
Infectious Entry Pathway of Enterovirus B Species


Abstract
:
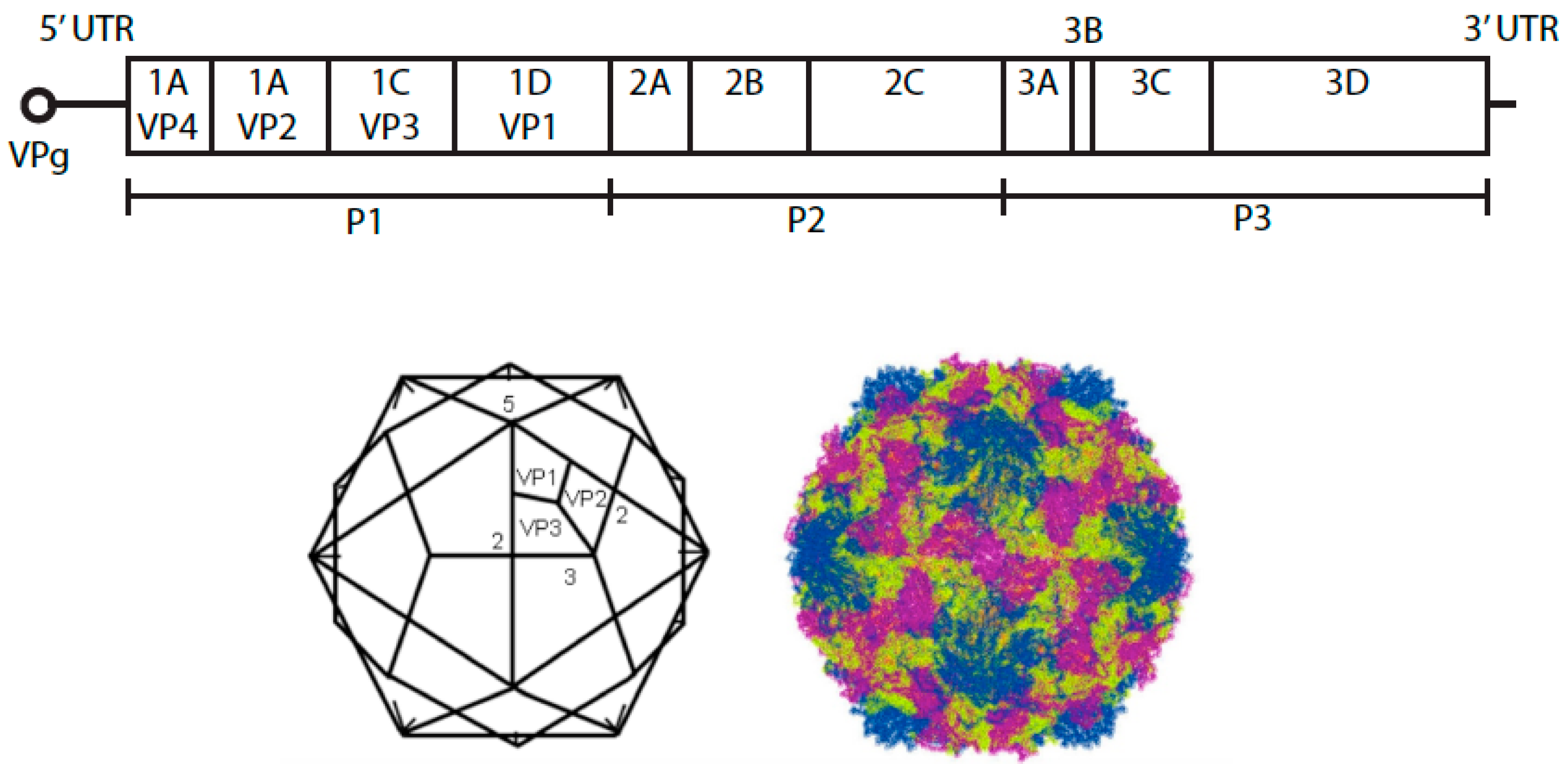
1. Species B Enteroviruses

1.1. Echovirus 1 (E1)
1.2. Coxsackievirus A9 (CVA9)
1.3. Coxsackie B Viruses
Entry of Coxsackie Virus B3
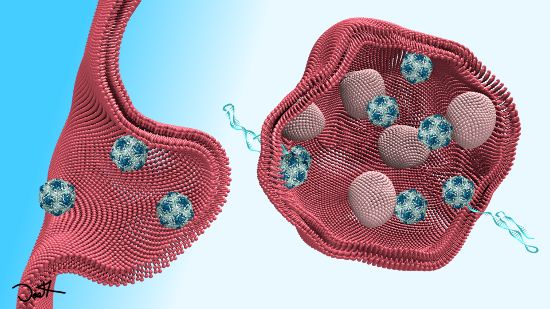
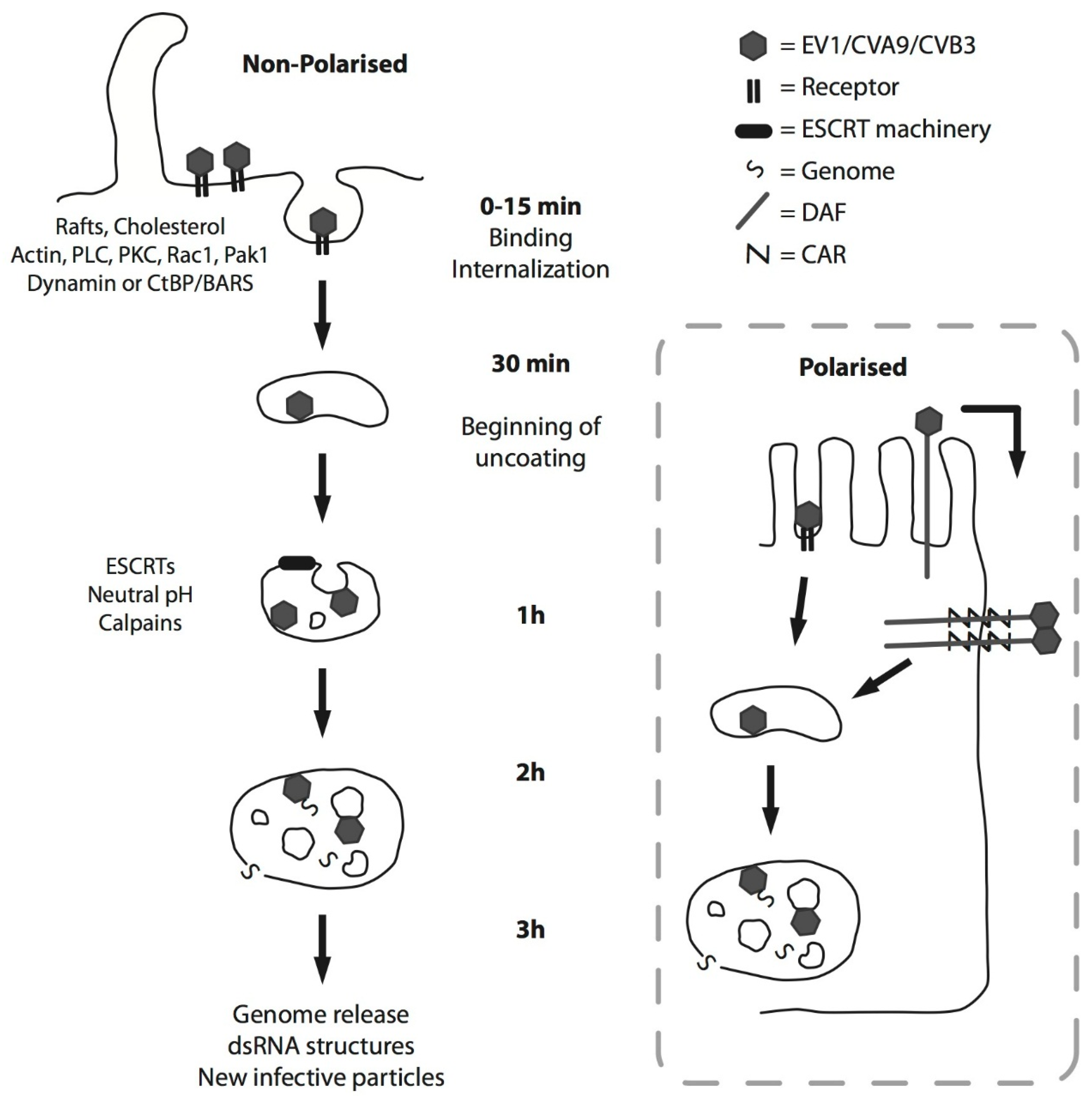
2. EV-B Infectious Entry Pathway

{kind=link}
{kind=link}
{kind=link}
| Target | Treatment | E1 | CVA9 | CVB3 | References |
|---|---|---|---|---|---|
| Clathrin | Chlorpromazine | − | − | +/− | [25,35,37,38,45] |
| Ap180 | − | − | [12,25] | ||
| DN Eps15 | +/− | − | − | [12,25,28,32,44,45] | |
| siClathrinHeavyChain | +/− | − | [45] | ||
| Clathrin RNAi | − | [37,38] | |||
| Dynamin/CtBP/BARS | Dynasore | +/− | + | +/− | [9,25,37,38,44,45] |
| DN Dynamin | +/− | + | +/− | [9,12,25,37,38,44,45] | |
| siDynamin | + | +/− | [37,38,44,45] | ||
| CtBP/BARS siRNA | + | [15,45] | |||
| Caveolin | siCaveolin | − | +/− | [37,38,45] | |
| DN Caveolin | +/− | − | +/− | [9,12,25,32,37,44,45] | |
| Cholesterol | MβCD | + | − | + | [5,12,25,32,37,38,45] |
| Progesterone+Nystatin | + | − | [12,25] | ||
| Filipin | +/− | +/− | [11,37,38,45] | ||
| Nystatin | + | − | [11,35,38] | ||
| Ketokonazole | + | [11] | |||
| U18666A | − | [11] | |||
| Cholesterol oxidase | + | [38] | |||
| Actin | Cytochalasin D | +/− | − | +/− | [8,9,12,25,32,38,45] |
| Latrunculin A | − | − | +/− | [12,25,32,38] | |
| Jasplakinolide | +/− | + | + | [8,9,12,25,45] | |
| Rac1 | NSC23766 | + | + | [24,32] | |
| DN Rac1 | + | + | [9,32,45] | ||
| siRac1 | + | [9] | |||
| Pak1 | IPA−3 | + | − | + | [24,45] |
| DN Pak1 | + | [9] | |||
| PLC | U−73122 | + | + | +/− | [9,24,44] |
| PKC | Bisindolylmaleimide | + | [8,12] | ||
| Safingol | + | [8,12] | |||
| PMA | + | [8] | |||
| Rottlerin | + | + | [38,42,45] | ||
| DN PKCa | + | [9] | |||
| Acidification | Bafilomycin A1 | − | − | +/− | [24,35,37,45] |
| NH4Cl | − | − | [25,37] | ||
| Microtubules | Nocodazole | − | − | + | [12,24,25,38] |
| Na+/H+ exchanger | EIPA | + | + | + | [9,24,25,38,42,45] |
| ESCRT | Hrs | + | + | [15,24] | |
| siTSG101 | +/− | [15] | |||
| siVps37A | + | [15] | |||
| siVps24 | + | [15] | |||
| DN Vps4 | + | + | [15,24] | ||
| PI3K | LY290042 | +/− | − | [9,24] | |
| Wortmannin | − | − | [24,25,38] | ||
| Calpain | Calpeptin | + | + | [43,44] | |
| Inhibitor1/2 | + | [44] | |||
| siCalpain(1/2) | + | +/− | [43,44] | ||
| ALLN | + | [43] | |||
| Inhibitor III | + | [43] | |||
| Tyrosine kinase | Genistein | + | + | [12,32,37,38,43] |
| Structure | Marker | E1 | CVA9 | CVB3 | References |
|---|---|---|---|---|---|
| Clathrin | Clathrin | − | [32] | ||
| Caveolae | Caveolin | + | − | + | [5,8,12,25,32,43] |
| Cholera Toxin B | Cholera Toxin B | +/− | + | [9,32,43] | |
| Macropinosome | Dextran | + | − | + | [9,25,44,45] |
| Early endosome | EEA1 | −/+ | − | [8,9,15,24,45] | |
| Rab5 | +/− | [42,43] | |||
| Recycling endosome | Transferrin | − | [5] | ||
| Late endosome | CI−MPR | − | (−1) | [5,9,32] | |
| LBPA | − | [15] | |||
| Rab7 | − | − | [15,24] | ||
| Late endosome/ | CD63 | − | (−1) | [9,15,32] | |
| Lysosome | Lamp1 | − | − | [15,24] | |
| Lamp2 | + | [45] | |||
| LysoTracker | − | − | [12,24] | ||
| Dil−LDL | − | − | [15,24] | ||
| Calpain | Calpain | + | + | [6,43] |

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roivainen, M.; Alfthan, G.; Jousilahti, P.; Kimpimäki, M.; Hovi, T.; Tuomilehto, J. Enterovirus infections as a possible risk factor for myocardial infarction. Circulation 1998, 98, 2534–2537. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, O.H.; Honkanen, H.; Pakkanen, O.; Oikarinen, S.; Hankaniemi, M.M.; Huhtala, H.; Ruokoranta, T.; Lecouturier, V.; André, P.; Harju, R.; et al. Coxsackievirus B1 is associated with induction of β-cell autoimmunity that portends type 1 diabetes. Diabetes 2014, 63, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Roivainen, M.; Klingel, K. Role of enteroviruses in the pathogenesis of type 1 diabetes. Diabetologia. 2009, 52, 995–996. [Google Scholar] [CrossRef] [PubMed]
- Xing, L. Structural and Functional Analysis of Integrin 2I Domain Interaction with Echovirus 1. J. Biol. Chem. 2004, 279, 11632–11638. [Google Scholar] [CrossRef] [PubMed]
- Marjomaki, V.; Pietiainen, V.; Matilainen, H.; Upla, P.; Ivaska, J.; Nissinen, L.; Reunanen, H.; Huttunen, P.; Hyypia, T.; Heino, J. Internalization of Echovirus 1 in Caveolae. J. Virol. 2002, 76, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Rintanen, N.; Karjalainen, M.; Alanko, J.; Paavolainen, L.; Mäki, A.; Nissinen, L.; Lehkonen, M.; Kallio, K.; Cheng, R.H.; Upla, P.; et al. Calpains promote α2β1 integrin turnover in nonrecycling integrin pathway. Mol. Biol. Cell. 2012, 23, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Pellinen, T.; Ivaska, J. Integrin traffic. J. Cell Sci. 2006, 119, 3723–3731. [Google Scholar] [CrossRef] [PubMed]
- Upla, P.; Marjomäki, V.; Kankaanpää, P.; Ivaska, J.; Hyypiä, T.; Van Der Goot, F.G.; Heino, J. Clustering induces a lateral redistribution of α2β1 integrin from membrane rafts to caveolae and subsequent protein kinase C-dependent internalization. Mol. Biol. Cell 2004, 15, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, M.; Kakkonen, E.; Upla, P.; Paloranta, H.; Kankaanpää, P.; Liberali, P.; Renkema, G.H.; Hyypiä, T.; Heino, J.; Marjomäki, V. A Raft-derived, Pak1-regulated entry participates in α2β1 integrin-dependent sorting to caveosomes. Mol. Biol. Cell 2008, 19, 2857–2869. [Google Scholar] [CrossRef] [PubMed]
- Damm, E.-M.; Pelkmans, L.; Kartenbeck, J.; Mezzacasa, A.; Kurzchalia, T.; Helenius, A. Clathrin- and caveolin-1-independent endocytosis: Entry of simian virus 40 into cells devoid of caveolae. J. Cell Biol. 2005, 168, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Siljamäki, E.; Rintanen, N.; Kirsi, M.; Upla, P.; Wang, W.; Karjalainen, M.; Ikonen, E.; Marjomäki, V. Cholesterol dependence of collagen and echovirus 1 trafficking along the novel α2β1 integrin internalization pathway. PLoS ONE 2013, 8, e55465. [Google Scholar] [CrossRef] [PubMed]
- Pietiäinen, V.; Marjomäki, V.; Upla, P.; Pelkmans, L.; Helenius, A.; Hyypiä, T. Echovirus 1 endocytosis into caveosomes requires lipid rafts, dynamin II, and signaling events. Mol. Biol. Cell 2004, 15, 4911–4925. [Google Scholar] [CrossRef] [PubMed]
- Liberali, P.; Kakkonen, E.; Turacchio, G.; Valente, C.; Spaar, A.; Perinetti, G.; Böckmann, R.A.; Corda, D.; Colanzi, A.; Marjomäki, V.; et al. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J. 2008, 27, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Schnatwinkel, C.; Christoforidis, S.; Lindsay, M.R.; Uttenweiler-Joseph, S.; Wilm, M.; Parton, R.G.; Zerial, M. The Rab5 effector Rabankyrin-5 regulates and coordinates different endocytic mechanisms. PLoS Biol. 2004, 2, E261. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, M.; Rintanen, N.; Lehkonen, M.; Kallio, K.; Mäki, A.; Hellström, K.; Siljamäki, V.; Upla, P.; Marjomäki, V. Echovirus 1 infection depends on biogenesis of novel multivesicular bodies. Cell Microbiol. 2011, 13, 1975–1995. [Google Scholar] [CrossRef] [PubMed]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Hahn, K.M.; Grinstein, S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Bergelson, J.M. Echovirus 7 entry into polarized intestinal epithelial cells requires clathrin and Rab7. MBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.; Hoflack, B.; Simons, K.; Mellman, I.; Kornfeld, S. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 1988, 52, 329–341. [Google Scholar] [CrossRef]
- Gruenberg, J.; Griffiths, G.; Howell, K.E. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J. Cell Biol. 1989, 108, 1301–1316. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Rusten, T.E.; Stenmark, H. Protein sorting into multivesicular endosomes. Curr. Opin. Cell. Biol. 2003, 15, 446–455. [Google Scholar] [CrossRef]
- Soonsawad, P.; Paavolainen, L.; Upla, P.; Weerachatyanukul, W.; Rintanen, N.; Espinoza, J.; McNerney, G.; Marjomäki, V.; Cheng, R.H. Permeability Changes of Integrin-Containing Multivesicular Structures Triggered by Picornavirus Entry. PLoS ONE 2014, 9, e108948. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, A.; Strauss, M.; Gold, S.; Wenham, H.; Jackson, T.; Chou, J.J.; Rowlands, D.J.; Stonehouse, N.J.; Hogle, J.M.; Tuthill, T.J. Capsid protein VP4 of human rhinovirus induces membrane permeability by the formation of a size-selective multimeric pore. PLoS Pathog. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Limpens, R.W.A.L.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.M.; Bárcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. MBio 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, M.; Waris, M.; Kajander, R.; Hyypia, T.; Marjomaki, V. Coxsackievirus A9 infects cells via nonacidic multivesicular bodies. J. Virol. 2014, 88, 5138–5151. [Google Scholar] [CrossRef] [PubMed]
- Heikkilä, O.; Susi, P.; Tevaluoto, T.; Härmä, H.; Marjomäki, V.; Hyypiä, T.; Kiljunen, S. Internalization of coxsackievirus A9 is mediated by β2-microglobulin, dynamin, and Arf6 but not by caveolin-1 or clathrin. J. Virol. 2010, 84, 3666–3681. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Triantafilou, M. Lipid raft microdomains: key sites for Coxsackievirus A9 infectious cycle. Virology 2003, 317, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Modlin, J.F.; Wieland-Alter, W.; Cunningham, J.A.; Crowell, R.L.; Finberg, R.W. Clinical coxsackievirus B isolates differ from laboratory strains in their interaction with two cell surface receptors. J. Infect. Dis. 1997, 175, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Shieh, J.T.; Pickles, R.J.; Okegawa, T.; Hsieh, J.T.; Bergelson, J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. USA 2001, 98, 15191–15196. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Mohanty, J.G.; Crowell, R.L.; St John, N.F.; Lublin, D.M.; Finberg, R.W. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J. Virol. 1995, 69, 1903–1906. [Google Scholar] [PubMed]
- Shieh, J.T.C.; Bergelson, J.M. Interaction with decay-accelerating factor facilitates coxsackievirus B infection of polarized epithelial cells. J. Virol. 2002, 76, 9474–9480. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Bergelson, J.M. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 2006, 124, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Milstone, A.M.; Petrella, J.; Sanchez, M.D.; Mahmud, M.; Whitbeck, J.C.; Bergelson, J.M. Interaction with coxsackievirus and adenovirus receptor, but not with decay-accelerating factor (DAF), induces A-particle formation in a DAF-binding coxsackievirus B3 isolate. J. Virol. 2005, 79, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Zautner, A.E.; Körner, U.; Henke, A.; Badorff, C.; Schmidtke, M. Heparan sulfates and coxsackievirus-adenovirus receptor: Each one mediates coxsackievirus B3 PD infection. J. Virol. 2003, 77, 10071–10077. [Google Scholar] [CrossRef] [PubMed]
- Zautner, A.E.; Jahn, B.; Hammerschmidt, E.; Wutzler, P.; Schmidtke, M. N- and 6-O-sulfated heparan sulfates mediate internalization of coxsackievirus B3 variant PD into CHO-K1 cells. J. Virol. 2006, 80, 6629–6636. [Google Scholar] [CrossRef] [PubMed]
- De Verdugo, U.R.; Selinka, H.C.; Huber, M.; Kramer, B.; Kellermann, J.; Hofschneider, P.H.; Kandolf, R. Characterization of a 100-kilodalton binding protein for the six serotypes of coxsackie B viruses. J. Virol. 1995, 69, 6751–6757. [Google Scholar] [PubMed]
- Patel, K.P.; Coyne, C.B.; Bergelson, J.M. Dynamin- and lipid raft-dependent entry of decay-accelerating factor (DAF)-binding and non-DAF-binding coxsackieviruses into nonpolarized cells. J. Virol. 2009, 83, 11064–11077. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Sadovsky, Y.; Coyne, C.B. Lipid raft- and SRC family kinase-dependent entry of coxsackievirus B into human placental trophoblasts. J. Virol. 2013, 87, 8569–8581. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, M.; Selinka, H.C.; Heim, A.; Jahn, B.; Tonew, M.; Kandolf, R.; Stelzner, A.; Zell, R. Attachment of coxsackievirus B3 variants to various cell lines: Mapping of phenotypic differences to capsid protein VP1. Virology 2000, 275, 77–88. [Google Scholar] [PubMed]
- Bordería, A.V.; Isakov, O.; Moratorio, G.; Henningsson, R.; Agüera-González, S.; Organtini, L.; Gnädig, N.F.; Blanc, H.; Alcover, A.; Hafenstein, S.; et al. Group Selection and Contribution of Minority Variants during Virus Adaptation Determines Virus Fitness and Phenotype. PLoS Pathog. 2015, 11, e1004838. [Google Scholar] [CrossRef] [PubMed]
- Carson, S.D.; Chapman, N.M.; Hafenstein, S.; Tracy, S. Variations of coxsackievirus B3 capsid primary structure, ligands, and stability are selected for in a coxsackievirus and adenovirus receptor-limited environment. J. Virol. 2011, 85, 3306–3314. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Shen, L.; Turner, J.R.; Bergelson, J.M. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2007, 2, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Bozym, R.A.; Morosky, S.A.; Kim, K.S.; Cherry, S.; Coyne, C.B. Release of intracellular calcium stores facilitates coxsackievirus entry into polarized endothelial cells. PLoS Pathog. 2010, 6, e1001135. [Google Scholar] [CrossRef] [PubMed]
- Upla, P.; Marjomäki, V.; Nissinen, L.; Nylund, C.; Waris, M.; Hyypiä, T.; Heino, J. Calpain 1 and 2 are required for RNA replication of echovirus 1. J. Virol. 2008, 82, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Krieger, S.E.; Kim, C.; Zhang, L.; Marjomaki, V.; Bergelson, J.M. Echovirus 1 entry into polarized Caco-2 cells depends on dynamin, cholesterol, and cellular factors associated with macropinocytosis. J. Virol. 2013, 16, 8884–8895. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marjomäki, V.; Turkki, P.; Huttunen, M. Infectious Entry Pathway of Enterovirus B Species. Viruses 2015, 7, 6387-6399. https://doi.org/10.3390/v7122945
Marjomäki V, Turkki P, Huttunen M. Infectious Entry Pathway of Enterovirus B Species. Viruses. 2015; 7(12):6387-6399. https://doi.org/10.3390/v7122945
Chicago/Turabian StyleMarjomäki, Varpu, Paula Turkki, and Moona Huttunen. 2015. "Infectious Entry Pathway of Enterovirus B Species" Viruses 7, no. 12: 6387-6399. https://doi.org/10.3390/v7122945
APA StyleMarjomäki, V., Turkki, P., & Huttunen, M. (2015). Infectious Entry Pathway of Enterovirus B Species. Viruses, 7(12), 6387-6399. https://doi.org/10.3390/v7122945