1. Introduction
Alphaviruses are arthropod-borne viruses that cause frequent epidemics in humans and other vertebrates. Sindbis virus (SINV) is the type member of the genus
Alphavirus that replicates in mammalian host and mosquito vector cells. It has a positive-sense, single-stranded RNA genome of 11,703 nucleotides with a cap at the 5′ end and a 3′ poly(A) tail. Nonstructural proteins (nsP1-nsP4) are translated from the 49S genomic RNA, whereas structural proteins capsid (CP), E3, E2, 6K, and El are translated as a polyprotein from a 26S subgenomic RNA [
1]. From the structural polyprotein precursor, CP is autoproteolytically cleaved, exposing an N-terminal signal sequence on E3 that translocates the glycoprotein precursor into the endoplasmic reticulum (ER). In the ER lumen, signalase cleavage removes 6K from pE2 (E3-E2) and E1 envelope proteins that are subsequently glycosylated and form heterodimers. These glycoprotein heterodimers trimerize to form glycoprotein spikes that are transported to the plasma membrane (PM) via the secretory pathway [
2,
3]. Furin cleavage followed by the release of E3 in the late Golgi primes the glycoprotein spikes for subsequent fusogenic activation during cell entry [
4]. CP binds genomic RNA in the cytoplasm to form nucleocapsid cores (NCs). Subsequently, virus particles bud from the plasma membrane (PM) where specific interactions between CP and the cytoplasmic domain of E2 (cdE2) drive envelopment and budding of virions [
5].
SINV virions are spherical (~70 nm diameter) and contain 240 copies each of CP, E1, and E2 arranged with icosahedral symmetry in a T = 4 lattice [
6]. A host-derived lipid bilayer membrane lies sandwiched between the outer glycoprotein shell and the inner nucleocapsid core (NC) that encapsidates the genomic RNA. Virions also contain sub-stoichiometric amounts of the small “6K” and “TF” proteins [
7]. There are two types of virus-induced membranous structures found in the infected cells: type I and type II cytopathic vacuoles (CPV-I and CPV-II) [
8,
9]. CPV-I (0.6 to 2.0 μm diameter) originates from endosomes and lysosomes and contains replication spherules that are the sites of viral RNA synthesis [
10]. CPV-II [
11] originates from the
trans-Golgi network ~4 h post-infection (p.i.) [
12,
13] and contains the E1/E2 glycoproteins with numerous NCs attached to its cytoplasmic face [
11,
12,
14]. Electron tomography studies have revealed that the E1/E2 glycoproteins are arranged in a helical array within CPV-II in a manner that resembles their organization on the viral envelope [
2]. CPV-IIs have been proposed earlier to be caused by over-loading of the secretory pathway by the highly expressed viral glycoproteins [
12]. Later it was also suggested that CPV-IIs promote the intracellular transport of the glycoproteins from the
trans-Golgi network to the PM and also the transport of NCs to the site of virus budding at the PM [
2]. Subsequently, NC buds through the PM by forming specific interactions with cdE2 [
5,
15]. The curvature of the preassembled NC, coupled with regularly spaced, strong interactions between the NC and the cytoplasmic domains of the E2 molecules, allows the membrane and embedded glycoprotein spikes to encircle the NC to form enveloped, fully mature virus particles [
6].
We and others have described fluorescent fusion proteins including CP [
16], E2 [
17,
18,
19,
20], and tetra cysteine-labeled structural proteins for virus entry and budding studies [
21]. Furthermore, generation of CPV-I in cells infected with Semliki Forest virus has been demonstrated by live-cell imaging coupled with transmission electron microscopy (TEM) [
22]. Several imaging studies have utilized fluorescent protein-tagged viruses and subviral particles in single-particle tracking to probe virus entry and assembly. Fluorescently tagged derivatives of Gag-containing human immunodeficiency virus (HIV)-1 virus-like particles were employed to demonstrate assembly, budding, and release of particles from live cells [
23]. Similar studies in hepatitis B virus (HBV) have found that the incorporation of only a few fluorescent protein-tagged envelope proteins is sufficient to generate functional, fluorescent virions and subviral particles that enter HBV receptor-positive cells [
24]. Furthermore, cryo-electron microscopy (cryoEM) reconstructions have been utilized to determine the organization of fluorescent proteins on purified virus particles. Using cryoEM methods it has been previously shown that green fluorescent protein (GFP)-tagged HBV core particles purified from a bacterial expression system retained icosahedral structure and displayed GFP on its surface [
25]. Likewise, a Herpes Simplex Virus 1 GFP-tagged UL17 minor capsid protein was used to determine its location in the capsid vertex-specific component using cryoEM studies [
26]. SINV with fluorescent protein labels on the E2 envelope protein has been employed to study virus assembly and budding in living cells. Previous correlative light and electron microscopy studies using fluorescent SINV have provided information about alphavirus budding. Such studies established that glycoprotein E2 is enriched on the PM in localized patches that also contain other viral structural proteins, from which capsid protein interacts with E2 protein for virus budding. This study also suggested that SINV induces reorganization of the PM and cytoskeleton, leading to virus budding from specialized sites [
18].
In the current study we characterized the structural stability of an FP-tagged virus and determined the arrangement of mCherry on the virus surface. We provide evidence for the structural stability of the FP-tagged virus and demonstrate that single-particle tracking can be employed to visualize SINV budding from live cells. By employing FP-tagged virus to study virus spread in mammalian cells, we observed that SINV buds from the PM and is associated with filopodial extensions that assist in the dispersal of virions. Comparison of wild-type and budding negative mutant viruses confirmed that fluorescent specks budding from filopodial extensions of mCherry-E2 virus-infected cells are individual virions. By treating infected cells with fusogenic low-pH media, we show that the nascent virions were able to fuse to the PM of filopodial extensions of the infected cells, and we provide evidence for the presence of virions on the outside of these filopodia. This FP-tagged virus can be employed as a tool in high-resolution live and fixed cell imaging coupled with other labeled host proteins and other components to study various aspects of the alphavirus lifecycle.
2. Materials and Methods
2.1. Cells and Viruses
Baby hamster kidney fibroblast cells (BHK-15) obtained from the American Type Culture Collection (ATCC) were maintained in minimal essential medium [
27] supplemented with 10% fetal bovine serum (FBS). All SINV cDNA clones were constructed using standard overlapping PCR mutagenesis from pToto64, a full-length cDNA clone of SINV, as previously described [
28]. Viruses were propagated in BHK-15 cells at 37 °C in Minimum Essential Medium (MEM) supplemented with 5% FBS in the presence of 5% CO
2 unless otherwise noted.
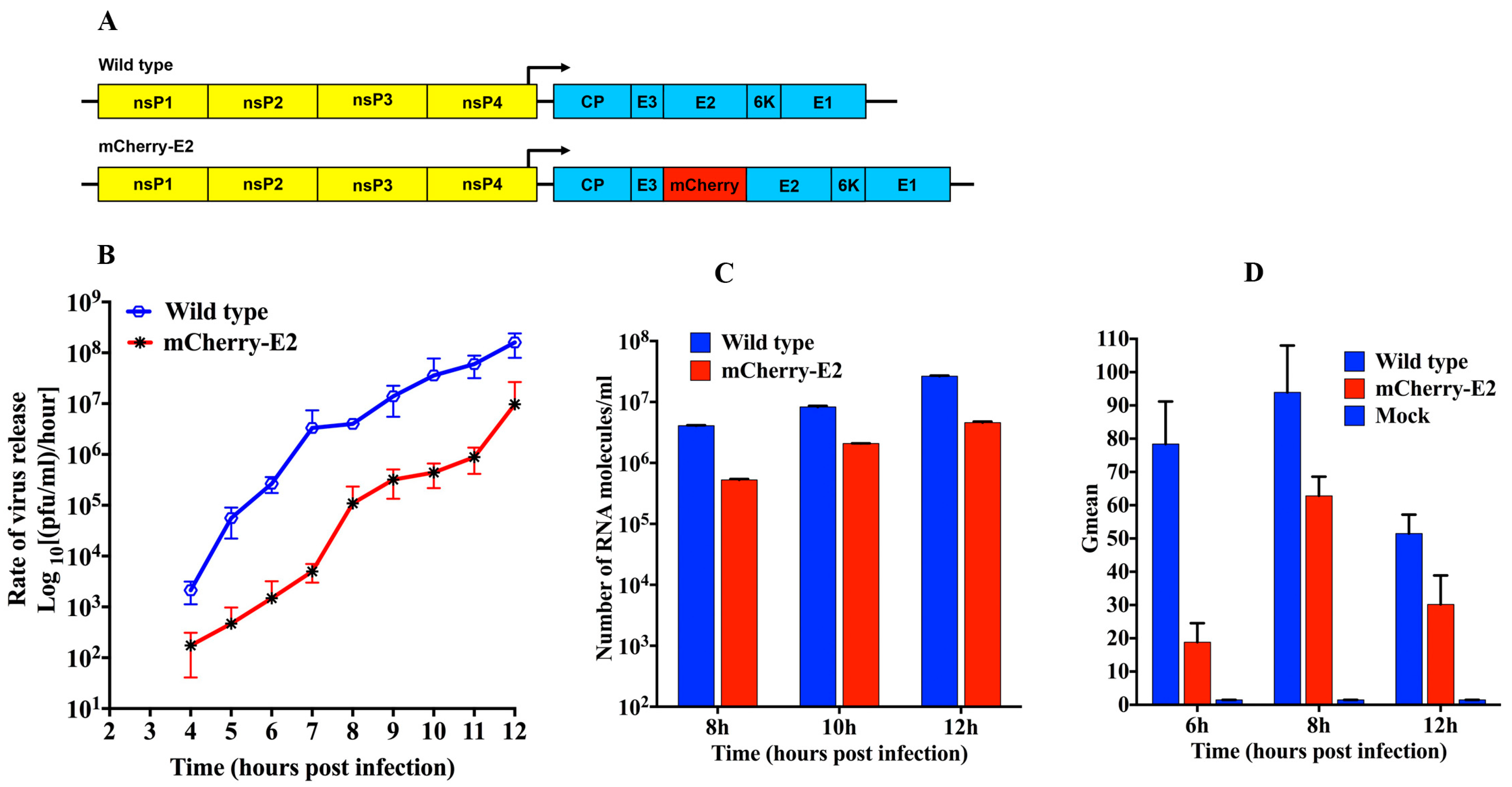
2.2. Construction and Characterization of FP-Tagged Virus and Mutants
Sequences that encode mCherry, with additional Ser residues at the N- and C-termini, were cloned after Ser
1 of E2 replacing E2 Val
2. Previously characterized cdE2 mutations (
400YAL
402/A3 and
416CC
417/A2) [
5] and an E1 (G91D) fusion loop mutation [
29] were generated by overlapping PCR and were cloned into the mCherry-E2 cDNA plasmid using
BssHII-
BsiWI restriction sites. The full-length WT and tagged cDNA clones were linearized with
SacI,
in vitro transcribed with SP6 RNA polymerase, and transfected into BHK-15 cells as previously described [
5]. Infectious virus produced from the transfected cells was quantified by standard plaque assay using medium over cells collected at 24 h post-electroporation. Plaque phenotypes and virus titers were determined by comparing the mutant with WT Toto64 plaques.
2.3. One-Step Growth Curve Analysis
One-step growth analyses were performed as described previously to measure growth kinetics of the mCherry-E2-tagged virus [
5]. BHK-15 cells in 35-mm culture dishes were infected with virus at a multiplicity of infection (MOI) of 5 for 1 h at room temperature. Infected cells were washed extensively with MEM and incubated further and culture media were harvested at every hour for 12 h. The amount of infectious virus in the virus supernatant was quantified by titration on BHK cells. All experiments were conducted in triplicate.
2.4. Quantitative Real Time RT-PCR
The number of virus particles released at different time points and total RNA molecules in the media over infected cells were determined by qRT-PCR as previously described [
30]. RNA was extracted from virus supernatants using the RNeasy kit (Quiagen, Valencia, CA, USA) according to the manufacturer’s instructions. qRT-PCR was performed using the SuperScript III Platinum SYBR Green One-Step qRT-PCR Kit (Invitrogen, Grand Island, NY, USA) with primers 5′-TTCCCTgTgTgCACgTACAT-3′ and 5′- TgAgCCCAACCAgAAgTTTT-3′, which bind to nucleotides 1044–1063 and nucleotides 1130–1149 of the SINV genome, respectively. Amplification reactions were carried out in triplicate in 25 μL sample volumes that contained a 5 μL aliquot of purified viral RNA [
5]. Cycling conditions were 4 min at 50 °C and 5 min at 95 °C, followed by 40 cycles of 5 s at 95 °C and 1 min at 60 °C. The number of molecules of viral RNA was determined using a standard curve of the cycle threshold values (CT) determined by qRT-PCR
versus the number of molecules of
in vitro transcribed genomic RNA using primers.
2.5. Flow Cytometry (FC)
Transport and cell surface expression of E2 in infected cells were assayed using FC and anti-E2 antibody. BHK-15 cells were infected with an MOI of 5. The cells were trypsinized at 6 h, 8 h, and 12 h post-transfection and resuspended in MEM supplemented with 10% FBS. Cells were washed two times with PBS supplemented with 1% FBS and incubated on ice for 1 h with a 1:50 dilution of anti-E2 127 monoclonal antibody. The cells were washed subsequently three times with PBS (1% FBS) and then incubated on ice in the dark for 30 min with a fluorescein-conjugated goat anti-mouse secondary antibody. The cells were washed thrice with PBS (1% FBS) and suspended in 500 μL of PBS and were analyzed on an FC500 flow cytometer (Beckman Coulter, Indianapolis, IN, USA) with the FlowJo software package. Control staining was performed with mock-transfected cells.
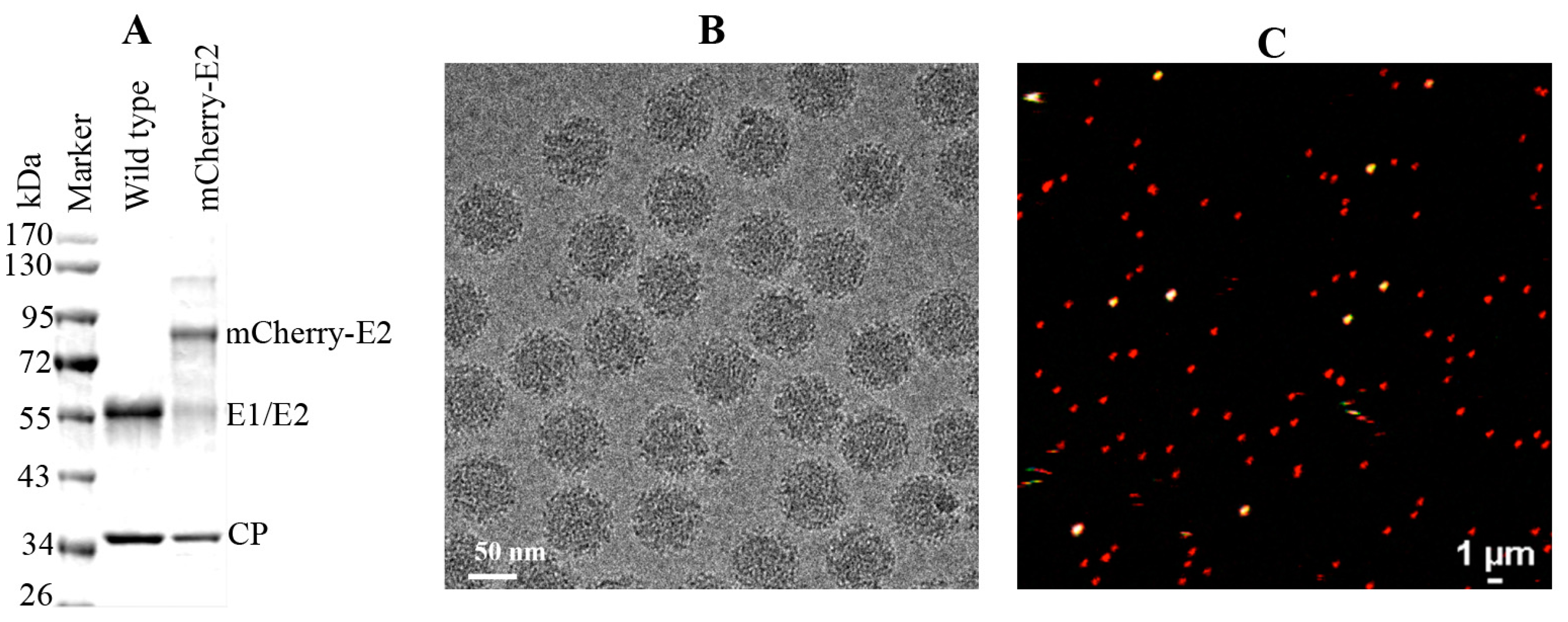
2.6. Virus Purification, Cryo-Electron Microscopy (cryoEM), and 3D Image Reconstruction
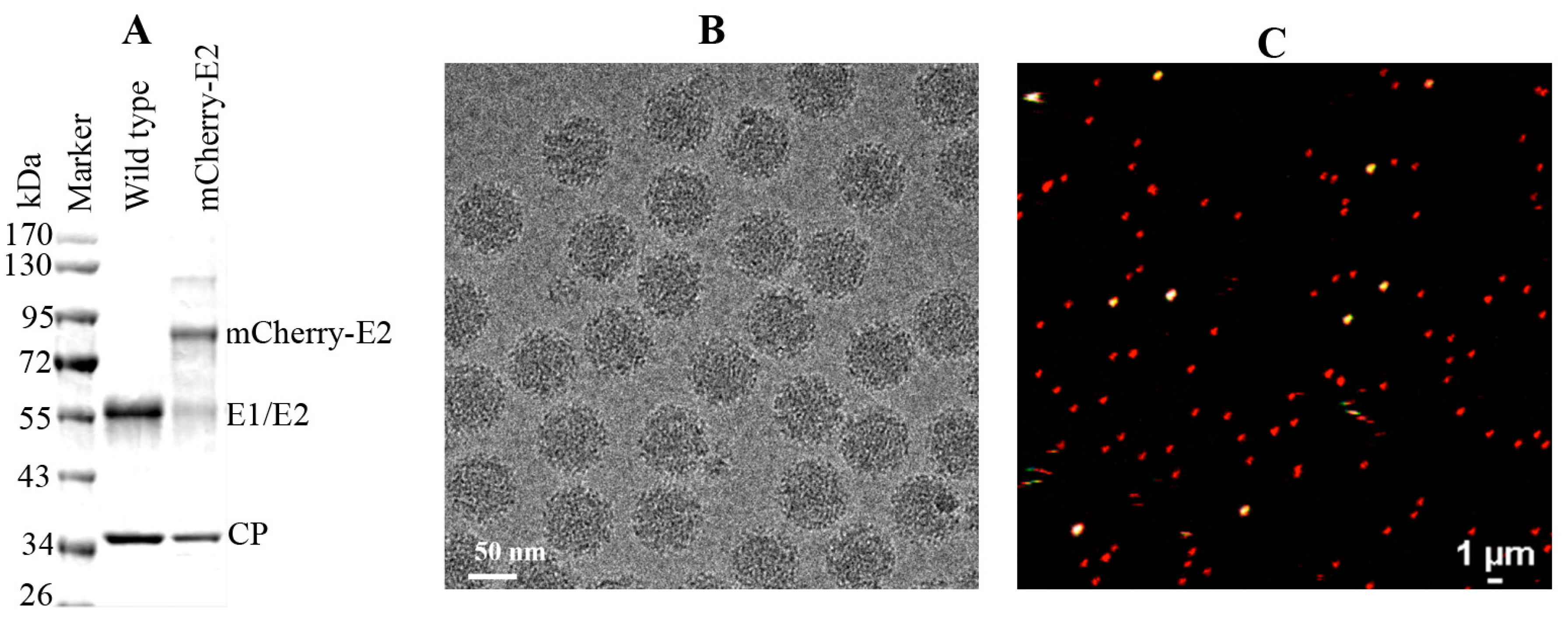
WT and mCherry-E2 viruses were purified according to standard virus purification protocols. Briefly, cell culture supernatants from SINV or mCherry-E2-tagged virus-infected BHK were collected at 12 h p.i. and the media were harvested and clarified by centrifugation for 15 min at 9000 × g. Virus particles were pelleted through a 27% sucrose cushion in a Beckman Ti-50.2 rotor at 38,000 rpm for 2 h. The virus pellets were resuspended and loaded onto a 0 to 30% continuous iodixanol gradient, in TNE (50 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA), and centrifuged at 38,000 rpm in a Beckman SW-41 rotor for 2 h. The virus band was extracted by syringe and buffer exchanged using TNE buffer and the presence of the mCherry-E2 tag was confirmed by SDS PAGE analysis.
Small (3 μL) aliquots of the purified mCherry-E2-tagged virus were vitrified for cryoEM via standard, rapid freeze-plunging procedures [
31] on Quantifoil holey grids (Quantifoil, Electron Microscopy Sciences , Hatfield, Pennsylvania, USA). Grids were then loaded into a multi-specimen holder and inserted into an FEI Polara microscope and maintained at liquid-nitrogen temperature. Micrographs were recorded on a 4K
2 Ultrascan CCD (Gatan, Inc., Pleasanton, CA, USA) at a nominal magnification of 51,000× under low-dose conditions (≈15 e/Å
2) with the microscope operated at 200 keV and the objective lens defocused between 0.9 and 4.7 μm underfocus. Micrographs that exhibited some astigmatism or specimen drift were eliminated from the data set. Individual virus particles were boxed from the remaining 103 micrographs with the program RobEM [
32].The Random model computation method [
33] was employed to generate an initial 3D map at ~25 Å resolution for the mCherry-E2 insertion mutant. This map was then used as the starting model to initiate orientation and origin determinations for the full set of 9235 particle images using the AUTO3DEM program suite [
33] to yield a final 3D map at 11 Å resolution. Graphical representations were generated with RobEM and Chimera [
34]. A SINV pseudo-atomic model [
6] was used to fit and interpret the mCherry-E2 virus reconstruction. The crystal structure of red fluorescent protein [
35] was used to model the densities not accounted for by the virus itself in the mutant. Optimal fitting of the red fluorescent protein model was achieved by rigid body refinement with the
Fit in Map module of Chimera [
34].
2.7. Live Cell Imaging
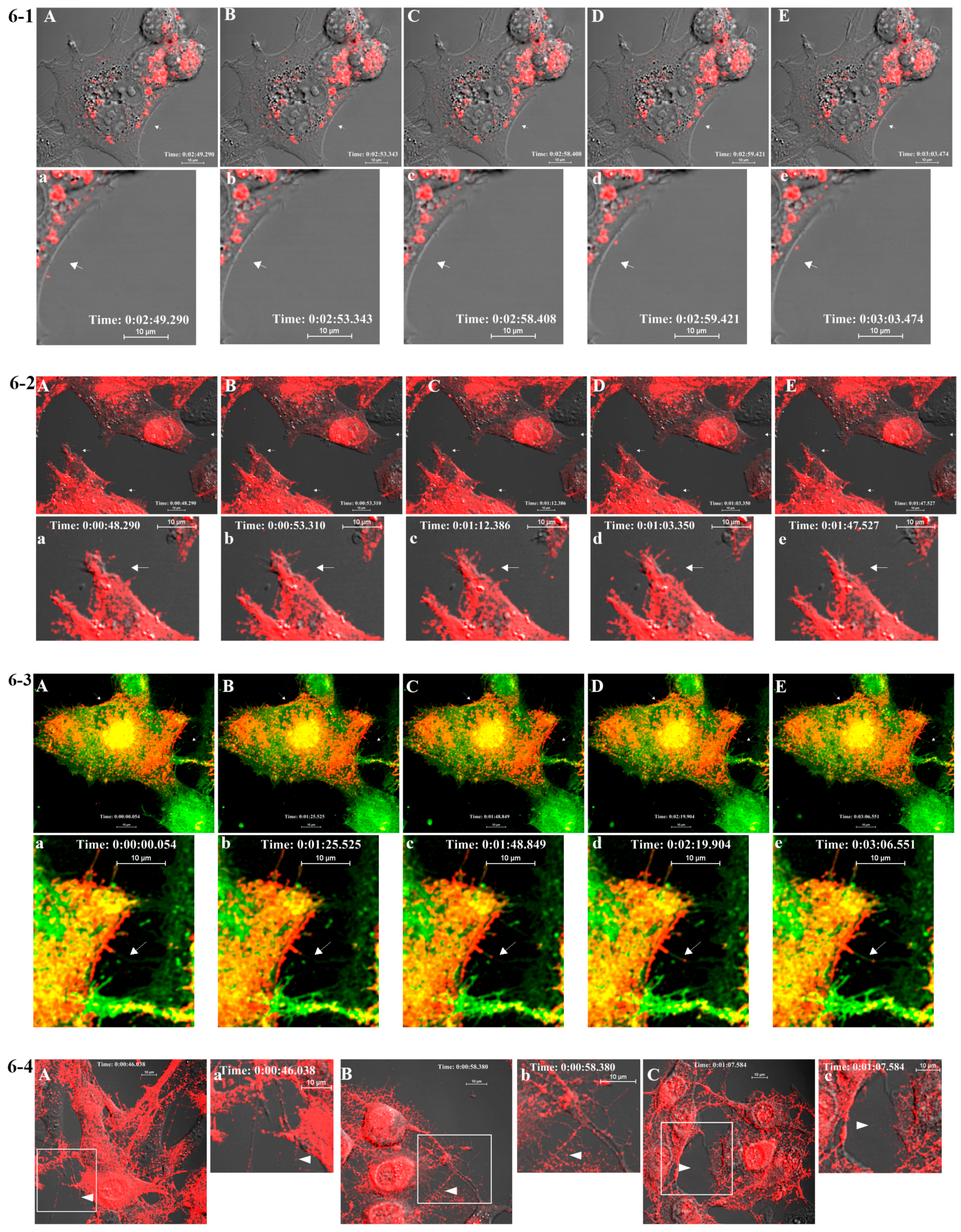
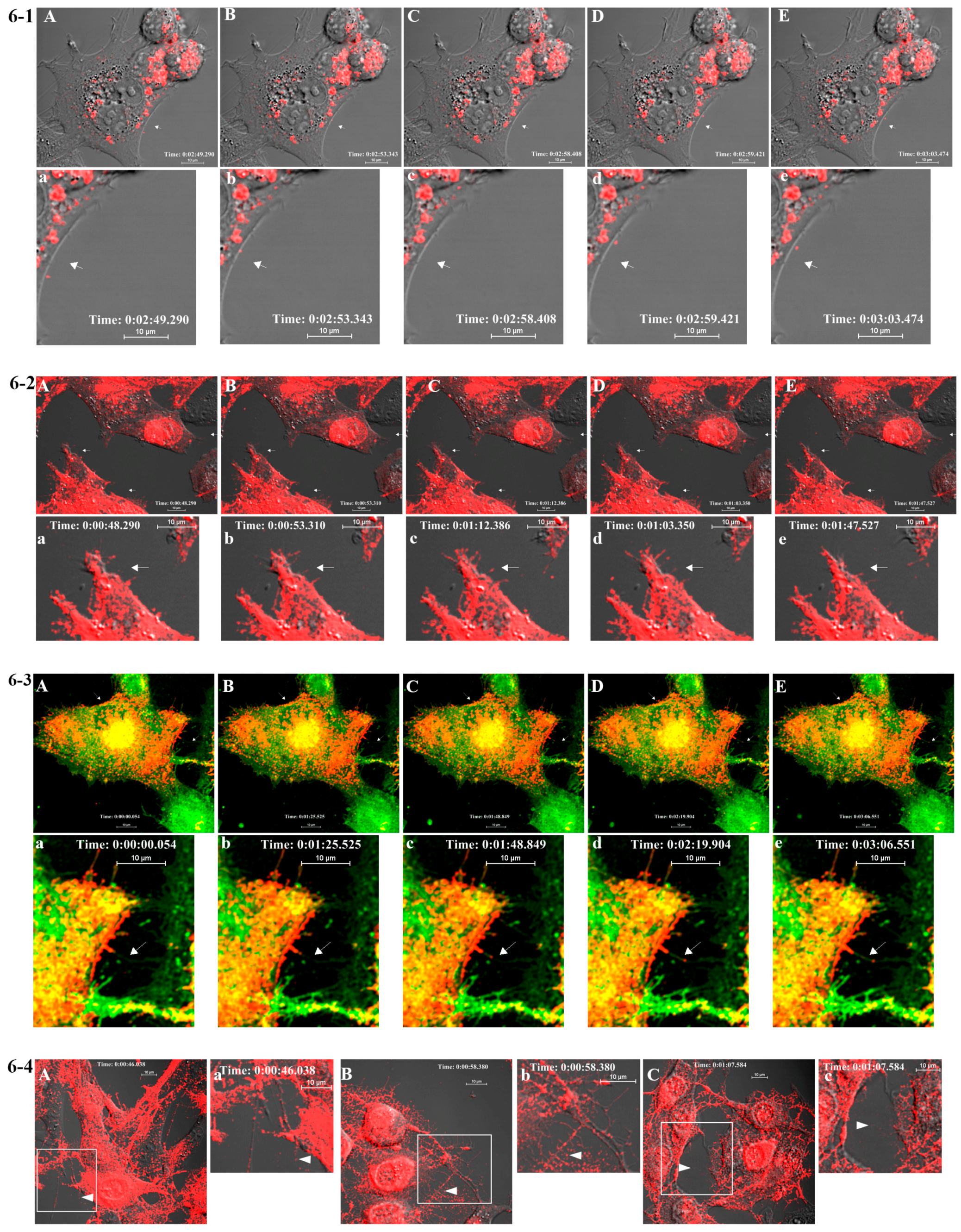
BHK-15 cells were seeded onto a four-chambered borosilicate cover glass (Fischer Scientific, Pittsburgh, PA, USA) and infected with fluorescent virus at an MOI of 50 at 25% confluence. Infected cells were imaged after media were replaced with Opti-MEM I Reduced-Serum Medium (Invitrogen) at specified time points. Live imaging-compatible stains were obtained from Invitrogen/Molecular Probes. These included Hoechst stain (nucleus) and BODIPY FL C5 ceramide (Golgi stain) and were used according to the manufacturer’s instructions in conjunction with mCherry-E2 virus. Fluorescent images were acquired at indicated temperatures using Nikon A1R confocal microscope (Nikon, Melville, NY, USA ) with 60×, 1.4 numerical aperture (NA) lens) using NIS Elements software (Nikon, Melville, NY, USA). Live imaging for 10–30 min periods was conducted using a heated 60× oil immersion objective (1.4 NA) in a live imaging chamber (Tokai Hit, Fujinomiya, Shizuoka Prefecture, Japan) supplied with 5% CO2 at 37 °C. The lasers and emission band passes used for imaging were as follows: blue, excitation: 405 nm, emission: 425–475 nm; green, excitation: 488 nm, emission: 500–550 nm; red, excitation: 561 nm, emission: 570–620 nm. Differential interference contrast images were collected from transmitted light along with fluorescent images for colocalization of viral proteins in the cellular organelles. NIS-Elements software was used for image acquisition and analysis. For generating videos, live images were collected at frame rates ranging from 0.8 to 1 frames per second (fps) for a time scale of 1–30 min, and time-lapse videos were generated from the acquired images at a frame rate of 5–7 fps using ImageJ (NIH Bethesda, Maryland, USA). To compare the size and fluorescent properties of purified mCherry-E2 virus, purified virus was mixed with 0.1 μm diameter fluorescent microspheres (TetraSpeck Beads, Invitrogen) and imaged on a cover glass using a Nikon A1R system with a 60x oil immersion objective (1.4 NA).
2.8. Immunofluorescence (IF) Analysis
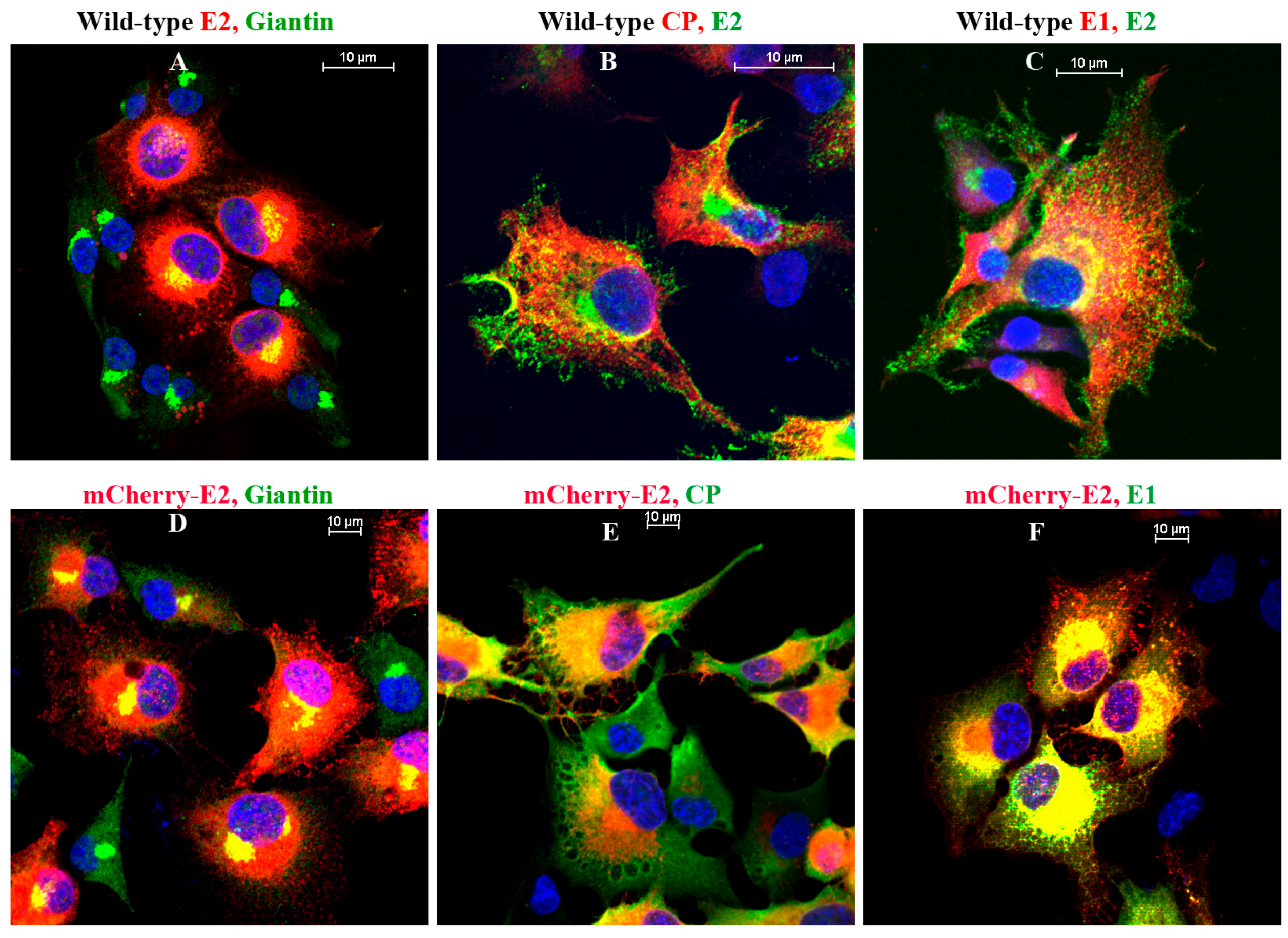
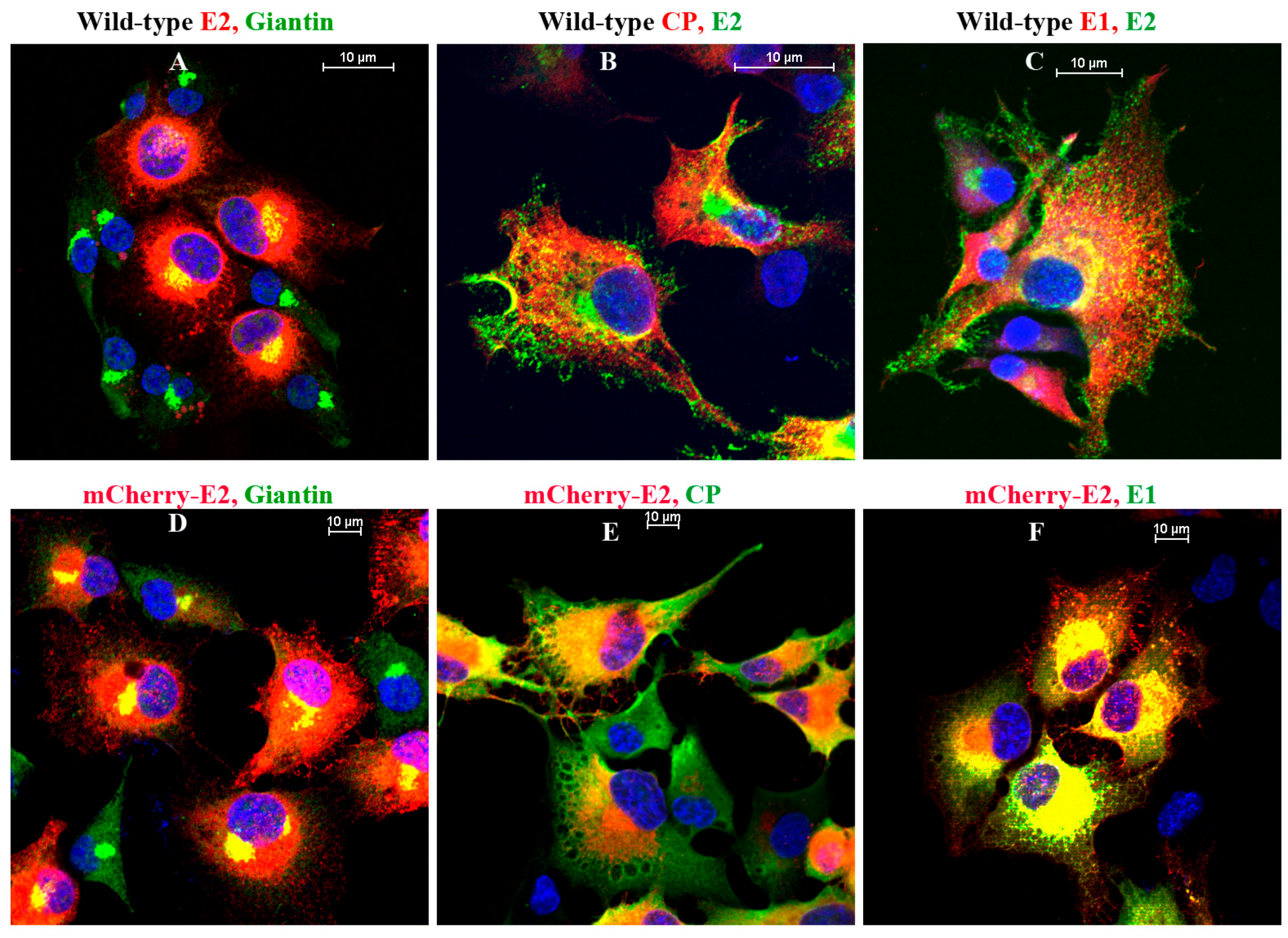
IF analyses were performed on BHK-15 cells grown on glass coverslips. Primary antibodies used in the experiments were Golgi-specific rabbit polyclonal anti-Giantin (Abcam, Cambridge, MA, USA), SINV-specific rabbit polyclonal anti-E1, anti-CP and mouse monoclonal anti-E2. Cells were fixed using 3.7% paraformaldehyde for 15 min at room temperature and permeabilized using 0.1% Triton × 100 in phosphate-buffered saline (PBS) for 5 min. The secondary antibodies used were fluorescein isothiocyanate (FITC) or tetramethyl rhodamine (TRITC)-conjugated goat anti-rabbit and goat anti-mouse antibodies [
36] in PBS with 10 mg/mL bovine serum albumin. Nuclei were stained using Hoechst stain (Invitrogen) according to the manufacturer’s instructions. Images were acquired using a Nikon A1R-MP confocal microscope at room temperature with a 60× oil objective and 1.4 NA. Images were processed using the NIS Elements software (Nikon) and the brightness and contrast were adjusted using nonlinear lookup tables.
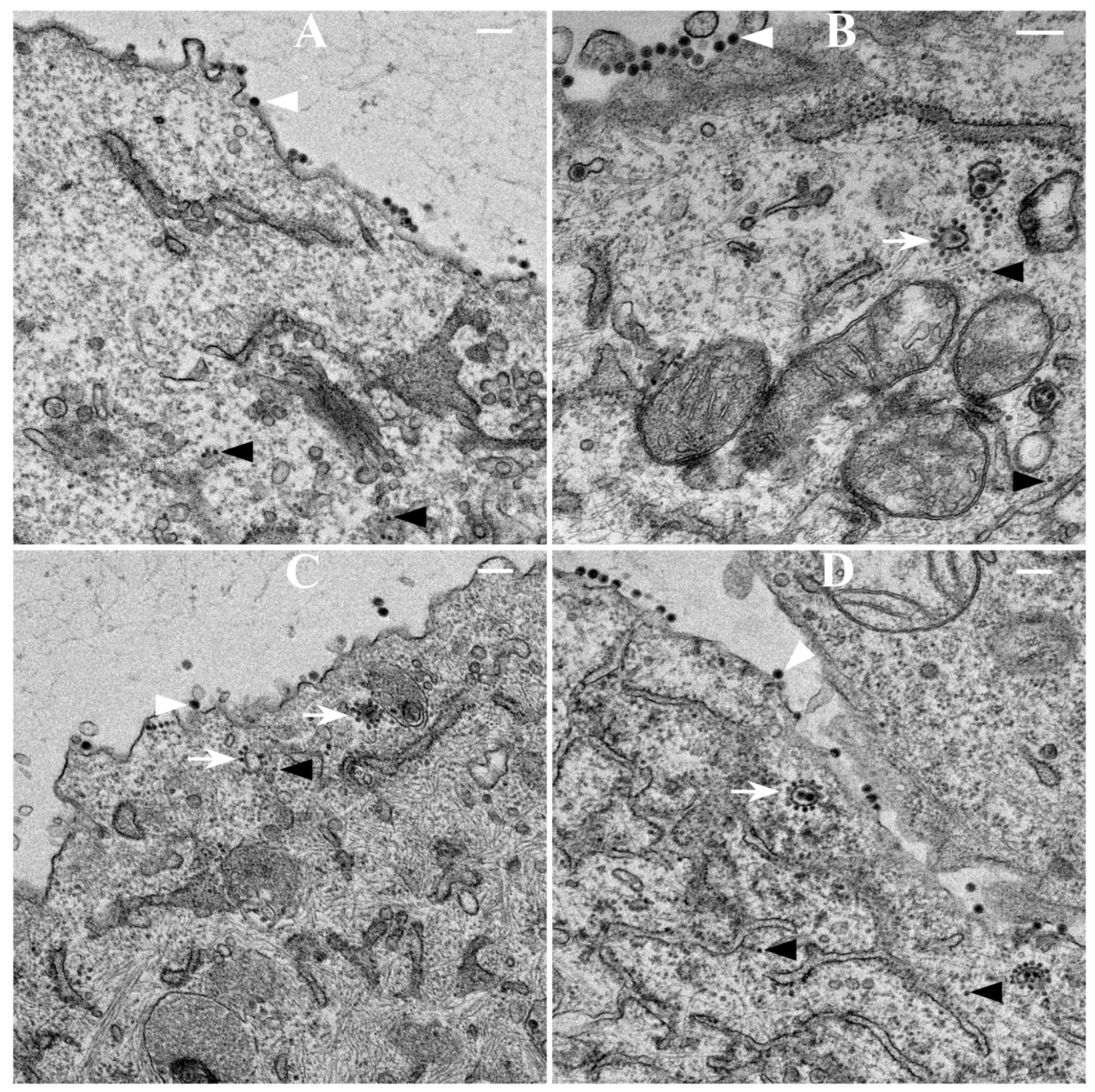
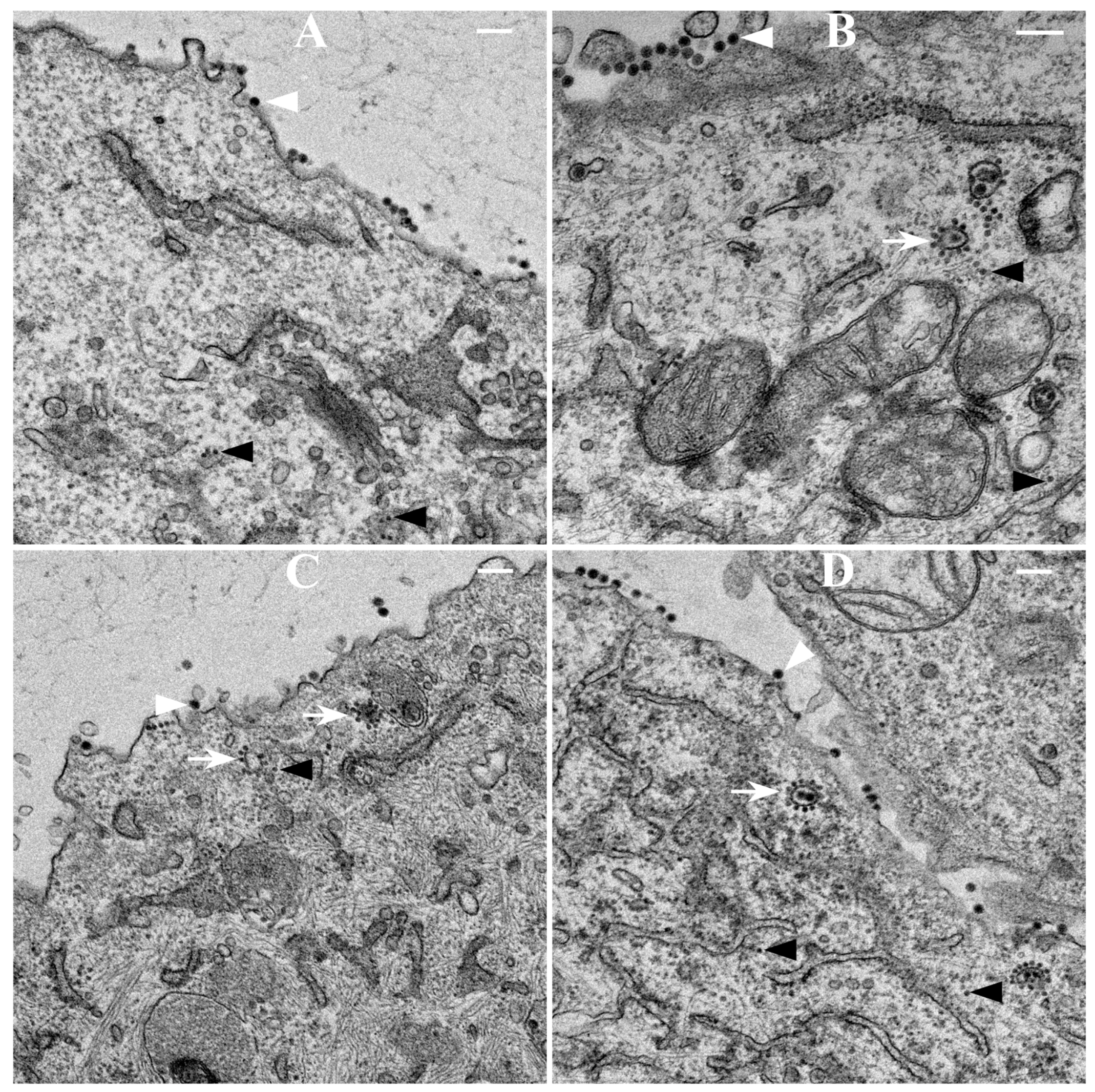
2.9. Thin-Section Transmission Electron Microscopy (TEM)
BHK-15 cells infected with wild-type or mCherry-E2-tagged SINV at an MOI 5 were fixed at 6 or 12 h p.i. Cells were fixed for three days in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, embedded in 2% agarose, post-fixed for 90 min in buffered 1% osmium tetroxide containing 0.8% potassium ferricyanide, and stained for 45 min in 2% uranyl acetate. They were then dehydrated with a graded series of ethanol, transferred into propylene oxide and embedded in EMbed-812 resin. Thin sections were cut on a Reichert-Jung Ultracut E ultramicrotome and stained with 2% uranyl acetate and lead citrate [
37]. Images were acquired in an FEI Tecnai G
2 20 electron microscope equipped with a LaB
6 source and operated at 100 keV (Life Science Microscopy Facility, Purdue University, West Lafayette, IN, USA).
4. Discussion
Single-particle tracking and real-time live imaging provide powerful tools for obtaining spatial and temporal resolution information. This contrasts with traditional modes of TEM and super resolution light microscopy that provide high spatial resolution but lack temporal resolution. In this study, we used live imaging coupled with an FP-tagged viral protein to analyze temporal aspects of alphavirus assembly in mammalian cells. We generated an FP-tagged virus with mCherry fused to the N-terminus of the E2 glycoprotein, which is known to tolerate insertions of the immunoglobulin-binding domains of protein L [
39] and fluorescent proteins [
17,
18,
19,
20]. In this study, mCherry was deemed to be an ideal tag based on its monomeric nature, photostability, fast maturation, and resistance to low pH [
41]. The mCherry tag has a low pK
a value of 4.5, and hence retains its fluorescence when it encounters the cellular secretory pathway [
42]. During maturation, E3 packs against the acid-sensitive region of E2, which maintains the A and B domains of E2 and the B domain to cover the E1 fusion loop, thus protecting the virus from premature fusion with other cellular membranes [
40]. After furin cleaves E3, acidification of the virus during entry causes E2 domain B to move away from its neutral pH position and exposes the fusion loop [
4]. We have demonstrated that the mCherry tag did not adversely affect any of these functions of E3 and E2.
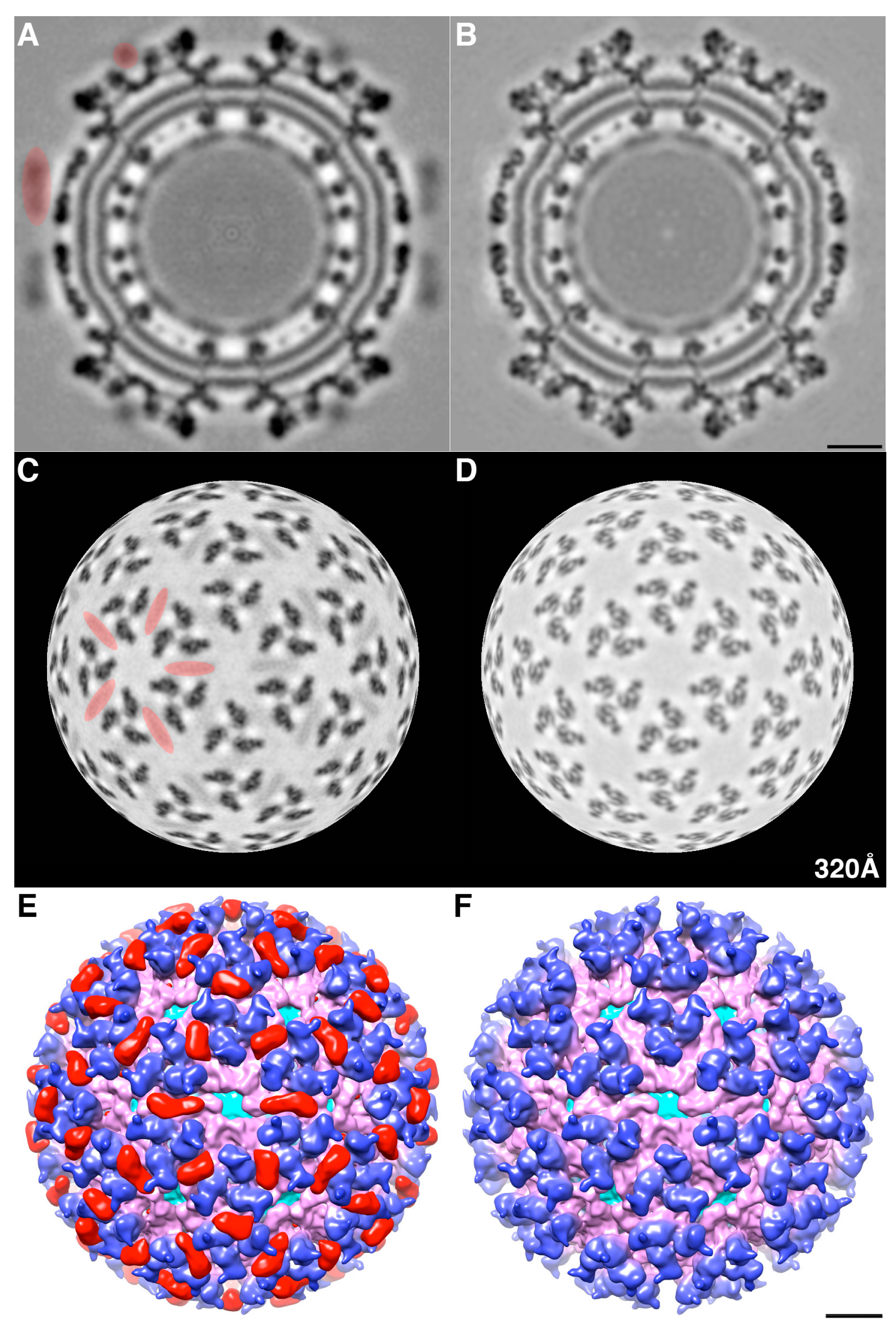
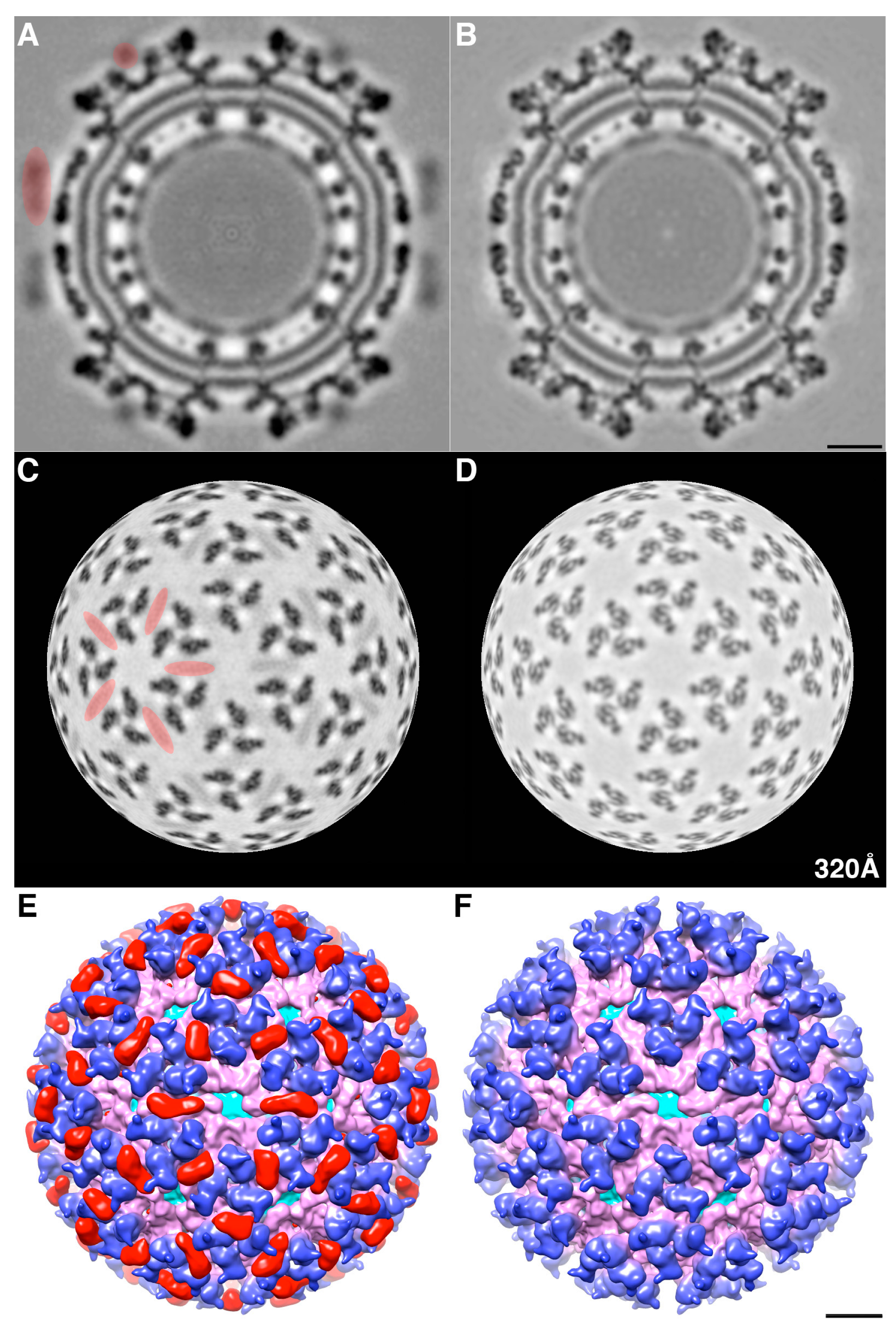
We determined the 3D cryoEM structure of the mCherry-E2 virus to assess the effects, if any, of the 236-residue insertion on the structural integrity of the virus and its potential to alter the virus lifecycle. The cryo-reconstruction of the mCherry-E2 virus at 11 Å resolution revealed that the overall size of the virion and the icosahedral arrangement of the E1, E2, and CP proteins remained essentially unaffected despite the presence of the large FP insertion. The 240 copies of the mCherry tag wedge tightly between neighboring spikes, and this arrangement causes a slight rearrangement of the spikes as well as the nucleocapsid protein (NCP) pentamers and hexamers and small conformational changes in the membrane bilayer. This confirms our previous observation that minor conformational adjustments of the viral glycoprotein spikes get transmitted radially to the NC via the strong interactions that occur between the inner NC and the outer glycoprotein layers [
6]. These small alterations, coupled with the delay in surface expression of the viral glycoproteins as demonstrated by flow cytometry analysis, contribute to the 10-fold reduction in mCherry-E2 virus growth. However, the presence of the mCherry tag did not affect the receptor binding or fusion functions of E2 and E1, respectively. The cryoEM structure of the FP-tagged SINV also confirmed that there is a 1:1 ratio of mCherry to E2 in every virion, which leads to a strong fluorescence signal and thus greatly facilitates single-particle tracking experiments.
We examined mCherry-E2 SINV in live cell imaging primarily to demonstrate virus assembly and budding in real-time using a FP-tagged virus that has been characterized as structurally stable. At times as early as 3 h p.i. we detected budding of FP-tagged virus particles from infected BHK cells, and we gleaned additional information about virus budding and dissemination by examining virus budding and entry mutants. By moving budding viral particles away from the PM of infected cells, the filopodia may act to suppress superinfection, possibly by reducing the re-attachment into the infected cells. As the first step to probe SINV entry and fusion in late endosomes and to study the mechanism of virus fusion, we generated a G91D fusion loop mutation in E1, which abrogates low-pH-triggered fusion and infection [
43]. The mCherry-E2 with the G91D fusion loop mutation in E1 was released from the transfected cells, but was unable to fuse and became trapped presumably in the endosome after entry. Using cdE2-NC interaction-deficient, non-budding mCherry-E2 mutants
400YAL
402/A3 and
416CC
417/A2 we show that non-budding, FP-tagged cdE2 mutations are sufficient to stop fluorescent particle budding from transfected cells. Additionally, using live imaging, we describe that virus budding occurs at the PM for both wild-type and mCherry-E2 virus by the interaction of surface glycoproteins that are transported to the PM via cytopathic vacuoles. We characterized these cytopathic vacuoles using TEM, and live imaging has shown that they contain E2 glycoproteins on their membranes. NCs were also found on the outer membrane of these vacuoles by TEM analysis. As the virus assembly sites are established on the PM, the budded virions utilize filopodial extensions for spreading away from the infected cells.
Similar to the WT SINV, in the mCherry-E2 virus construct, the furin cleavage occurs after the E3 coding sequence, but before mCherry-E2. Data from our virus characterization and imaging experiments of the mCherry-E2 virus suggest that the presence of mCherry after the furin cleavage site on pE2 does not cause significant virus assembly and entry defects. While the E3 protein is cleaved in the Golgi from pE2 to yield the mature E2 protein, E3 stays associated with Venezuelan equine encephalitic virus (VEEV) even after furin cleavage, as evidenced from the cryoEM structure of mature VEEV [
44]. Although, in this structure, densities could be attributed to the two alpha-helices of E3, due to disconnected densities, a high resolution E3 density map was not obtained for the cryoEM map of mature VEEV containing cleaved E3. Nevertheless, the observed E3 density decorating the outermost portion of E2 above subdomains A and B was similar to the position of E3 in the pE2 cleavage-impaired, immature SINV mutant virus [
45]. These observations have suggested that E3 functions to maintain the relative orientation between E2 subdomains A and B, so as to protect the E1 fusion loop from premature exposure to the host membranes [
4,
40]. However, E3 does not stay associated with mature SINV after cleavage [
6,
40]. In our FP-tagged virus, the mCherry density is buried between neighboring glycoprotein spikes and does not occupy the position of E3 over the E2 acid-sensitive region. We hypothesize that this property of the FP-tagged virus is possibly because of the flexible linker region between E3 and E2 (between E3 and mCherry in the FP-tagged virus) that allows sufficient movement of E3 to still maintain its position on E2 to protect the acid-sensitive region of E2 during glycoprotein maturation of the mCherry-E2 virus. Thus, our cryoEM structure explains the unusual stability of the FP-tagged virus.
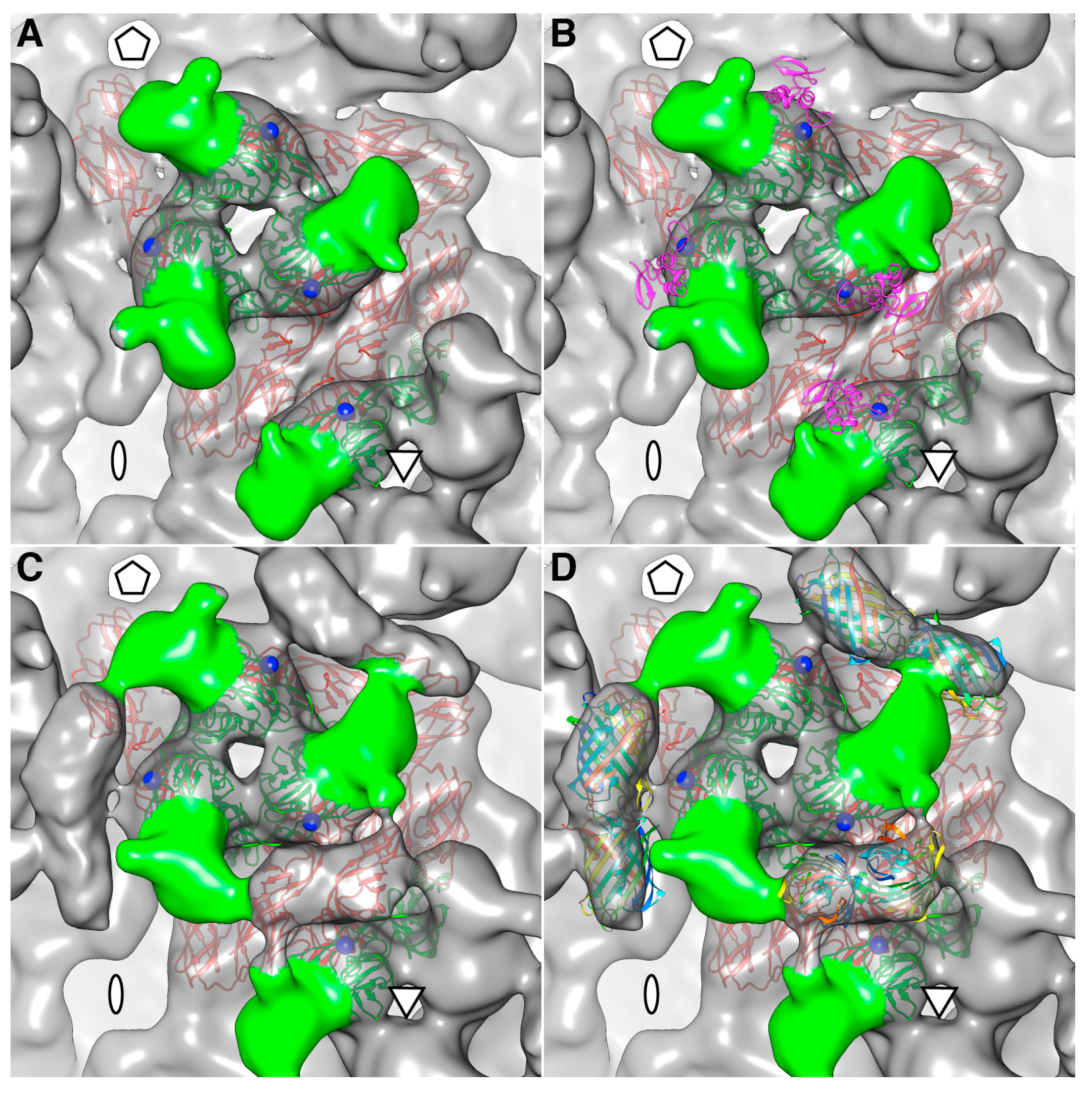
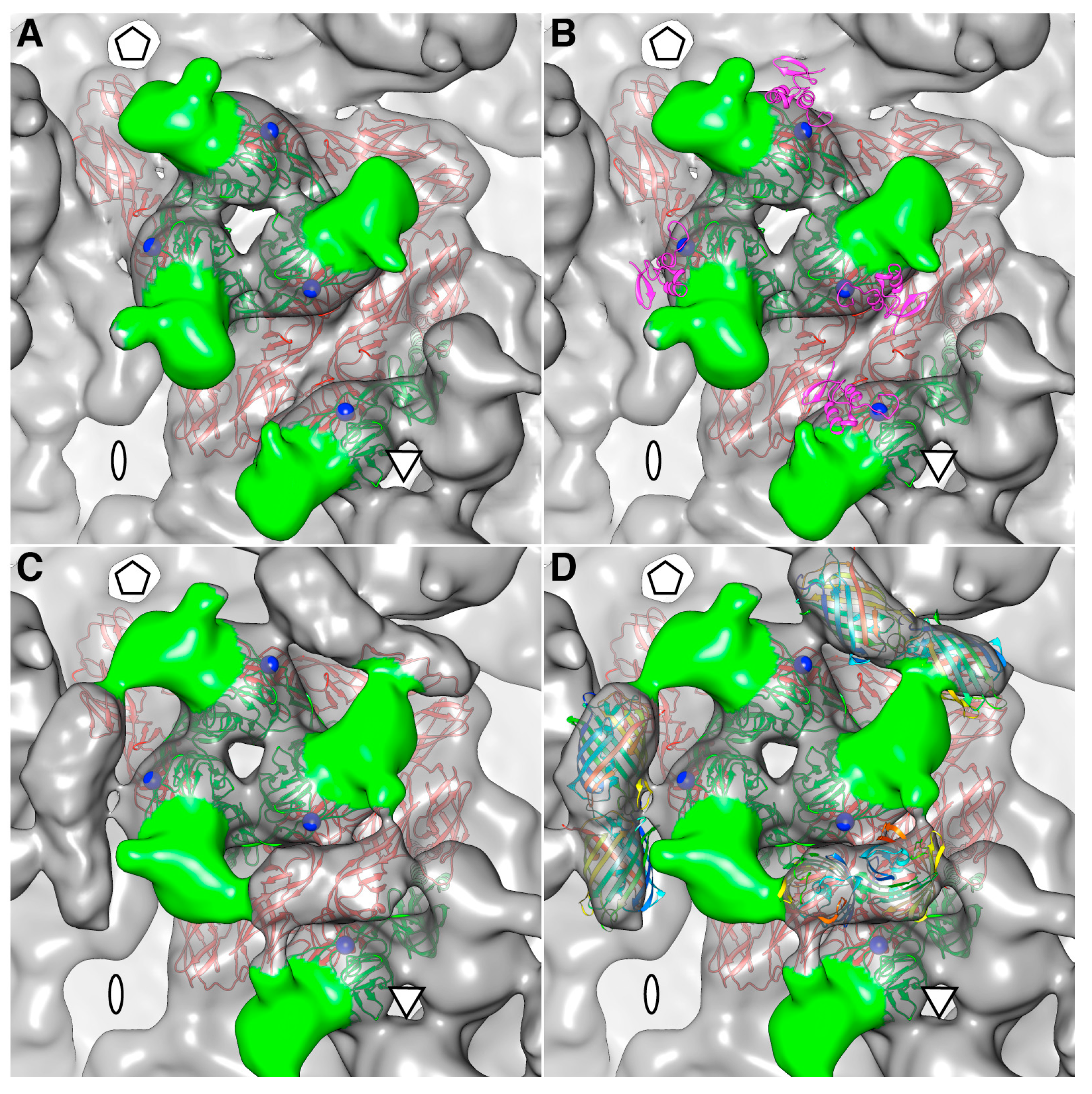
The density map of the mCherry-E2 virus reveals strong density extending from the N-terminus of E2 which is absent in the wild-type virus (
Figure 4C). This density of mCherry can be seen near the five-fold axis between adjacent spikes in a close-up surface view of the virus. The shape and volume of the extra density closely fit the red fluorescent protein (RFP) crystal structure of a dimer but come from two different adjacent E2 molecules (
Figure 4D) from two different spikes. Additionally, mCherry appears to make several contacts with the glycoprotein spikes, possibly adding to the stability of the tag. Similar observations were reported for a cryoEM density map of HSV-1 with a GFP-labeled UL17 capsid protein where the freedom of movement of the GFP tag was restricted due to the contact between the GFP tag and capsid density that was sufficient to prevent delocalization of the tag density but without abrogating formation of the capsid vertex-specific component heterodimer [
26]. The mCherry tag thus gives additional stability for the FP-tagged virus and explains the accommodation of 240 copies of the mCherry molecule without increasing the diameter of the particles.
Correlative light and electron microscopy (CLEM) studies using fluorescently tagged SINV have indicated the importance of filopodial extensions as preferred sites for alphavirus production, and they appear to mediate cell-cell virus particle transfer [
18]. By live imaging, Martinez
et al. have shown that long cellular extensions are involved in alphavirus cell-to-cell particle transfer [
18]. Importantly, using fluorescent SINV virions, we demonstrate single-particle budding that spread from infected cells via filopodial extensions. Such virus budding was absent from cells transfected with RNA from an FP-tagged, cdE2 budding mutant. Using live imaging experiments that utilize FP-tagged viruses, we demonstrate that, in infected BHK cells, fluorescent vesicles containing glycoproteins are transported to the PM. These vesicles presumably originate from late Golgi and we provide evidence for the association of glycoprotein E2 with Golgi using a live imaging-compatible Golgi stain.
In mammalian cells, the viral glycoproteins reside on the membranes of ER, Golgi, CPV-II, and PM, and the virus eventually buds from the PM. We showed that the released FP-tagged virus could be immobilized onto the PM of the infected cells by low pH-mediated fusion at pH 5, confirming that the virus particles are outside the cell. The budded virions that fuse to the PM lost their fluorescence when the cells were treated at pH 4, which is below the pK
a of mCherry. Along with the fused virions, fluorescent glycoprotein spikes present on the PM also lost their fluorescence whereas the mCherry-E2 molecules inside the infected cells were protected from the low pH 4 treatment of the cell. Consistent with our previous findings [
5,
15], we show that the interaction of NC with the cell-surface glycoproteins generates virions that get propelled by the filopodial extensions, and we hypothesize that this process facilitates viral dissemination while preventing superinfection. SINV attachment factors such as heparan sulfate [
46] and entry receptors such as NRAMP (divalent metal, ion transporter natural resistance-associated macrophage protein) [
47] have been shown to enhance viral infection. These host factors and the mechanism of receptor-mediated endocytosis can be further investigated with this FP-tagged virus. Exploiting the FP-tagged mutant viruses generated in this study in conjunction with live imaging-compatible stains and labeled host proteins, high-resolution live imaging studies are ongoing with an aim to understand the various molecular interactions between viral glycoproteins and host proteins that are required for productive alphavirus receptor binding, entry, and fusion. Such high-resolution live imaging studies will provide new spatial and temporal information regarding various steps in the alphavirus lifecycle.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}