
Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions


Abstract
:1. Introduction

2. Characteristics of an Ideal Cell Carrier Oncolytic Virotherapy

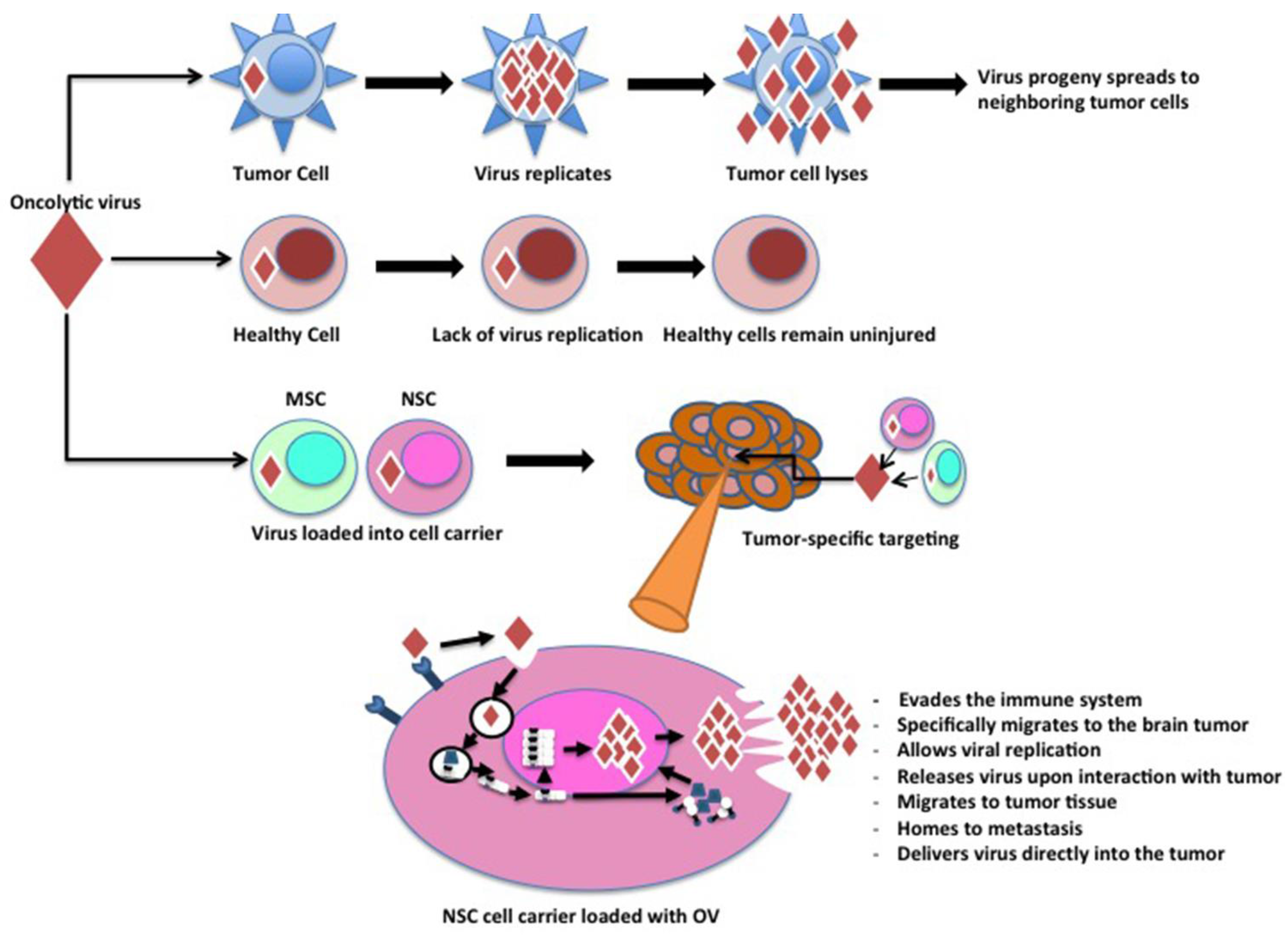{kind=link}
{kind=link}
| Type of Carrier | Advantages | Disadvantages | Reference |
|---|---|---|---|
| Transformed Cancer Cells | |||
| Solid Tumors | Often stimulate antitumor immunity. Support rapid replication of the viruses they carry. Easy to inject. | Large size limits which tumor forms they can treat. Can cause new metastases. Administer low amounts of virus because of immediate immune responses upon injection. | [18,19,20,21] |
| Hematopoietic and lymphoid tumors | Kinesis via the circulatory system. | Rapid proliferation rate can lead to de novo tumors. Elicit immune response, reducing amount of virus delivered. | [15] |
| Xenogeneic/allogeneic | Injected cells are destroyed, preventing de novo metastases. | Immune response is profound, limits delivery because or side effects and rejection of injected cells. | [6] |
| Immune Cells | |||
| T cells | Home to metastases. Activated at tumor cite, release virus specifically into tumor. Do not elicit immune response. | Strong preference to be loaded with reoviruses. Usually refractory to viral infection in vivo. | [22,23,24] |
| Activated T Cells | Increased ability to take up viruses. Efficacy of viral treatments increases. | Activation is lengthy and tedious. Do not support all viruses. | [25,26] |
| CIKs | Home to tumors. Release high amounts of viruses upon reaching the tumor. Can affect a variety of tumor types. | Requires expansion of primary leukocytes using cytokines in vivo. | [27] |
| Progenitor Cells | |||
| Blood outgrowth endothelial cells | Very targeted delivery because of ability to become incorporated into tumor neovasculature Divide successfully and rapidly in vivo. | Cells are not immortal, new cells must be isolated from clinical samples. Currently unknown if they can support infection with replicating therapeutics. | [28] |
| Mesenchymal Stem Cells | Migrate to the tumor tissue. Allow viral replication. Release virus upon interaction with tumor. Evade the immune system. | High amount of non-specific migration in some cancers. Must be harvested from bone marrow. | [29,30] |
| Neural Stem Cells | Specifically migrate to brain tumors. Allow viral replication. Evade the immune system. | Require stereotactical extraction of cells from the subventricular zone. | [31,32,33] |
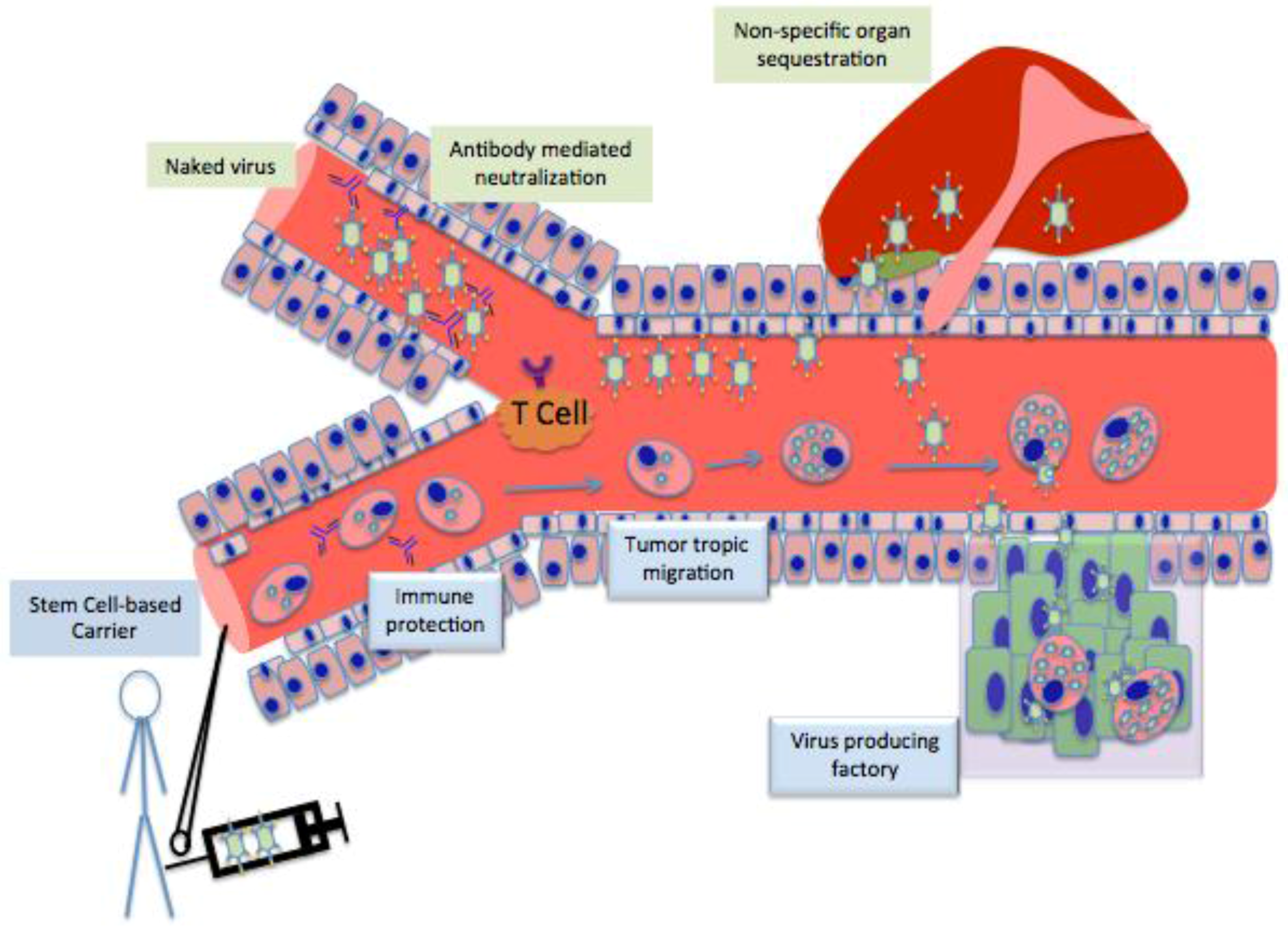
3. Stem cell as Cell Carrier
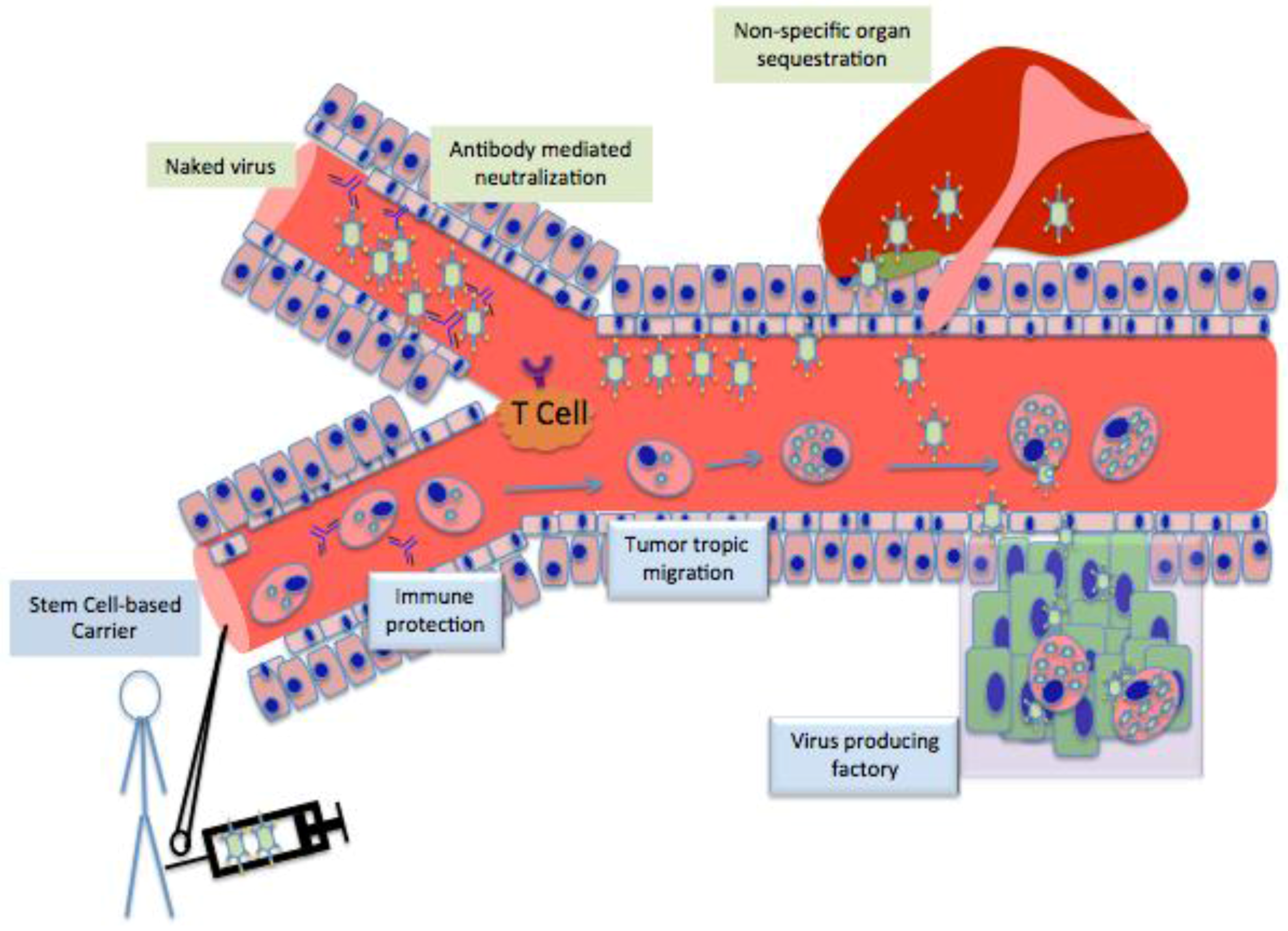
4. Different Stem Cells as Cell Carrier for Oncolytic Vitrotherpy
| Type of Stem Cell Carrier | Species of Origin | Type of Virus | Type of Cancer Treated | Result | Reference |
|---|---|---|---|---|---|
| Bone marrow-derived mesenchymal stromal cells | Human | Oncolytic Adenovirus | Pancreatic | Capsid modification leads to enhancement of therapeutic viral loading onto MSC-based cell carriers (Engineered 5/3 fiber chimerism adenoviruses enter MSCs at a 35-to 3310-fold rate compared to adenovirus 5 wild type capsid.) | [59] |
| Bone marrow-derived mesenchymal stem cells | Human | Osteocalcin promoter-directed Ad-hOC-E1 oncolytic adenovirus | Renal Cell Carcinoma | Injection of pharmaceutical inducible MSC carrying oncolytic adenovirus combined with vitamin D3 treatment induced effective viral delivery to RCC tumors and significant tumor regression. These were significantly greater than those of injection of carrier-free Ad-hOC-E1. | [60] |
| Bone marrow-derived mesenchymal stem cells | Human | Adenovirus carrying the IFN-β gene | Glioblastoma | MSCs home to tumors in murine models. MSCs loaded with therapeutic virus injected intra-arterially prolonged median survival of animals. | [61] |
| Bone marrow-derived mesenchymal progenitor cells | Human | Adenovirus Ad5/3 | Ovarian | MSCs home to ovarian tumors, allow virus to replicate, and prolong survival in vivo. (Median survival time of 34 days for mice treated with PBS controls, 44 days for the uninfected MPC transplanted controls, but 69 days in the oncolytic virus-infected MPCs group.) | [62] |
| Bone marrow-derived mesenchymal stem cells | Human | Adenovirus Delta24-RGD | Glioblastoma | Carotid injections of MSCs loaded with therapeutic eradicated tumors, halted tumor growth, and prolonged survival. (Increase in median survival from 42 days to 75.5 days in murine in vivo models with autotrophic patient derived glioma.) | [63] |
| Mesenchymal stem cells derived from ovarian cancer patients (ovMSC) | Human | Measles | Ovarian | Migration of ovMSCs to tumors was comparable to that of MSCs derived from healthy donors. Delivery of virus in vivo using mice passively immune to measles yielded similar results upon treatment with MSCs and ovMSCs, both of which elicited longer survival than naked measles virus injection alone. (Median survival for PBS control is 36 days, 37 days for MV injection, but 82 days for MSC/MV injection.) | [64] |
| Bone marrow-derived mesenchymal stem cells | Human | Measles | Hepatocellular Carcinoma | Systemically delivered MSCs homed to HCC tumors implanted in the liver. MSCs effectively transferred MVs via heterofusion. The therapy inhibited tumor growth in passively immunized SCID mice, which did not occur upon naked MV injections. | [34] |
| Immortalized fetal brain-derived neural stem cells | Human | Adenoviral vector CRAd-S-pk7 | Glioblastoma Multiforme | Viral loaded NSC therapy, when delivered prior to, rather than after conventional therapy prompts 30% longer survival in mice with autotrophic patient-derived glioma compared to application after therapy. (Adenoviral loaded NSC injections in conjunction with XRT-TMZ treatments increased median murine survival 46% compared to XRT-TMZ alone.) | [65] |
| Immortalized neural stem cell type HB1.F3-CD derived from fetal brain | Human | Adenoviral vector CRAd-S-pk7 | Glioblastoma Multiforme | Virus delivered via NSC carrier was localized within the injected hemisphere. NSC carrier cells handed off the therapeutic virus to tumors within 5 days post-injection in vivo in mice with autotrophic patient-derived glioma. | [66] |
| Human fetal brain-derived neural Stem Cells | Human | Adenovirus | Glioblastoma Multiforme | NSCs are superior viral cell carriers to MSCs in targeting glioma. NSCs release virus at an amount a log higher than MSCs (p < 0.001). NSCs injected intracranially in an orthotropic glioma model increased the survival of tumor bearing animals more robustly than MSCs (median survival for NSCs 68.5 days against 44 days for MSCs, p < 0.002) | [31] |
5. What Next for Stem Cell-Based Cell Carrier?
6. Limitations and How to Overcome Them
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Yang, X.; Chen, E.; Jiang, H.; Muszynski, K.; Harris, R.D.; Giardina, S.L.; Gromeier, M.; Mitra, G.; Soman, G. Evaluation of ires-mediated, cell-type-specific cytotoxicity of poliovirus using a colorimetric cell proliferation assay. J. Virol. Methods 2009, 155, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Breton, C.; Barber, G.N.; Russell, S.J.; Peng, K.W. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum. Gene Ther. 2012, 23, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Willmon, C.; Harrington, K.; Kottke, T.; Prestwich, R.; Melcher, A.; Vile, R. Cell carriers for oncolytic viruses: Fed ex for cancer therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Power, A.T.; Wang, J.; Falls, T.J.; Paterson, J.M.; Parato, K.A.; Lichty, B.D.; Stojdl, D.F.; Forsyth, P.A.; Atkins, H.; Bell, J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic viruses for cancer therapy: Overcoming the obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Alvarez, B.; Rodriguez, R.M.; Calvanese, V.; Blanco-Gelaz, M.A.; Suhr, S.T.; Ortega, F.; Otero, J.; Cibelli, J.B.; Moore, H.; Fraga, M.F.; et al. Epigenetic mechanisms regulate mhc and antigen processing molecules in human embryonic and induced pluripotent stem cells. PLoS ONE 2010, 5, e10192. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Alvarez-Vallina, L.; Crittenden, M.; Gough, M.; Chong, H.; Diaz, R.M.; Vassaux, G.; Lemoine, N.; Vile, R. Cells as vehicles for cancer gene therapy: The missing link between targeted vectors and systemic delivery? Hum. Gene Ther. 2002, 13, 1263–1280. [Google Scholar] [CrossRef] [PubMed]
- Chester, J.; Ruchatz, A.; Gough, M.; Crittenden, M.; Chong, H.; Cosset, F.L.; Diaz, R.M.; Harrington, K.; Alvarez-Vallina, L.; Vile, R. Tumor antigen-specific induction of transcriptionally targeted retroviral vectors from chimeric immune receptor-modified t cells. Nat. Biotechnol. 2002, 20, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; et al. Dc-sign, a dendritic cell-specific hiv-1-binding protein that enhances trans-infection of t cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Cole, C.; Qiao, J.; Kottke, T.; Diaz, R.M.; Ahmed, A.; Sanchez-Perez, L.; Brunn, G.; Thompson, J.; Chester, J.; Vile, R.G. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific t cells. Nat. Med. 2005, 11, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Southam, C.M. Present status of oncolytic virus studies. Trans. N. Y. Acad. Sci. 1960, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quintanilla, J.; He, D.; Wakimoto, H.; Alemany, R.; Shah, K. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Iankov, I.D.; Blechacz, B.; Liu, C.; Schmeckpeper, J.D.; Tarara, J.E.; Federspiel, M.J.; Caplice, N.; Russell, S.J. Infected cell carriers: A new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Tsai, V.; Johnson, D.E.; Rahman, A.; Wen, S.F.; LaFace, D.; Philopena, J.; Nery, J.; Zepeda, M.; Maneval, D.C.; Demers, G.W.; et al. Impact of human neutralizing antibodies on antitumor efficacy of an oncolytic adenovirus in a murine model. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7199–7206. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Kovesdi, I.; Bruder, J.T. Effective repeat administration with adenovirus vectors to the muscle. Gene Ther. 2000, 7, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Peichev, M.; Naiyer, A.J.; Pereira, D.; Zhu, Z.; Lane, W.J.; Williams, M.; Oz, M.C.; Hicklin, D.J.; Witte, L.; Moore, M.A.; et al. Expression of vegfr-2 and ac133 by circulating human cd34(+) cells identifies a population of functional endothelial precursors. Blood 2000, 95, 952–958. [Google Scholar] [PubMed]
- Quirici, N.; Soligo, D.; Caneva, L.; Servida, F.; Bossolasco, P.; Deliliers, G.L. Differentiation and expansion of endothelial cells from human bone marrow cd133(+) cells. Br. J. Haematol. 2001, 115, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Jevremovic, D.; Gulati, R.; Hennig, I.; Diaz, R.M.; Cole, C.; Kleppe, L.; Cosset, F.L.; Simari, R.D.; Vile, R.G. Use of blood outgrowth endothelial cells as virus-producing vectors for gene delivery to tumors. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H494–H500. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Morishita, N.; Mitsuoka, C.; Kitajima, S.; Hamada, K.; Lee, K.M.; Kawabata, M.; Fujisawa, M.; Shirakawa, T. Intravenous injection of irradiated tumor cell vaccine carrying oncolytic adenovirus suppressed the growth of multiple lung tumors in a mouse squamous cell carcinoma model. J. Gene Med. 2011, 13, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Thorne, S.H.; Liang, W.C.; Sampath, P.; Schmidt, T.; Sikorski, R.; Beilhack, A.; Contag, C.H. Targeting localized immune suppression within the tumor through repeat cycles of immune cell-oncolytic virus combination therapy. Mol. Ther. 2010, 18, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.J.; Prestwich, R.J.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Vile, R.G.; et al. Dendritic cells and t cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.H.; Jerebtsova, M.; Ray, P.E. Liver bypass significantly increases the transduction efficiency of recombinant adenoviral vectors in the lung, intestine, and kidney. Hum. Gene Ther. 2000, 11, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Parimi, V.; O'Malley, M.E.; Thirunavukarasu, P.; Sathaiah, M.; Austin, F.; Bartlett, D.L. The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene Ther. 2010, 17, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, D.C.; Charlton, D.; Henderson, D.R. Pre-existent adenovirus antibody inhibits systemic toxicity and antitumor activity of cn706 in the nude mouse lncap xenograft model: Implications and proposals for human therapy. Hum. Gene Ther. 2000, 11, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Verneris, M.R.; Ito, M.; Shizuru, J.A.; Negrin, R.S. Expansion of cytolytic cd8(+) natural killer t cells with limited capacity for graft-versus-host disease induction due to interferon gamma production. Blood 2001, 97, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Weisdorf, D.J.; Solovey, A.; Hebbel, R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Investig. 2000, 105, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Thorne, S.H.; Negrin, R.S.; Contag, C.H. Synergistic antitumor effects of immune cell-viral biotherapy. Science 2006, 311, 1780–1784. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hu, G.; Su, J.; Li, W.; Chen, Q.; Shou, P.; Xu, C.; Chen, X.; Huang, Y.; Zhu, Z.; et al. Mesenchymal stem cells: A new strategy for immunosuppression and tissue repair. Cell Res. 2010, 20, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U.; Tyler, M.A.; Thaci, B.; Alexiades, N.G.; Han, Y.; Ulasov, I.V.; Lesniak, M.S. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 2011, 8, 1559–1572. [Google Scholar] [CrossRef] [PubMed]
- Einstein, O.; Ben-Hur, T. The changing face of neural stem cell therapy in neurologic diseases. Arch. Neurol. 2008, 65, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Aligholi, H.; Hassanzadeh, G.; Azari, H.; Rezayat, S.M.; Mehr, S.E.; Akbari, M.; Attari, F.; Khaksarian, M.; Gorji, A. A new and safe method for stereotactically harvesting neural stem/progenitor cells from the adult rat subventricular zone. J. Neurosci. Methods 2014, 225, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U.; Thaci, B.; Tobias, A.L.; Auffinger, B.; Zhang, L.; Cheng, Y.; Kim, C.K.; Yunis, C.; Han, Y.; Alexiades, N.G.; et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl. Cancer Inst. 2013, 105, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Aboody, K.S.; Brown, A.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M.; et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, S.; Pirola, B.; Pollo, B.; Magrassi, L.; Bruzzone, M.G.; Rigamonti, D.; Galli, R.; Selleri, S.; Di Meco, F.; De Fraja, C.; et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nat. Med. 2000, 6, 447–450. [Google Scholar] [PubMed]
- Herrlinger, U.; Woiciechowski, C.; Sena-Esteves, M.; Aboody, K.S.; Jacobs, A.H.; Rainov, N.G.; Snyder, E.Y.; Breakefield, X.O. Neural precursor cells for delivery of replication-conditional hsv-1 vectors to intracerebral gliomas. Mol. Ther. J. Am. Soc. Gene Ther. 2000, 1, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Studeny, M.; Marini, F.C.; Dembinski, J.L.; Zompetta, C.; Cabreira-Hansen, M.; Bekele, B.N.; Champlin, R.E.; Andreeff, M. Mesenchymal stem cells: Potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J. Natl. Cancer Inst. 2004, 96, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Najbauer, J.; Annala, A.J.; Garcia, E.; Metz, M.Z.; Gutova, M.; Polewski, M.D.; Gilchrist, M.; Glackin, C.A.; Kim, S.U.; et al. Human neural stem cell tropism to metastatic breast cancer. Stem Cells 2012, 30, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, D.; Zhou, J.; Cheng, Y.; Liang, T.; Zhang, G. Characteristics of human amniotic fluid mesenchymal stem cells and their tropism to human ovarian cancer. PLoS ONE 2015, 10, e0123350. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Huang, B.; Zhang, T.Y.; Miao, P.H.; Tang, G.P.; Tabata, Y.; Gao, J.Q. Mesenchymal stem cells as a novel carrier for targeted delivery of gene in cancer therapy based on nonviral transfection. Mol. Pharm. 2012, 9, 2698–2709. [Google Scholar] [CrossRef] [PubMed]
- Mader, E.K.; Maeyama, Y.; Lin, Y.; Butler, G.W.; Russell, H.M.; Galanis, E.; Russell, S.J.; Dietz, A.B.; Peng, K.W. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7246–7255. [Google Scholar] [CrossRef] [PubMed]
- Carbajal, K.S.; Schaumburg, C.; Strieter, R.; Kane, J.; Lane, T.E. Migration of engrafted neural stem cells is mediated by cxcl12 signaling through cxcr4 in a viral model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2010, 107, 11068–11073. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.O.; Koeder, D.; Messing, M.; Mueller, F.J.; Aboody, K.S.; Kim, S.U.; Black, P.M.; Carroll, R.S.; Westphal, M.; Lamszus, K. Vascular endothelial growth factor-stimulated cerebral microvascular endothelial cells mediate the recruitment of neural stem cells to the neurovascular niche. Brain Res. 2009, 1268, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Heese, O.; Disko, A.; Zirkel, D.; Westphal, M.; Lamszus, K. Neural stem cell migration toward gliomas in vitro. Neuro-Oncology 2005, 7, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Lee, J.; Fine, H.A. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J. Clin. Investig. 2004, 113, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Hossain, A.; Takezaki, T.; Fueyo, J.; Gumin, J.; Gao, F.; Nwajei, F.; Marini, F.C.; Andreeff, M.; Kuratsu, J.; et al. Tgf-beta mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 2013, 73, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Takenaga, K. Angiogenic signaling aberrantly induced by tumor hypoxia. Front. Biosci. 2011, 16, 31–48. [Google Scholar] [CrossRef]
- Zhao, D.; Najbauer, J.; Garcia, E.; Metz, M.Z.; Gutova, M.; Glackin, C.A.; Kim, S.U.; Aboody, K.S. Neural stem cell tropism to glioma: Critical role of tumor hypoxia. Mol. Cancer Res. MCR 2008, 6, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U.; Thaci, B.; Alexiades, N.G.; Han, Y.; Qian, S.; Liu, F.; Balyasnikova, I.V.; Ulasov, I.Y.; Aboody, K.S.; Lesniak, M.S. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Rattigan, Y.; Hsu, J.M.; Mishra, P.J.; Glod, J.; Banerjee, D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp. Cell Res. 2010, 316, 3417–3424. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. Vsv-tumor selective replication and protein translation. Oncogene 2005, 24, 7710–7719. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Kim, C.; Wu, W.; Shin, D.M.; Bryndza, E.; Kucia, M.; Ratajczak, J. The role of innate immunity in trafficking of hematopoietic stem cells-an emerging link between activation of complement cascade and chemotactic gradients of bioactive sphingolipids. Adv. Exp. Med. Biol. 2012, 946, 37–54. [Google Scholar] [PubMed]
- Abumaree, M.; Al Jumah, M.; Pace, R.A.; Kalionis, B. Immunosuppressive properties of mesenchymal stem cells. Stem Cell Rev. 2012, 8, 375–392. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Thanunchai, M.; Hongeng, S.; Thitithanyanont, A. Mesenchymal stromal cells and viral infection. Stem Cells Int. 2015, 2015, 860950. [Google Scholar] [CrossRef] [PubMed]
- Mammolenti, M.; Gajavelli, S.; Tsoulfas, P.; Levy, R. Absence of major histocompatibility complex class i on neural stem cells does not permit natural killer cell killing and prevents recognition by alloreactive cytotoxic t lymphocytes in vitro. Stem Cells 2004, 22, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U.; Rolle, C.E.; Tyler, M.A.; Han, Y.; Sengupta, S.; Wainwright, D.A.; Balyasnikova, I.V.; Ulasov, I.V.; Lesniak, M.S. Bone marrow mesenchymal stem cells loaded with an oncolytic adenovirus suppress the anti-adenoviral immune response in the cotton rat model. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Hammer, K.; Kazcorowski, A.; Liu, L.; Behr, M.; Schemmer, P.; Herr, I.; Nettelbeck, D.M. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int. J. Cancer. J. Int. Cancer 2015, 137, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.C.; Sung, S.Y.; Liao, C.H.; Wu, H.C.; Hsieh, C.L. Vitamin d3-inducible mesenchymal stem cell-based delivery of conditionally replicating adenoviruses effectively targets renal cell carcinoma and inhibits tumor growth. Mol. Pharm. 2012, 9, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Nakamizo, A.; Marini, F.; Amano, T.; Khan, A.; Studeny, M.; Gumin, J.; Chen, J.; Hentschel, S.; Vecil, G.; Dembinski, J.; et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005, 65, 3307–3318. [Google Scholar] [PubMed]
- Komarova, S.; Kawakami, Y.; Stoff-Khalili, M.A.; Curiel, D.T.; Pereboeva, L. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol. Cancer Ther. 2006, 5, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Yong, R.L.; Shinojima, N.; Fueyo, J.; Gumin, J.; Vecil, G.G.; Marini, F.C.; Bogler, O.; Andreeff, M.; Lang, F.F. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus delta24-rgd to human gliomas. Cancer Res. 2009, 69, 8932–8940. [Google Scholar] [CrossRef] [PubMed]
- Mader, E.K.; Butler, G.; Dowdy, S.C.; Mariani, A.; Knutson, K.L.; Federspiel, M.J.; Russell, S.J.; Galanis, E.; Dietz, A.B.; Peng, K.W. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase i clinical trial in ovarian cancer. J. Transl. Med. 2013, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tobias, A.L.; Thaci, B.; Auffinger, B.; Rincon, E.; Balyasnikova, I.V.; Kim, C.K.; Han, Y.; Zhang, L.; Aboody, K.S.; Ahmed, A.U.; et al. The timing of neural stem cell-based virotherapy is critical for optimal therapeutic efficacy when applied with radiation and chemotherapy for the treatment of glioblastoma. Stem Cells Transl. Med. 2013, 2, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Thaci, B.; Ahmed, A.U.; Ulasov, I.V.; Tobias, A.L.; Han, Y.; Aboody, K.S.; Lesniak, M.S. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: Targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012, 19, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, G.; Fox, J.; Ashton, B.; Middleton, J. Concise review: Mesenchymal stem cells: Their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells 2007, 25, 2739–2749. [Google Scholar] [CrossRef] [PubMed]
- Sonabend, A.M.; Ulasov, I.V.; Tyler, M.A.; Rivera, A.A.; Mathis, J.M.; Lesniak, M.S. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells 2008, 26, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Stoff-Khalili, M.A.; Rivera, A.A.; Mathis, J.M.; Banerjee, N.S.; Moon, A.S.; Hess, A.; Rocconi, R.P.; Numnum, T.M.; Everts, M.; Chow, L.T.; et al. Mesenchymal stem cells as a vehicle for targeted delivery of crads to lung metastases of breast carcinoma. Breast Cancer Res. Treat. 2007, 105, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Ji, T.; Chen, P.; Li, X.; Fang, Y.; Gao, Q.; Liao, S.; You, L.; Xu, H.; Ma, Q.; et al. Mesenchymal stem cells as carriers and amplifiers in crad delivery to tumors. Mol. Cancer 2011, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, J.L.; Spaeth, E.L.; Fueyo, J.; Gomez-Manzano, C.; Studeny, M.; Andreeff, M.; Marini, F.C. Reduction of nontarget infection and systemic toxicity by targeted delivery of conditionally replicating viruses transported in mesenchymal stem cells. Cancer Gene Ther. 2010, 17, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bergelson, J.M. Adenovirus receptors. J. Virol. 2005, 79, 12125–12131. [Google Scholar] [CrossRef] [PubMed]
- Hakkarainen, T.; Sarkioja, M.; Lehenkari, P.; Miettinen, S.; Ylikomi, T.; Suuronen, R.; Desmond, R.A.; Kanerva, A.; Hemminki, A. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Hum. Gene Ther. 2007, 18, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Duebgen, M.; Martinez-Quintanilla, J.; Tamura, K.; Hingtgen, S.; Redjal, N.; Wakimoto, H.; Shah, K. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J. Natl. Cancer Inst. 2014, 106, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Kauer, T.M.; Figueiredo, J.L.; Hingtgen, S.; Shah, K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 2012, 15, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Cattaneo, E. Neural stem cell systems: Physiological players or in vitro entities? Nat. Rev. Neurosci. 2010, 11, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Tyler, M.A.; Ulasov, I.V.; Sonabend, A.M.; Nandi, S.; Han, Y.; Marler, S.; Roth, J.; Lesniak, M.S. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2009, 16, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Zhu, Z.B.; Tyler, M.A.; Han, Y.; Rivera, A.A.; Khramtsov, A.; Curiel, D.T.; Lesniak, M.S. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum. Gene Ther. 2007, 18, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Oreffo, R.O.; Cooper, C.; Mason, C.; Clements, M. Mesenchymal stem cells: Lineage, plasticity, and skeletal therapeutic potential. Stem Cell Rev. 2005, 1, 169–178. [Google Scholar] [CrossRef]
- Aboody, K.S.; Najbauer, J.; Danks, M.K. Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther. 2008, 15, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kane, J.R.; Young, J.S.; Chang, A.L.; Kanojia, D.; Qian, S.; Spencer, D.A.; Ahmed, A.U.; Lesniak, M.S. Neural stem cell-mediated delivery of oncolytic adenovirus. Curr. Protoc. Hum. Genet. [CrossRef]
- Alexiades, N.G.; Auffinger, B.; Kim, C.K.; Hasan, T.; Lee, G.; Deheeger, M.; Tobias, A.L.; Kim, J.; Balyasnikova, I.; Lesniak, M.S.; et al. Mmp14 as a novel downstream target of vegfr2 in migratory glioma-tropic neural stem cells. Stem Cell Res. 2015, 15, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Amariglio, N.; Hirshberg, A.; Scheithauer, B.W.; Cohen, Y.; Loewenthal, R.; Trakhtenbrot, L.; Paz, N.; Koren-Michowitz, M.; Waldman, D.; Leider-Trejo, L.; et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 2009, 6, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lam, D.H.; Goh, S.S.; Lee, E.X.; Zhao, Y.; Tay, F.C.; Chen, C.; Du, S.; Balasundaram, G.; Shahbazi, M.; et al. Tumor tropism of intravenously injected human-induced pluripotent stem cell-derived neural stem cells and their gene therapy application in a metastatic breast cancer model. Stem Cells 2012, 30, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Lujan, E.; Chanda, S.; Ahlenius, H.; Sudhof, T.C.; Wernig, M. Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2527–2532. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Zheng, Q.; Wu, J.; Xu, Z.; Wang, L.; Li, W.; Zhang, H.; Zhao, X.Y.; Liu, L.; Wang, Z.; et al. Direct reprogramming of sertoli cells into multipotent neural stem cells by defined factors. Cell Res. 2012, 22, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Thier, M.; Worsdorfer, P.; Lakes, Y.B.; Gorris, R.; Herms, S.; Opitz, T.; Seiferling, D.; Quandel, T.; Hoffmann, P.; Nothen, M.M.; et al. Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell 2012, 10, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Chiocca, E.A. Switching a replication-defective adenoviral vector into a replication-competent, oncolytic adenovirus. J. Virol. 2014, 88, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Thu, M.S.; Najbauer, J.; Kendall, S.E.; Harutyunyan, I.; Sangalang, N.; Gutova, M.; Metz, M.Z.; Garcia, E.; Frank, R.T.; Kim, S.U.; et al. Iron labeling and pre-clinical mri visualization of therapeutic human neural stem cells in a murine glioma model. PLoS ONE 2009, 4, e7218. [Google Scholar] [CrossRef] [PubMed]
- Morshed, R.A.; Gutova, M.; Juliano, J.; Barish, M.E.; Hawkins-Daarud, A.; Oganesyan, D.; Vazgen, K.; Yang, T.; Annala, A.; Ahmed, A.U.; et al. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using mri-based tracking and histological reconstruction. Cancer Gene Ther. 2015, 22, 55–61. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Hall, R.R.; Lesniak, M.S.; Ahmed, A.U. Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions. Viruses 2015, 7, 6200-6217. https://doi.org/10.3390/v7122921
Kim J, Hall RR, Lesniak MS, Ahmed AU. Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions. Viruses. 2015; 7(12):6200-6217. https://doi.org/10.3390/v7122921
Chicago/Turabian StyleKim, Janice, Robert R. Hall, Maciej S. Lesniak, and Atique U. Ahmed. 2015. "Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions" Viruses 7, no. 12: 6200-6217. https://doi.org/10.3390/v7122921
APA StyleKim, J., Hall, R. R., Lesniak, M. S., & Ahmed, A. U. (2015). Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions. Viruses, 7(12), 6200-6217. https://doi.org/10.3390/v7122921