
Natural Products as Tools for Defining How Cellular Metabolism Influences Cellular Immune and Inflammatory Function during Chronic Infection

Abstract
:
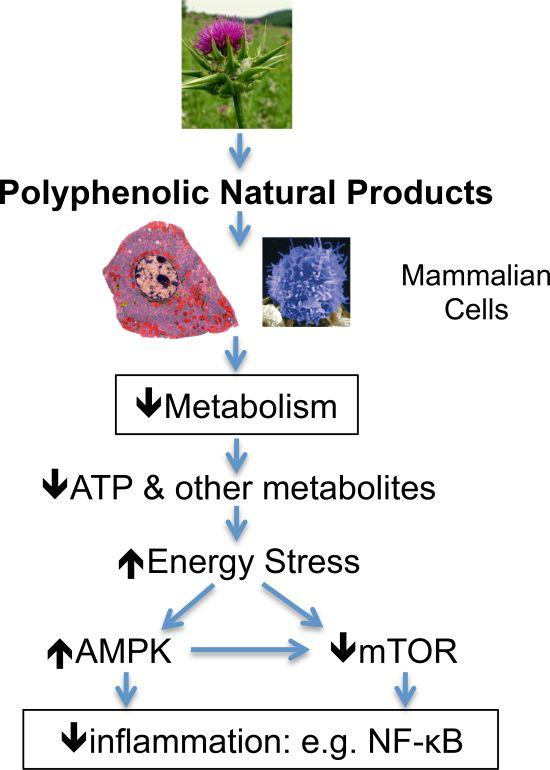{kind=link}
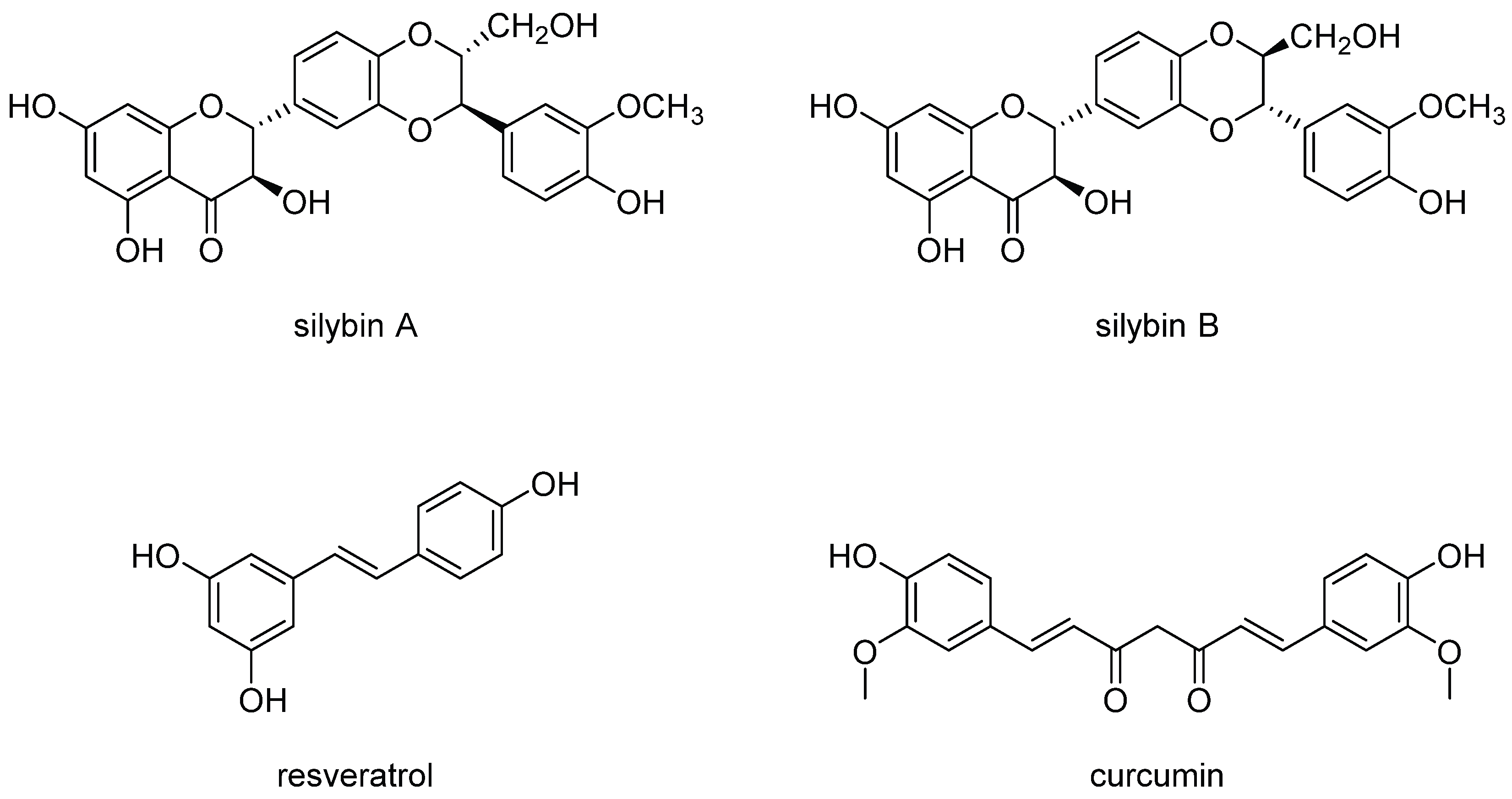
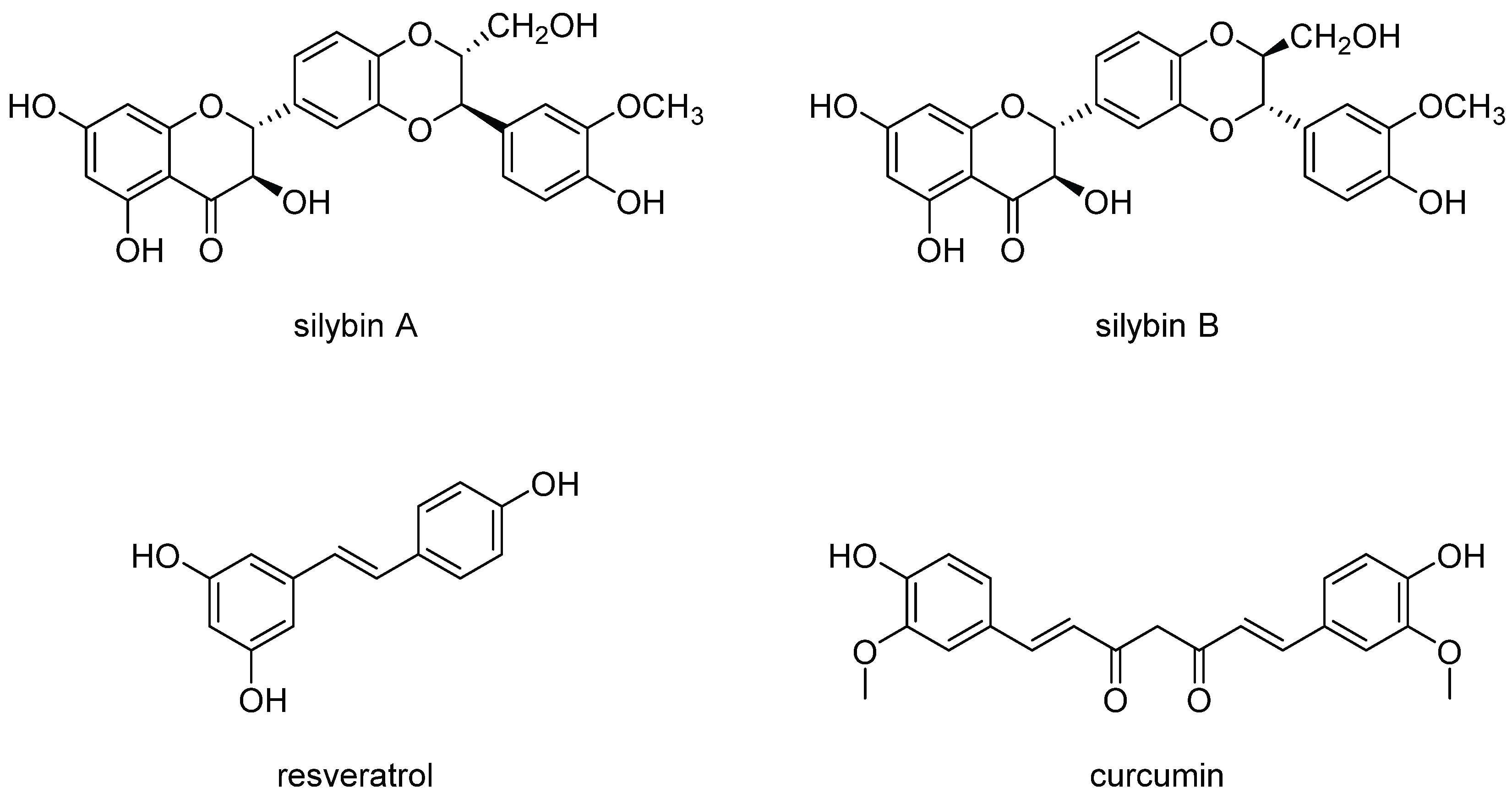{kind=link}
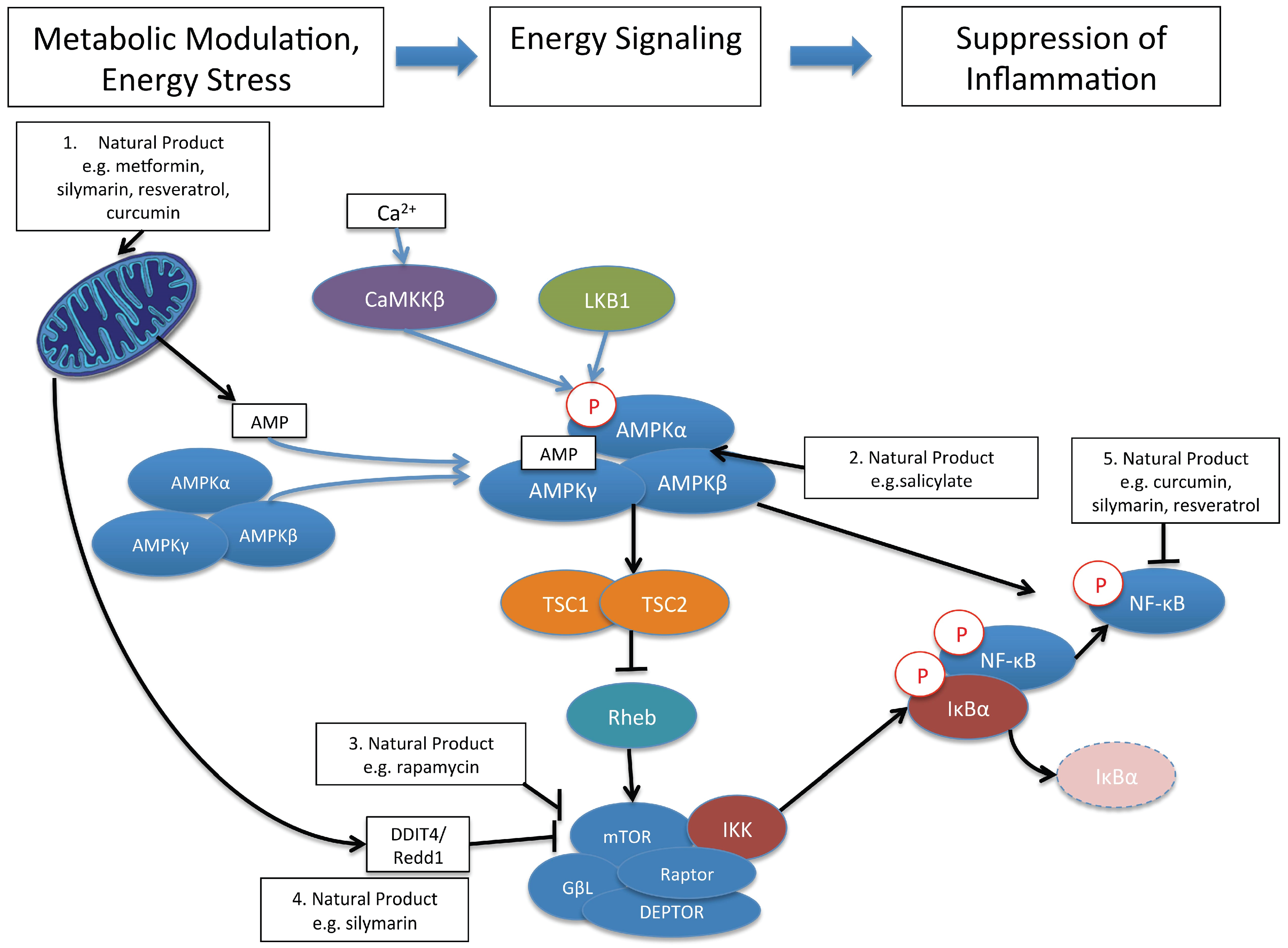{kind=link}
1. Introduction
2. Inflammation
3. Chronic Inflammation: The Case for HCV
4. Chronic Inflammation and the Road to HCV-Induced Liver Disease
5. Chronic Inflammation: The Case for HIV
6. Metabolic Modulators That Impact Immune and Inflammatory Responses
7. Immune Cell Activation
8. Natural Products to Quell Chronic Inflammation in HCV and HIV Infection
9. AMPK, and mTOR: Cellular Signaling Pathways That Link Cellular Metabolism to Function
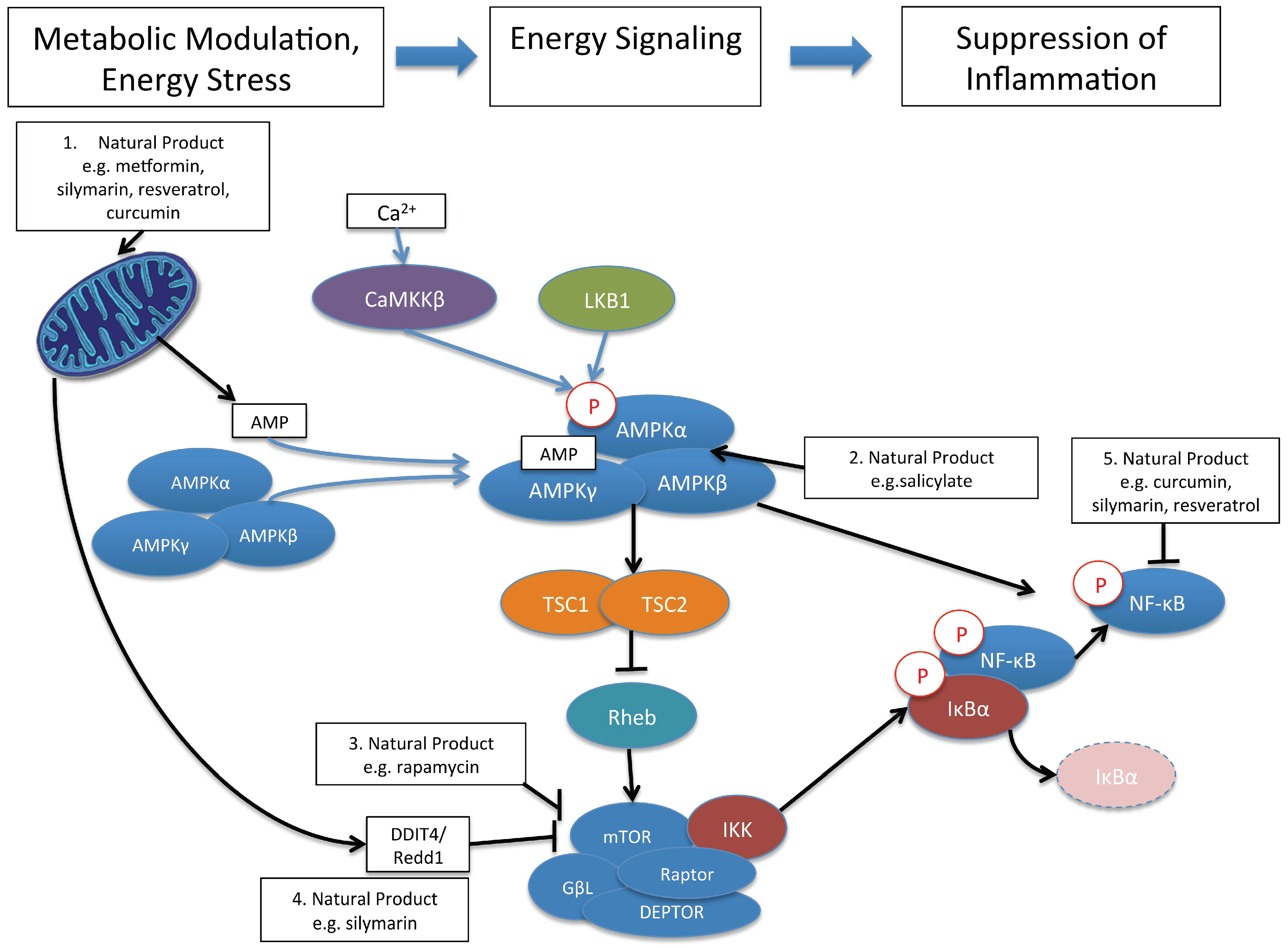
10. Control of Immune and Inflammatory Responses by Natural Product Modulation of Metabolic Signaling by AMPK and mTOR
Acknowledgments
Conflicts of Interest
References
- American Society of Pharmacognosy. Available online: http://www.pharmacognosy.us/ (accessed on 25 November 2015).
- Society for Medicinal Plant and Natural Product Research. Available online: http://www.ga-online.org/about_en.html (accessed on 25 November 2015).
- Pal, S. Complementary and alternative medicine: An overview. Curr. Sci. 2002, 82, 518–524. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Edlin, B.R. Perspective: Test and treat this silent killer. Nature 2011, 474, s18–s19. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A. Smallpox: Can we still learn from the journey to eradication? Indian J. Med. Res. 2013, 137, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Moutou, F. The second eradication: Rinderpest. Bull. Soc. Pathol. Exot. 2014, 107, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Available online: http://www.cdc.gov/polio/progress/index.htm (accessed on 25 November 2015).
- Smith, B.D.; Morgan, R.L.; Beckett, G.A.; Falck-Ytter, Y.; Holtzman, D.; Teo, C.G.; Jewett, A.; Baack, B.; Rein, D.B.; Patel, N.; et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. Morb. Mortal. Wkly. Rep. 2012, 61, 1–32. [Google Scholar]
- Li, K.; Chen, Z.; Kato, N.; Gale, M., Jr.; Lemon, S.M. Distinct poly(IC) and virus-activated signaling pathways leading to interferon-β production in hepatocytes. J. Biol. Chem. 2005, 280, 16739–16747. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.J.; Khabar, K.S.; Rezeiq, M.; Gretch, D.R. Elevated levels of interleukin-8 in serum are associated with hepatitis C virus infection and resistance to interferon therapy. J. Virol. 2001, 75, 6209–6211. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.; Wagoner, J.; Lovelace, E.S.; Thirstrup, D.; Mohar, I.; Smith, W.; Giugliano, S.; Li, K.; Crispe, I.N.; Rosen, H.R.; et al. Independent, parallel pathways to CXCL10 induction in HCV-infected hepatocytes. J. Hepatol. 2013, 59, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Cheent, K.; Khakoo, S.I. Natural killer cells and hepatitis C: Action and reaction. Gut 2011, 60, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Naftaly, M.; Friedman, S.L. Current status of novel antifibrotic therapies in patients with chronic liver disease. Ther. Adv. Gastroenterol. 2011, 4, 391–417. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, S.; Kriss, M.; Golden-Mason, L.; Dobrinskikh, E.; Stone, A.E.; Soto-Gutierrez, A.; Mitchell, A.; Khetani, S.R.; Yamane, D.; Stoddard, M.; et al. Hepatitis C virus infection induces autocrine interferon signaling by human liver endothelial cells and release of exosomes, which inhibits viral replication. Gastroenterology 2015, 148, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Lohse, N.; Hansen, A.B.; Pedersen, G.; Kronborg, G.; Gerstoft, J.; Sorensen, H.T.; Vaeth, M.; Obel, N. Survival of persons with and without HIV infection in Denmark, 1995–2005. Ann. Intern. Med. 2007, 146, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ipp, H.; Zemlin, A.E.; Erasmus, R.T.; Glashoff, R.H. Role of inflammation in HIV-1 disease progression and prognosis. Crit. Rev. Clin. Lab. Sci. 2014, 51, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Effros, R.B.; Fletcher, C.V.; Gebo, K.; Halter, J.B.; Hazzard, W.R.; Horne, F.M.; Huebner, R.E.; Janoff, E.N.; Justice, A.C.; Kuritzkes, D.; et al. Aging and infectious diseases: Workshop on HIV infection and aging: What is known and future research directions. Clin. Infect. Dis. 2008, 47, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin. Infect. Dis. 2011, 53, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Phillips, A.N. HIV infection, antiretroviral treatment, ageing, and non-aids related morbidity. BMJ 2009, 338. [Google Scholar] [CrossRef] [PubMed]
- D’Ettorre, G.; Paiardini, M.; Ceccarelli, G.; Silvestri, G.; Vullo, V. HIV-associated immune activation: From bench to bedside. AIDS Res. Hum. Retrovir. 2011, 27, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Valdez, H.; Connick, E.; Smith, K.Y.; Lederman, M.M.; Bosch, R.J.; Kim, R.S.; St Clair, M.; Kuritzkes, D.R.; Kessler, H.; Fox, L.; et al. Limited immune restoration after 3 years’ suppression of HIV-1 replication in patients with moderately advanced disease. AIDS 2002, 16, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Kelley, C.F.; Kitchen, C.M.; Hunt, P.W.; Rodriguez, B.; Hecht, F.M.; Kitahata, M.; Crane, H.M.; Willig, J.; Mugavero, M.; Saag, M.; et al. Incomplete peripheral CD4+ cell count restoration in HIV-infected patients receiving long-term antiretroviral treatment. Clin. Infect. Dis. 2009, 48, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, J.V.; Hultin, L.E.; McKeating, J.A.; Johnson, T.D.; Owens, B.; Jacobson, L.P.; Shih, R.; Lewis, J.; Wiley, D.J.; Phair, J.P.; et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J. Infect. Dis. 1999, 179, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.N.; Pillay, D.; Miners, A.H.; Bennett, D.E.; Gilks, C.F.; Lundgren, J.D. Outcomes from monitoring of patients on antiretroviral therapy in resource-limited settings with viral load, CD4 cell count, or clinical observation alone: A computer simulation model. Lancet 2008, 371, 1443–1451. [Google Scholar] [CrossRef]
- Deeks, S.G.; Verdin, E.; McCune, J.M. Immunosenescence and HIV. Curr. Opin. Immunol. 2012, 24, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Robbins, G.K.; Spritzler, J.G.; Chan, E.S.; Asmuth, D.M.; Gandhi, R.T.; Rodriguez, B.A.; Skowron, G.; Skolnik, P.R.; Shafer, R.W.; Pollard, R.B.; et al. Incomplete reconstitution of T cell subsets on combination antiretroviral therapy in the AIDS clinical trials group protocol 384. Clin. Infect. Dis. 2009, 48, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Jamieson, B.D.; Hultin, L.E.; Hultin, P.M.; Effros, R.B.; Detels, R. Premature aging of T cells is associated with faster HIV-1 disease progression. J. Acquir. Immune Defic. Syndr. 2009, 50, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Keating, R.; Hertz, T.; Wehenkel, M.; Harris, T.L.; Edwards, B.A.; McClaren, J.L.; Brown, S.A.; Surman, S.; Wilson, Z.S.; Bradley, P.; et al. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat. Immunol. 2013, 14, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L. Metabolism in T cell activation and differentiation. Curr. Opin. Immunol. 2010, 22, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vazquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chi, H. AMPK helps T cells survive nutrient starvation. Immunity 2015, 42, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.; Ballester, I.; Lopez-Posadas, R.; Suarez, M.D.; Zarzuelo, A.; Martinez-Augustin, O.; Sanchez de Medina, F. Effects of flavonoids and other polyphenols on inflammation. Crit. Rev. Food Sci. Nutr. 2011, 51, 331–362. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Banerjee, S.; Sil, P.C. The beneficial role of curcumin on inflammation, diabetes and neurodegenerative disease: A recent update. Food Chem. Toxicol. 2015, 83, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, J.L.; Chung, J.H. Metabolic effects of resveratrol: Addressing the controversies. Cell. Mol. Life Sci. 2015, 72, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Graf, T.N.; Wani, M.C.; Agarwal, R.; Kroll, D.J.; Oberlies, N.H. Gram-scale purification of flavonolignan diastereoisomers from silybum marianum (milk thistle) extract in support of preclinical in vivo studies for prostate cancer chemoprevention. Planta Med. 2007, 73, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Seeff, L.B.; Curto, T.M.; Szabo, G.; Everson, G.T.; Bonkovsky, H.L.; Dienstag, J.L.; Shiffman, M.L.; Lindsay, K.L.; Lok, A.S.; di Bisceglie, A.M.; et al. Herbal product use by persons enrolled in the hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) trial. Hepatology 2008, 47, 605–612. [Google Scholar] [CrossRef] [PubMed]
- McCord, A. Milk thistle may help improve liver health in people with HIV and hepatitis C. Proj. Inf. Perspect. 2008, 18. [Google Scholar]
- Polyak, S.J.; Morishima, C.; Lohmann, V.; Pal, S.; Lee, D.Y.; Liu, Y.; Graf, T.N.; Oberlies, N.H. Identification of hepatoprotective flavonolignans from silymarin. Proc. Natl. Acad. Sci. USA 2010, 107, 5995–5999. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.J.; Morishima, C.; Shuhart, M.C.; Wang, C.C.; Liu, Y.; Lee, D.Y. Inhibition of T-cell inflammatory cytokines, hepatocyte NF-κB signaling, and HCV infection by standardized silymarin. Gastroenterology 2007, 132, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.J.; Ferenci, P.; Pawlotsky, J.M. Hepatoprotective and antiviral functions of silymarin components in hepatitis C virus infection. Hepatology 2013, 57, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Wagoner, J.; Morishima, C.; Graf, T.N.; Oberlies, N.H.; Teissier, E.; Pecheur, E.I.; Tavis, J.E.; Polyak, S.J. Differential in vitro effects of intravenous versus oral formulations of silibinin on the HCV life cycle and inflammation. PLoS ONE 2011, 6, e16464. [Google Scholar] [CrossRef] [PubMed]
- Wagoner, J.; Negash, A.; Kane, O.J.; Martinez, L.E.; Nahmias, Y.; Bourne, N.; Owen, D.M.; Grove, J.; Brimacombe, C.; McKeating, J.A.; et al. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology 2010, 51, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Beinhardt, S.; Rasoul-Rockenschaub, S.; Maieron, A.; Steindl-Munda, P.; Hofer, H.; Ferenci, P. Intravenous silibinin-therapy in patients with chronic hepatitis C in the transplant setting. J. Hepatol. 2012, 56, S77–S78. [Google Scholar] [CrossRef]
- Beinhardt, S.; Rasoul-Rockenschaub, S.; Scherzer, T.M.; Ferenci, P. Silibinin monotherapy prevents graft infection after orthotopic liver transplantation in a patient with chronic hepatitis C. J. Hepatol. 2011, 54, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Scherzer, T.M.; Kerschner, H.; Rutter, K.; Beinhardt, S.; Hofer, H.; Schoniger-Hekele, M.; Holzmann, H.; Steindl-Munda, P. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology 2008, 135, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Neumann, U.P.; Biermer, M.; Eurich, D.; Neuhaus, P.; Berg, T. Successful prevention of hepatitis C virus (HCV) liver graft reinfection by silibinin mono-therapy. J. Hepatol. 2010, 52, 951–952. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.; Lovelace, E.S.; Elahi, S.; Maurice, N.J.; Wagoner, J.; Dragavon, J.; Mittler, J.E.; Kraft, Z.; Stamatatos, L.; Horton, H.; et al. Silibinin inhibits HIV-1 infection by reducing cellular activation and proliferation. PLoS ONE 2012, 7, e41832. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, E.S.; Wagoner, J.; MacDonald, J.; Bammler, T.; Bruckner, J.; Brownell, J.; Beyer, R.P.; Zink, E.M.; Kim, Y.M.; Kyle, J.E.; et al. Silymarin suppresses cellular inflammation by inducing reparative stress signaling. J. Nat. Prod. 2015, 78, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Morishima, C.; Shuhart, M.C.; Wang, C.C.; Paschal, D.M.; Apodaca, M.C.; Liu, Y.; Sloan, D.D.; Graf, T.N.; Oberlies, N.H.; Lee, D.Y.; et al. Silymarin inhibits in vitro T-cell proliferation and cytokine production in hepatitis C virus infection. Gastroenterology 2010, 138, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Gbetal, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The NF-κB activation pathway: Its regulation and role in inflammation and cell survival. Cancer J. Sci. Am. 1998, 4, S92–S99. [Google Scholar] [PubMed]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Rice, N.R.; MacKichan, M.L.; Israel, A. The precursor of NF-κB p50 has IκB-like functions. Cell 1992, 71, 243–253. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IκB of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Ruland, J. Aberrant NF-κB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.I.; Katz, H.; Berlin, V. Rapt1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc. Natl. Acad. Sci. USA 1994, 91, 12574–12578. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Del Giudice, G.; Lattanzi, M.; Valiante, N.M.; Praestgaard, J.; Huang, B.; Lonetto, M.A.; Maecker, H.T.; Kovarik, J.; Carson, S.; et al. mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.; Margineantu, D.H.; Sweet, I.R.; Polyak, S.J. Inhibition of HIV by Legalon-SIL is independent of its effect on cellular metabolism. Virology 2014, 449, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Colturato, C.P.; Constantin, R.P.; Maeda, A.S., Jr.; Yamamoto, N.S.; Bracht, A.; Ishii-Iwamoto, E.L.; Constantin, J. Metabolic effects of silibinin in the rat liver. Chem.-Biol. Interact. 2012, 195, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Detaille, D.; Sanchez, C.; Sanz, N.; Lopez-Novoa, J.M.; Leverve, X.; El-Mir, M.Y. Interrelation between the inhibition of glycolytic flux by silibinin and the lowering of mitochondrial ROS production in perifused rat hepatocytes. Life Sci. 2008, 82, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Guigas, B.; Naboulsi, R.; Villanueva, G.R.; Taleux, N.; Lopez-Novoa, J.M.; Leverve, X.M.; El-Mir, M.Y. The flavonoid silibinin decreases glucose-6-phosphate hydrolysis in perfused rat hepatocytes by an inhibitory effect on glucose-6-phosphatase. Cell. Physiol. Biochem. 2007, 20, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin inhibits cytokine-induced nuclear factor κB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Shimizu, M.; Sakai, H.; Yasuda, Y.; Terakura, D.; Baba, A.; Ohno, T.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Preventive effects of curcumin on the development of azoxymethane-induced colonic preneoplastic lesions in male c57bl/ksj-db/db obese mice. Nutr. Cancer 2012, 64, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kubota, S.; Ozawa, Y.; Kurihara, T.; Sasaki, M.; Yuki, K.; Miyake, S.; Noda, K.; Ishida, S.; Tsubota, K. Roles of AMP-activated protein kinase in diabetes-induced retinal inflammation. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9142–9148. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.J.; Chen, G.; Zhang, W.; Hu, X.; Liu, Y.; Zhou, Q.; Zhu, L.X.; Zhao, Y.F. Curcumin dually inhibits both mammalian target of rapamycin and nuclear factor-kappab pathways through a crossed phosphatidylinositol 3-kinase/Akt/ IκB kinase complex signaling axis in adenoid cystic carcinoma. Mol. Pharmacol. 2011, 79, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.S.; Lee, H.J.; Kim, S.H.; Lee, E.O. Piceatannol suppresses breast cancer cell invasion through the inhibition of MMP-9: Involvement of PI3K/AKT and NF-κB pathways. J. Agric. Food Chem. 2012, 60, 4083–4089. [Google Scholar] [CrossRef] [PubMed]
- Hawke, R.L.; Schrieber, S.J.; Soule, T.A.; Wen, Z.; Smith, P.C.; Reddy, K.R.; Wahed, A.S.; Belle, S.H.; Afdhal, N.H.; Navarro, V.J.; et al. Silymarin ascending multiple oral dosing phase I study in noncirrhotic patients with chronic hepatitis C. J. Clin. Pharmacol. 2010, 50, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsson, H.I.; Thakur, P.; Herold, K.F.; Hobart, E.A.; Ramsey, N.B.; Periole, X.; de Jong, D.H.; Zwama, M.; Yilmaz, D.; Hall, K.; et al. Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem. Biol. 2014, 9, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; DiStefano, P.S.; Fields, S.; et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [Google Scholar] [CrossRef] [PubMed]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar] [CrossRef] [PubMed]
- Pilkington, L.I.; Wagoner, J.; Polyak, S.J.; Barker, D. Enantioselective synthesis, stereochemical correction, and biological investigation of the rodgersinine family of 1,4-benzodioxane neolignans. Org. Lett. 2015, 17, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Coan, K.E.; Shoichet, B.K. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008, 130, 9606–9612. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Owen, S.C.; Doak, A.K.; Wassam, P.; Shoichet, M.S.; Shoichet, B.K. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 2012, 7, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lovelace, E.S.; Polyak, S.J. Natural Products as Tools for Defining How Cellular Metabolism Influences Cellular Immune and Inflammatory Function during Chronic Infection. Viruses 2015, 7, 6218-6232. https://doi.org/10.3390/v7122933
Lovelace ES, Polyak SJ. Natural Products as Tools for Defining How Cellular Metabolism Influences Cellular Immune and Inflammatory Function during Chronic Infection. Viruses. 2015; 7(12):6218-6232. https://doi.org/10.3390/v7122933
Chicago/Turabian StyleLovelace, Erica S., and Stephen J. Polyak. 2015. "Natural Products as Tools for Defining How Cellular Metabolism Influences Cellular Immune and Inflammatory Function during Chronic Infection" Viruses 7, no. 12: 6218-6232. https://doi.org/10.3390/v7122933
APA StyleLovelace, E. S., & Polyak, S. J. (2015). Natural Products as Tools for Defining How Cellular Metabolism Influences Cellular Immune and Inflammatory Function during Chronic Infection. Viruses, 7(12), 6218-6232. https://doi.org/10.3390/v7122933