CrossRef]
- Kira, J.; Nakamura, M.; Sawada, T.; Koyanagi, Y.; Ohori, N.; Itoyama, Y.; Yamamoto, N.; Sakaki, Y.; Goto, I. Antibody titers to HTLV-I-p40tax protein and gag-env hybrid protein in HTLV-I-associated myelopathy/tropical spastic paraparesis: Correlation with increased HTLV-I proviral DNA load. J. Neurol. Sci. 1992, 107, 98–104. [Google Scholar] [CrossRef]
- Ishihara, S.; Okayama, A.; Stuver, S.; Horinouchi, H.; Shioiri, S.; Murai, K.; Kubota, T.; Yamashita, R.; Tachibana, N.; Tsubouchi, H.; et al. Association of HTLV-I antibody profile of asymptomatic carriers with proviral DNA levels of peripheral blood mononuclear cells. J. Acquir. Immune Defic. Syndr. 1994, 7, 199–203. [Google Scholar] [PubMed]
- Hanon, E.; Hall, S.; Taylor, G.P.; Saito, M.; Davis, R.; Tanaka, Y.; Usuku, K.; Osame, M.; Weber, J.N.; Bangham, C.R. Abundant tax protein expression in CD4+ T cells infected with human T-cell lymphotropic virus type I (HTLV-I) is prevented by cytotoxic T lymphocytes. Blood 2000, 95, 1386–1392. [Google Scholar] [PubMed]
- Lezin, A.; Gillet, N.; Olindo, S.; Signate, A.; Grandvaux, N.; Verlaeten, O.; Belrose, G.; de Carvalho Bittencourt, M.; Hiscott, J.; Asquith, B.; et al. Histone deacetylase mediated transcriptional activation reduces proviral loads in HTLV-1 associated myelopathy/tropical spastic paraparesis patients. Blood 2007, 110, 3722–3728. [Google Scholar] [CrossRef] [PubMed]
- Lezin, A.; Olindo, S.; Belrose, G.; Signate, A.; Cesaire, R.; Smadja, D.; Macallan, D.; Asquith, B.; Bangham, C.; Bouzar, A.; et al. Gene activation therapy: From the BLV model to HAM/TSP patients. Front. Biosci. (Schol. Ed.) 2009, 1, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Belrose, G.; Gross, A.; Olindo, S.; Lezin, A.; Dueymes, M.; Komla-Soukha, I.; Smadja, D.; Tanaka, Y.; Willems, L.; Mesnard, J.M.; et al. Effects of valproate on Tax and HBZ expression in HTLV-1 and HAM/TSP T lymphocytes. Blood 2011, 118, 2483–2491. [Google Scholar] [CrossRef] [PubMed]
- Afonso, P.V.; Mekaouche, M.; Mortreux, F.; Toulza, F.; Moriceau, A.; Wattel, E.; Gessain, A.; Bangham, C.R.; Dubreuil, G.; Plumelle, Y.; et al. Highly active antiretroviral treatment against STLV-1 infection combining reverse transcriptase and HDAC inhibitors. Blood 2010, 116, 3802–3808. [Google Scholar] [CrossRef] [PubMed]
- Asquith, B.; Mosley, A.J.; Heaps, A.; Tanaka, Y.; Taylor, G.P.; McLean, A.R.; Bangham, C.R. Quantification of the virus-host interaction in human T lymphotropic virus I infection. Retrovirology 2005, 2, 75. [Google Scholar] [CrossRef] [PubMed]
- Hilburn, S.; Rowan, A.; Demontis, M.A.; MacNamara, A.; Asquith, B.; Bangham, C.R.; Taylor, G.P. In vivo expression of human T-lymphotropic virus type 1 basic leucine-zipper protein generates specific CD8+ and CD4+ T-lymphocyte responses that correlate with clinical outcome. J. Infect. Dis. 2011, 203, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Macnamara, A.; Rowan, A.; Hilburn, S.; Kadolsky, U.; Fujiwara, H.; Suemori, K.; Yasukawa, M.; Taylor, G.; Bangham, C.R.; Asquith, B. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. 2010, 6, e1001117. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, M.; Inoue, J.; Yoshida, M.; Seiki, M. Post-transcriptional regulator (rex) of HTLV-1 initiates expression of viral structural proteins but suppresses expression of regulatory proteins. EMBO J. 1988, 7, 519–523. [Google Scholar] [PubMed]
- Nicot, C.; Dundr, M.; Johnson, J.M.; Fullen, J.R.; Alonzo, N.; Fukumoto, R.; Princler, G.L.; Derse, D.; Misteli, T.; Franchini, G. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat. Med. 2004, 10, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Lemasson, I.; Polakowski, N.J.; Laybourn, P.J.; Nyborg, J.K. Transcription regulatory complexes bind the human T-cell leukemia virus 5′ and 3′ long terminal repeats to control gene expression. Mol. Cell. Biol. 2004, 24, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Gitlin, S.D.; Browning, C.M.; Lane, B.R.; Clark, N.M.; Shah, N.; Rainier, S.; Markovitz, D.M. GLI-2 modulates retroviral gene expression. J. Virol. 2001, 75, 2301–2313. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kubota, R.; Tara, M.; Izumo, S.; Osame, M. Existence of escape mutant in HTLV-I tax during the development of adult T-cell leukemia. Blood 2001, 97, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Ohashi, T.; Nishikawa, K.; Nishitsuji, H.; Kurihara, K.; Hasegawa, A.; Furuta, R.A.; Fujisawa, J.; Tanaka, Y.; Hanabuchi, S.; et al. Repression of tax expression is associated both with resistance of human T-cell leukemia virus type 1-infected T cells to killing by tax-specific cytotoxic T lymphocytes and with impaired tumorigenicity in a rat model. J. Virol. 2004, 78, 3827–3836. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, R.; Aboud, M.; Jeang, K.T. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene 2005, 24, 5976–5985. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Fujii, M. Distinct functions of HTLV-1 Tax1 from HTLV-2 Tax2 contribute key roles to viral pathogenesis. Retrovirology 2009, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Basbous, J.; Bazarbachi, A.; Granier, C.; Devaux, C.; Mesnard, J.M. The central region of human T-cell leukemia virus type 1 Tax protein contains distinct domains involved in subunit dimerization. J. Virol. 2003, 77, 13028–13035. [Google Scholar] [CrossRef] [PubMed]
- Peloponese, J.M., Jr.; Iha, H.; Yedavalli, V.R.; Miyazato, A.; Li, Y.; Haller, K.; Benkirane, M.; Jeang, K.T. Ubiquitination of human T-cell leukemia virus type 1 tax modulates its activity. J. Virol. 2004, 78, 11686–11695. [Google Scholar] [CrossRef] [PubMed]
- Peloponese, J.M., Jr.; Yasunaga, J.; Kinjo, T.; Watashi, K.; Jeang, K.T. Peptidylproline cis-trans-isomerase Pin1 interacts with human T-cell leukemia virus type 1 tax and modulates its activation of NF-kappaB. J. Virol. 2009, 83, 3238–3248. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.S.; Ward, M.D.; Fryrear, K.A.; Semmes, O.J. Site-specific phosphorylation differentiates active from inactive forms of the human T-cell leukemia virus type 1 Tax oncoprotein. J. Biol. Chem. 2006, 281, 31705–31712. [Google Scholar] [CrossRef] [PubMed]
- Chiari, E.; Lamsoul, I.; Lodewick, J.; Chopin, C.; Bex, F.; Pique, C. Stable ubiquitination of human T-cell leukemia virus type 1 tax is required for proteasome binding. J. Virol. 2004, 78, 11823–11832. [Google Scholar] [CrossRef] [PubMed]
- Lodewick, J.; Lamsoul, I.; Polania, A.; Lebrun, S.; Burny, A.; Ratner, L.; Bex, F. Acetylation of the human T-cell leukemia virus type 1 Tax oncoprotein by p300 promotes activation of the NF-kappaB pathway. Virology 2009, 386, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Ryo, A.; Yamamoto, N. The prolyl isomerase Pin1 stabilizes the human T-cell leukemia virus type 1 (HTLV-1) Tax oncoprotein and promotes malignant transformation. Biochem. Biophys. Res. Commun. 2009, 381, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Marriott, S.J.; Semmes, O.J. Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 2005, 24, 5986–5995. [Google Scholar] [CrossRef] [PubMed]
- Rosin, O.; Koch, C.; Schmitt, I.; Semmes, O.J.; Jeang, K.T.; Grassmann, R. A human T-cell leukemia virus Tax variant incapable of activating NF-kappaB retains its immortalizing potential for primary T-lymphocytes. J. Biol. Chem. 1998, 273, 6698–6703. [Google Scholar] [CrossRef] [PubMed]
- Robek, M.D.; Ratner, L. Immortalization of CD4(+) and CD8(+) T lymphocytes by human T-cell leukemia virus type 1 Tax mutants expressed in a functional molecular clone. J. Virol. 1999, 73, 4856–4865. [Google Scholar] [PubMed]
- Grossman, W.J.; Kimata, J.T.; Wong, F.H.; Zutter, M.; Ley, T.J.; Ratner, L. Development of leukemia in mice transgenic for the tax gene of human T-cell leukemia virus type I. Proc. Natl. Acad. Sci. USA 1995, 92, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Sawa, H.; Lewis, M.J.; Orba, Y.; Sheehy, N.; Yamamoto, Y.; Ichinohe, T.; Tsunetsugu-Yokota, Y.; Katano, H.; Takahashi, H.; et al. Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat. Med. 2006, 12, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Ohsugi, T.; Kumasaka, T.; Okada, S.; Urano, T. The Tax protein of HTLV-1 promotes oncogenesis in not only immature T cells but also mature T cells. Nat. Med. 2007, 13, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Tamiya, S.; Takemoto, S.; Yamaguchi, K.; Takatsuki, K. HTLV-I provirus in the clinical subtypes of ATL. Leukemia 1997, 11 (Suppl. 3), 67–69. [Google Scholar] [PubMed]
- Tamiya, S.; Matsuoka, M.; Etoh, K.; Watanabe, T.; Kamihira, S.; Yamaguchi, K.; Takatsuki, K. Two types of defective human T-lymphotropic virus type I provirus in adult T-cell leukemia. Blood 1996, 88, 3065–3073. [Google Scholar] [PubMed]
- Miyazaki, M.; Yasunaga, J.; Taniguchi, Y.; Tamiya, S.; Nakahata, T.; Matsuoka, M. Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5' long terminal repeat during oncogenesis. J. Virol. 2007, 81, 5714–5723. [Google Scholar] [CrossRef] [PubMed]
- Haller, K.; Kibler, K.V.; Kasai, T.; Chi, Y.H.; Peloponese, J.M.; Yedavalli, V.S.; Jeang, K.T. The N-terminus of rodent and human MAD1 confers species-specific stringency to spindle assembly checkpoint. Oncogene 2006, 25, 2137–2147. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Munger, K. Mechanisms of genomic instability in human cancer: Insights from studies with human papillomavirus oncoproteins. Int. J. Cancer 2004, 109, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Satou, Y.; Yasunaga, J.; Fujisawa, J.; Matsuoka, M. Transcriptional control of spliced and unspliced human T-cell leukemia virus type 1 bZIP factor (HBZ) gene. J. Virol. 2008, 82, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- Arpin-Andre, C.; Laverdure, S.; Barbeau, B.; Gross, A.; Mesnard, J.M. Construction of a reporter vector for analysis of bidirectional transcriptional activity of retrovirus LTR. Plasmid 2014, 74, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Duc Dodon, M.; Mesnard, J.M.; Barbeau, B. [Adult T-cell leukemia induced by HTLV-1: Before and after HBZ]. Med. Sci. (Paris) 2010, 26, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Vicente, C.; Cools, J. The genomic landscape of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1226–1227. [Google Scholar] [CrossRef] [PubMed]
- Lemasson, I.; Lewis, M.R.; Polakowski, N.; Hivin, P.; Cavanagh, M.H.; Thebault, S.; Barbeau, B.; Nyborg, J.K.; Mesnard, J.M. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J. Virol. 2007, 81, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Basbous, J.; Arpin, C.; Gaudray, G.; Piechaczyk, M.; Devaux, C.; Mesnard, J.M. The HBZ factor of human T-cell leukemia virus type I dimerizes with transcription factors JunB and c-Jun and modulates their transcriptional activity. J. Biol. Chem. 2003, 278, 43620–43627. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Ohshima, T.; Isono, O.; Shimotohno, K. HTLV-1 HBZ suppresses AP-1 activity by impairing both the DNA-binding ability and the stability of c-Jun protein. Oncogene 2005, 24, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Hivin, P.; Basbous, J.; Raymond, F.; Henaff, D.; Arpin-Andre, C.; Robert-Hebmann, V.; Barbeau, B.; Mesnard, J.M. The HBZ-SP1 isoform of human T-cell leukemia virus type I represses JunB activity by sequestration into nuclear bodies. Retrovirology 2007, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Thebault, S.; Basbous, J.; Hivin, P.; Devaux, C.; Mesnard, J.M. HBZ interacts with JunD and stimulates its transcriptional activity. FEBS Lett. 2004, 562, 165–170. [Google Scholar] [CrossRef]
- Zhao, T.; Yasunaga, J.; Satou, Y.; Nakao, M.; Takahashi, M.; Fujii, M.; Matsuoka, M. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-kappaB. Blood 2009, 113, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Peloponese, J.M.; Yeung, M.L.; Jeang, K.T. Modulation of nuclear factor-kappaB by human T cell leukemia virus type 1 Tax protein: Implications for oncogenesis and inflammation. Immunol. Res. 2006, 34, 1–12. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, L.; Lovly, C.M.; Ratner, L. The role of NF-{kappa}B-1 and NF-{kappa}B-2-mediated resistance to apoptosis in lymphomas. Proc. Natl. Acad. Sci. USA 2006, 103, 9220–9225. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.; Zimmerman, B.; Li, M.; Lairmore, M.D.; Green, P.L. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood 2008, 112, 3788–3797. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, A.S.; Villaudy, J.; Gazzolo, L.; Castellazzi, M.; Mesnard, J.M.; Duc Dodon, M. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT). Retrovirology 2007, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, B.; Mesnard, J.M. Does chronic infection in retroviruses have a sense? Trends Microbiol. 2015, 23, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Mortreux, F.; Leclercq, I.; Gabet, A.S.; Leroy, A.; Westhof, E.; Gessain, A.; Wain-Hobson, S.; Wattel, E. Somatic mutation in human T-cell leukemia virus type 1 provirus and flanking cellular sequences during clonal expansion in vivo. J. Natl. Cancer Inst. 2001, 93, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Mortreux, F.; Gabet, A.S.; Wattel, E. Molecular and cellular aspects of HTLV-1 associated leukemogenesis in vivo. Leukemia 2003, 17, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Vernin, C.; Thenoz, M.; Pinatel, C.; Gessain, A.; Gout, O.; Delfau-Larue, M.H.; Nazaret, N.; Legras-Lachuer, C.; Wattel, E.; Mortreux, F. HTLV-1 bZIP factor HBZ promotes cell proliferation and genetic instability by activating OncomiRs. Cancer Res. 2014, 74, 6082–6093. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Yasunaga, J.; Zhao, T.; Yoshida, M.; Miyazato, P.; Takai, K.; Shimizu, K.; Ohshima, K.; Green, P.L.; Ohkura, N.; et al. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011, 7, e1001274. [Google Scholar] [CrossRef] [PubMed]
- Gazon, H.; Lemasson, I.; Polakowski, N.; Cesaire, R.; Matsuoka, M.; Barbeau, B.; Mesnard, J.M.; Peloponese, J.M., Jr. Human T-cell leukemia virus type 1 (HTLV-1) bZIP factor requires cellular transcription factor JunD to upregulate HTLV-1 antisense transcription from the 3′ long terminal repeat. J. Virol. 2012, 86, 9070–9078. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.H.; Edwards, A.J.; Cruickshank, J.K.; Rudge, P.; Dalgleish, A.G. In vivo cellular tropism of human T-cell leukemia virus type 1. J. Virol. 1990, 64, 5682–5687. [Google Scholar] [PubMed]
- Karube, K.; Ohshima, K.; Tsuchiya, T.; Yamaguchi, T.; Kawano, R.; Suzumiya, J.; Utsunomiya, A.; Harada, M.; Kikuchi, M. Expression of FoxP3, a key molecule in CD4CD25 regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br. J. Haematol. 2004, 126, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Gartenhaus, R.; Kuzel, T. HTLV-I associated leukemia/lymphoma: Epidemiology, biology, and treatment. Cancer Treat. Res. 2001, 104, 75–88. [Google Scholar] [PubMed]
- Yamada, Y.; Tomonaga, M. The current status of therapy for adult T-cell leukaemia-lymphoma in Japan. Leuk Lymphoma 2003, 44, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, Y.; Iwanaga, M.; Imaizumi, Y.; Tawara, M.; Joh, T.; Kohno, T.; Yamada, Y.; Kamihira, S.; Ikeda, S.; Miyazaki, Y.; et al. Long-term study of indolent adult T-cell leukemia-lymphoma. Blood 2010, 115, 4337–4343. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.; Hermine, O. Treatment with a combination of zidovudine and alpha-interferon in naive and pretreated adult T-cell leukemia/lymphoma patients. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 13 (Suppl. 1), S186–S190. [Google Scholar] [CrossRef] [PubMed]
- Nasr, R.; el Hajj, H.; Kfoury, Y.; de The, H.; Hermine, O.; Bazarbachi, A. Controversies in targeted therapy of adult T cell leukemia/lymphoma: ON target or OFF target effects? Viruses 2011, 3, 750–769. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.; Suarez, F.; Fields, P.; Hermine, O. How I treat adult T-cell leukemia/lymphoma. Blood 2011, 118, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Ikeda, S.; Murata, K.; Maeda, T.; Atogami, S.; Sohda, H.; Momita, S.; Jubashi, T.; Yamada, Y.; Mine, M.; et al. Characteristics of chemotherapy-induced clinical remission in long survivors with aggressive adult T-cell leukemia/lymphoma. Leuk Res. 1993, 17, 157–166. [Google Scholar] [CrossRef]
- Taguchi, H.; Kinoshita, K.I.; Takatsuki, K.; Tomonaga, M.; Araki, K.; Arima, N.; Ikeda, S.; Uozumi, K.; Kohno, H.; Kawano, F.; et al. An intensive chemotherapy of adult T-cell leukemia/lymphoma: CHOP followed by etoposide, vindesine, ranimustine, and mitoxantrone with granulocyte colony-stimulating factor support. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 12, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Tobinai, K. Current management of adult T-cell leukemia/lymphoma. Oncology (Williston Park) 2009, 23, 1250–1256. [Google Scholar] [PubMed]
- Makiyama, J.; Imaizumi, Y.; Tsushima, H.; Taniguchi, H.; Moriwaki, Y.; Sawayama, Y.; Imanishi, D.; Taguchi, J.; Hata, T.; Tsukasaki, K.; et al. Treatment outcome of elderly patients with aggressive adult T cell leukemia-lymphoma: Nagasaki University Hospital experience. Int. J. Hematol. 2014, 100, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Kawano, N.; Yoshida, S.; Kuriyama, T.; Tahara, Y.; Yamashita, K.; Nagahiro, Y.; Kawano, J.; Koketsu, H.; Toyofuku, A.; Manabe, T.; et al. Clinical Features and Treatment Outcomes of 81 Patients with Aggressive Type Adult T-cell Leukemia-lymphoma at a Single Institution over a 7-year Period (2006–2012). Intern. Med. 2015, 54, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Utsunomiya, A.; Fukuda, H.; Shibata, T.; Fukushima, T.; Takatsuka, Y.; Ikeda, S.; Masuda, M.; Nagoshi, H.; Ueda, R.; et al. VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J. Clin. Oncol. 2007, 25, 5458–5464. [Google Scholar] [CrossRef] [PubMed]
- Mercieca, J.; Matutes, E.; Dearden, C.; MacLennan, K.; Catovsky, D. The role of pentostatin in the treatment of T-cell malignancies: Analysis of response rate in 145 patients according to disease subtype. J. Clin. Oncol. 1994, 12, 2588–2593. [Google Scholar] [PubMed]
- Tsukasaki, K.; Tobinai, K.; Shimoyama, M.; Kozuru, M.; Uike, N.; Yamada, Y.; Tomonaga, M.; Araki, K.; Kasai, M.; Takatsuki, K.; et al. Deoxycoformycin-containing combination chemotherapy for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study (JCOG9109). Int. J. Hematol. 2003, 77, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Uike, N.; Choi, I.; Tokoro, A.; Goto, T.; Yufu, Y.; Kozuru, M.; Tobinai, K. Adult T-cell leukemia-lymphoma successfully treated with 2-chlorodeoxyadenosine. Intern. Med. 1998, 37, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Tobinai, K.; Uike, N.; Saburi, Y.; Chou, T.; Etoh, T.; Masuda, M.; Kawano, F.; Matsuoka, M.; Taguchi, H.; Makino, T.; et al. Phase II study of cladribine (2-chlorodeoxyadenosine) in relapsed or refractory adult T-cell leukemia-lymphoma. Int. J. Hematol. 2003, 77, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; Nakahara, K.; Takatsuka, Y.; Shimotakahara, S.; Utsunomiya, A.; Hanada, S.; Tokunaga, M.; Arima, T. [Successful treatment of chemotherapy-resistant adult T cell leukemia/lymphoma by irinotecan hydrochloride (CPT-11)]. Rinsho Ketsueki 1994, 35, 42–48. [Google Scholar] [PubMed]
- Ohno, R.; Masaoka, T.; Shirakawa, S.; Sakamoto, S.; Hirano, M.; Hanada, S.; Yasunaga, K.; Yokomaku, S.; Mitomo, Y.; Nagai, K.; et al. Treatment of adult T-cell leukemia/lymphoma with MST-16, a new oral antitumor drug and a derivative of bis(2,6-dioxopiperazine). The MST-16 Study Group. Cancer 1993, 71, 2217–2221. [Google Scholar] [CrossRef]
- Taguchi, T.; Ohta, K.; Hotta, T.; Shirakawa, S.; Masaoka, T.; Kimura, I. [Menogaril (TUT-7) late phase II study for malignant lymphoma, adult T-cell leukemia and lymphoma (ATLL)]. Gan To Kagaku Ryoho 1997, 24, 1263–1271. [Google Scholar] [PubMed]
- Tsukasaki, K.; Tomonaga, M. ATRA, NF-kappaB and ATL. Leuk Res. 2001, 25, 407–408. [Google Scholar] [CrossRef]
- Toshima, M.; Nagai, T.; Izumi, T.; Tarumoto, T.; Takatoku, M.; Imagawa, S.; Komatsu, N.; Ozawa, K. All-trans-retinoic acid treatment for chemotherapy-resistant acute adult T-cell leukemia. Int. J. Hematol. 2000, 72, 343–345. [Google Scholar] [PubMed]
- Hasegawa, H.; Yamada, Y.; Tsukasaki, K.; Mori, N.; Tsuruda, K.; Sasaki, D.; Usui, T.; Osaka, A.; Atogami, S.; Ishikawa, C.; et al. LBH589, a deacetylase inhibitor, induces apoptosis in adult T-cell leukemia/lymphoma cells via activation of a novel RAIDD-caspase-2 pathway. Leukemia 2011, 25, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Tobinai, K. Clinical Trials and Treatment of ATL. Leuk Res. Treat. 2012, 2012, 101754. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Matsuda, T.; Tadano, M.; Kinjo, T.; Yamada, Y.; Tsukasaki, K.; Ikeda, S.; Yamasaki, Y.; Tanaka, Y.; Ohta, T.; et al. Apoptosis induced by the histone deacetylase inhibitor FR901228 in human T-cell leukemia virus type 1-infected T-cell lines and primary adult T-cell leukemia cells. J. Virol. 2004, 78, 4582–4590. [Google Scholar] [CrossRef] [PubMed]
- Blaheta, R.A.; Cinatl, J., Jr. Anti-tumor mechanisms of valproate: A novel role for an old drug. Med. Res. Rev. 2002, 22, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Bezecny, P. Histone deacetylase inhibitors in glioblastoma: Pre-clinical and clinical experience. Med. Oncol. 2014, 31, 985. [Google Scholar] [CrossRef] [PubMed]
- Lagneaux, L.; Gillet, N.; Stamatopoulos, B.; Delforge, A.; Dejeneffe, M.; Massy, M.; Meuleman, N.; Kentos, A.; Martiat, P.; Willems, L.; et al. Valproic acid induces apoptosis in chronic lymphocytic leukemia cells through activation of the death receptor pathway and potentiates TRAIL response. Exp. Hematol. 2007, 35, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Belrose, G.; Gazon, H.; Meniane, J.-C.; Olindo, S.; Mesnard, J.-M.; Peloponese, J.-M.; Cesaire, R. Effects of valproate on Tax and HBZ expression in ex vivo cultured ATL cells. Retrovirology 2014, 11 (Suppl. 1), P39. [Google Scholar] [CrossRef]
- Mosley, A.J.; Meekings, K.N.; McCarthy, C.; Shepherd, D.; Cerundolo, V.; Mazitschek, R.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R. Histone deacetylase inhibitors increase virus gene expression but decrease CD8+ cell antiviral function in HTLV-1 infection. Blood 2006, 108, 3801–3807. [Google Scholar] [CrossRef] [PubMed]
- Olindo, S.; Belrose, G.; Gillet, N.; Rodriguez, S.; Boxus, M.; Verlaeten, O.; Asquith, B.; Bangham, C.; Signate, A.; Smadja, D.; et al. Safety of long-term treatment of HAM/TSP patients with valproic acid. Blood 2011, 118, 6306–6309. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, J.L.; Janik, J.E.; Stewart, D.M.; Jaffe, E.S.; Stetler-Stevenson, M.; Shih, J.H.; Fleisher, T.A.; Turner, M.; Urquhart, N.E.; Wharfe, G.H.; et al. Safety, efficacy, and pharmacokinetics/pharmacodynamics of daclizumab (anti-CD25) in patients with adult T-cell leukemia/lymphoma. Clin. Immunol. 2014, 155, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Yoshie, O.; Fujisawa, R.; Nakayama, T.; Harasawa, H.; Tago, H.; Izawa, D.; Hieshima, K.; Tatsumi, Y.; Matsushima, K.; Hasegawa, H.; et al. Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood 2002, 99, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Utsunomiya, A.; Tobinai, K.; Tsukasaki, K.; Uike, N.; Uozumi, K.; Yamaguchi, K.; Yamada, Y.; Hanada, S.; Tamura, K.; et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J. Clin. Oncol. 2010, 28, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K. [Mogamulizumab for the treatment of ATL and PTCL]. Gan To Kagaku Ryoho 2015, 42, 553–557. [Google Scholar] [PubMed]
- Ishida, T.; Jo, T.; Takemoto, S.; Suzushima, H.; Uozumi, K.; Yamamoto, K.; Uike, N.; Saburi, Y.; Nosaka, K.; Utsunomiya, A.; et al. Dose-intensified chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: A randomized phase II study. Br. J. Haematol. 2015, 169, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, I.; Sueki, Y.; Yamamoto, T.; Nozaki, Y.; Nakajima, K.; Mitsumori, T.; Kirito, K. Adult T cell leukemia-lymphoma with allo-HSCT after treatment for pulmonary involvement with Mogamulizumab. Rinsho Ketsueki 2015, 56, 210–215. [Google Scholar] [PubMed]
- Utsunomiya, A.; Miyazaki, Y.; Takatsuka, Y.; Hanada, S.; Uozumi, K.; Yashiki, S.; Tara, M.; Kawano, F.; Saburi, Y.; Kikuchi, H.; et al. Improved outcome of adult T cell leukemia/lymphoma with allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001, 27, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Okamura, J.; Uike, N.; Utsunomiya, A.; Tanosaki, R. Allogeneic stem cell transplantation for adult T-cell leukemia/lymphoma. Int. J. Hematol. 2007, 86, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Tanosaki, R.; Uike, N.; Utsunomiya, A.; Saburi, Y.; Masuda, M.; Tomonaga, M.; Eto, T.; Hidaka, M.; Harada, M.; Choi, I.; et al. Allogeneic hematopoietic stem cell transplantation using reduced-intensity conditioning for adult T cell leukemia/lymphoma: Impact of antithymocyte globulin on clinical outcome. Biol. Blood Marrow Transplant. 2008, 14, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Hishizawa, M.; Kanda, J.; Utsunomiya, A.; Taniguchi, S.; Eto, T.; Moriuchi, Y.; Tanosaki, R.; Kawano, F.; Miyazaki, Y.; Masuda, M.; et al. Transplantation of allogeneic hematopoietic stem cells for adult T-cell leukemia: A nationwide retrospective study. Blood 2010, 116, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Kanda, J.; Chiou, L.W.; Szabolcs, P.; Sempowski, G.D.; Rizzieri, D.A.; Long, G.D.; Sullivan, K.M.; Gasparetto, C.; Chute, J.P.; Morris, A.; et al. Immune recovery in adult patients after myeloablative dual umbilical cord blood, matched sibling, and matched unrelated donor hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2012, 18, 1664.e1–1676.e1. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.; Cwynarski, K.; Boumendil, A.; Finel, H.; Fields, P.; Raj, K.; Nagler, A.; Mohty, M.; Sureda, A.; Dreger, P.; et al. Outcome of patients with HTLV-1-associated adult T-cell leukemia/lymphoma after SCT: A retrospective study by the EBMT LWP. Bone Marrow Transplant. 2014, 49, 1266–1268. [Google Scholar] [CrossRef] [PubMed]
- Umino, A.; Nakagawa, M.; Utsunomiya, A.; Tsukasaki, K.; Taira, N.; Katayama, N.; Seto, M. Clonal evolution of adult T-cell leukemia/lymphoma takes place in the lymph nodes. Blood 2011, 117, 5473–5478. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R.; Cook, L.B.; Melamed, A. HTLV-1 clonality in adult T-cell leukaemia and non-malignant HTLV-1 infection. Semin. Cancer Biol. 2014, 26, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Komatsu, N.; Bandobashi, K.; Taniguchi, A.; Kuwayama, Y.; Togitani, K.; Koeffler, H.P.; Taguchi, H. Histone deacetylase inhibitors induce growth arrest and apoptosis of HTLV-1-infected T-cells via blockade of signaling by nuclear factor kappaB. Leuk Res. 2008, 32, 287–296. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesnard, J.-M.; Barbeau, B.; Césaire, R.; Péloponèse, J.-M. Roles of HTLV-1 basic Zip Factor (HBZ) in Viral Chronicity and Leukemic Transformation. Potential New Therapeutic Approaches to Prevent and Treat HTLV-1-Related Diseases. Viruses 2015, 7, 6490-6505. https://doi.org/10.3390/v7122952
Mesnard J-M, Barbeau B, Césaire R, Péloponèse J-M. Roles of HTLV-1 basic Zip Factor (HBZ) in Viral Chronicity and Leukemic Transformation. Potential New Therapeutic Approaches to Prevent and Treat HTLV-1-Related Diseases. Viruses. 2015; 7(12):6490-6505. https://doi.org/10.3390/v7122952
Chicago/Turabian StyleMesnard, Jean-Michel, Benoit Barbeau, Raymond Césaire, and Jean-Marie Péloponèse. 2015. "Roles of HTLV-1 basic Zip Factor (HBZ) in Viral Chronicity and Leukemic Transformation. Potential New Therapeutic Approaches to Prevent and Treat HTLV-1-Related Diseases" Viruses 7, no. 12: 6490-6505. https://doi.org/10.3390/v7122952
APA StyleMesnard, J.-M., Barbeau, B., Césaire, R., & Péloponèse, J.-M. (2015). Roles of HTLV-1 basic Zip Factor (HBZ) in Viral Chronicity and Leukemic Transformation. Potential New Therapeutic Approaches to Prevent and Treat HTLV-1-Related Diseases. Viruses, 7(12), 6490-6505. https://doi.org/10.3390/v7122952