
Potential for Improving Potency and Specificity of Reovirus Oncolysis with Next-Generation Reovirus Variants


Abstract
1. Introduction
1.1. Reovirus Naturally Infects the Small Intestine
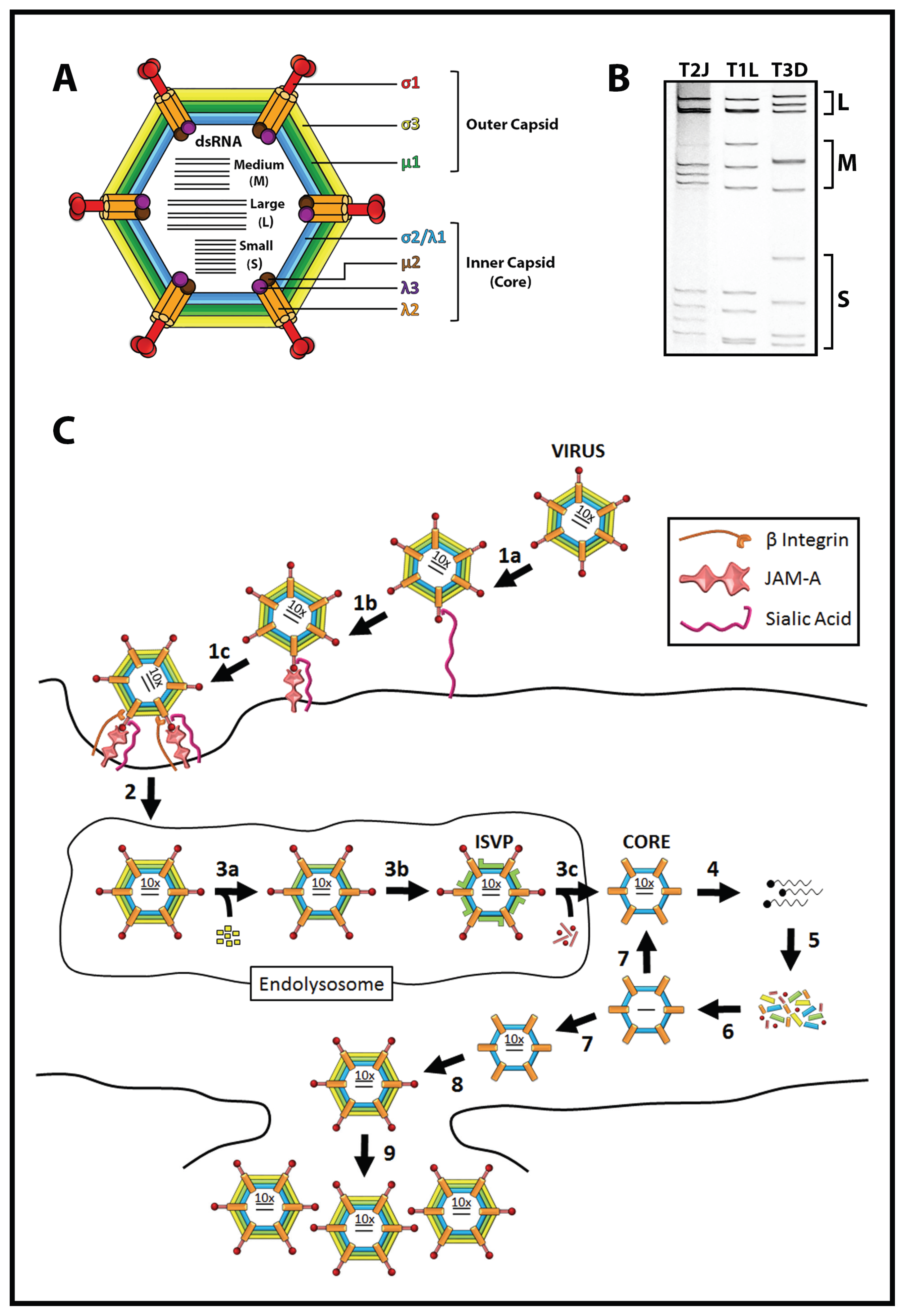
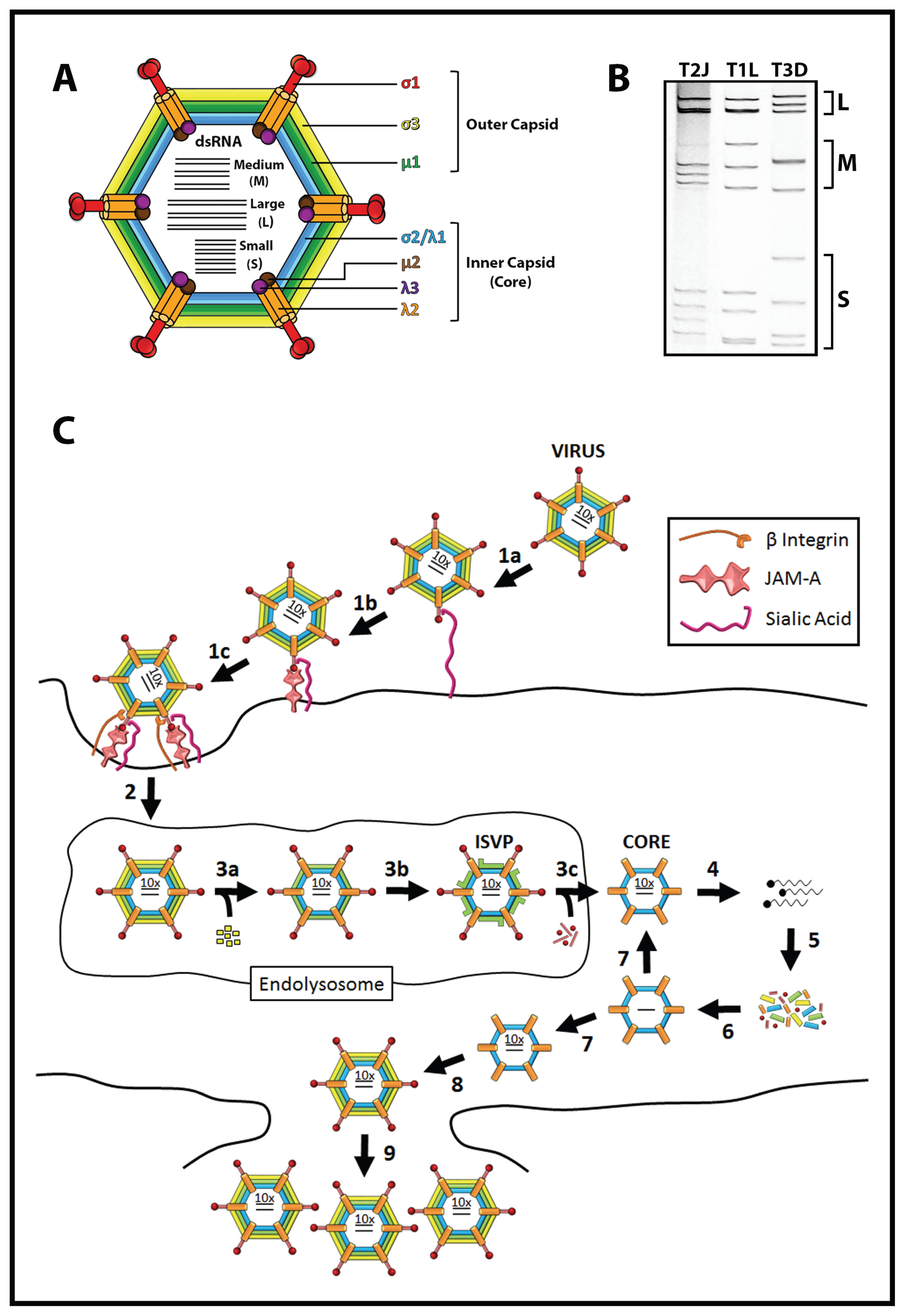
1.2. Reovirus Is a Promising Cancer Therapy
1.3. Optimizing Reovirus Oncolysis
2. Cell Attachment
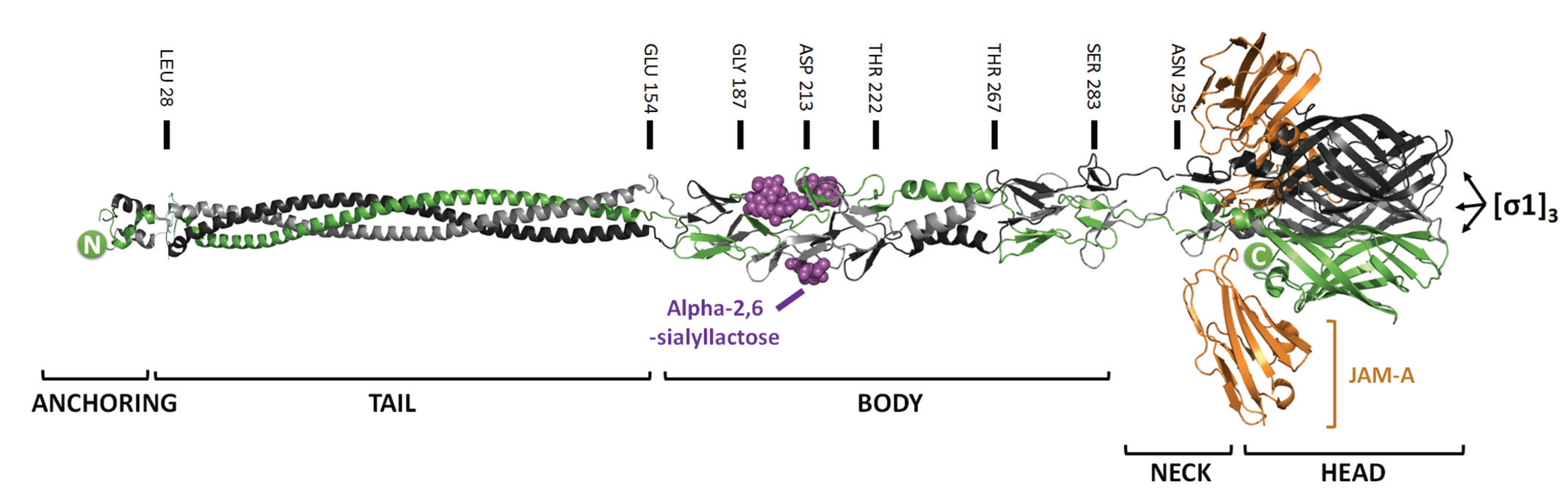
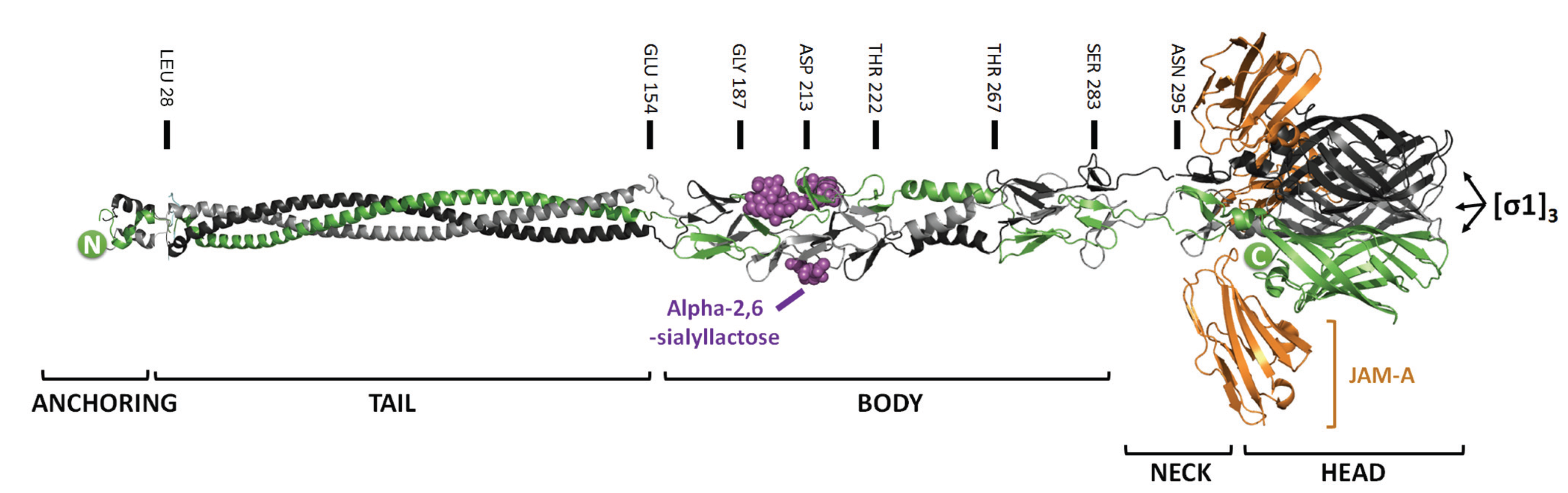
2.1. Mutations that Modulate Sialic Acid Binding

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Variant Name | Mutations (Amino Acid Change) 1 | Domain Function | Phenotype | |
|---|---|---|---|---|---|
| Improving Potency | Attachment | T3DSA− | σ1 (R202W) | Sialic-binding | Reduced infection in some cells (e.g., HeLa) and in vivo pathogenesis in neonatal immunocompromised mice |
| VeroAV | σ1 (Q78P; N198K) | Trimerization; Sialic-binding | Enhanced binding to Vero Cells | ||
| µ1 (E89G; A114V) | |||||
| Jin-1 | σ1 (T193M; Q336R) | Sialic-binding; JAM-A binding | Infectious towards JAM-A deficient cells | ||
| Jin-2 | σ1 (G187R; Q336R) | Sialic-binding; JAM-A binding | |||
| Jin-3 | σ1 (G196R) | Sialic-binding | |||
| T3D-S1His | σ1 ((His)6 tag @ C-terminus) | Additional binding domain added | Ability to replicate in JAM-A-deficient U118 cells that express (His)6-specific antibody fragment | ||
| Uncoating and Onset of Infection | NA | µ1 (A305L), (A276V), (D371N), (Q456R), (P497S), (L185S), or (E89Q) | µ1-µ1 interactions | Altered rates of ISVP → ISVP* and core production | |
| Y354H | σ3 (Y354H) | C-terminal surface exposed domain | Enhanced disassembly and resistance to E64 protease inhibitor. Enhanced replication, dissemination and pathogenesis in immunocompromised mice | ||
| T3v1 | λ1 (N138D) | Inner face of virion core | Enhanced particle infectivity and oncolytic activity in vivo | ||
| λ2 (M1101I) | Flap domain that open/close | ||||
| λ3 (P400S) | Core-facing surface | ||||
| T3v2 | σ1 (S18I) | Virion-anchoring domain | |||
| Improving Specificity/Safety | Attachment | HTR1 (AV-Reo) | σ1 (L116P; V127A; Q251STOP; I300M) σ3 (S177F; H251L) | Trimerization; JAM-A binding | Reduced toxicity in vivo |
| Antiviral Response | P4L-12 | σ3 (G198E; M221I) | Increased IFN-sensitivity. Improved specificity towards IFN-deficient Ras-transformed cells | ||
| µ1 (P315S; T449A) | |||||
| µNS (V705A) | |||||
| λ2 (T636M) | Methyltransferase domain | ||||
| NA | µ2 (P208) | Unknown | Important in repression of interferon signaling | ||
| NA | σ3 (R236), (R239), (K291), or (K293) | dsRNA binding domain | |||
| ts453 | σ3 (N16K) | µ1 association domain | Increased dsRNA binding and IFN resistance |
2.2. Modifications that Alter Binding to JAM-A or Promote JAM-A Independent Attachment
3. Virus Disassembly, Membrane Penetration, and Establishment of Infection
3.1. Reovirus Uncoating Contributes to Specificity and Potency of Oncolysis
3.2. Mutations in σ3 Promote Reovirus Uncoating
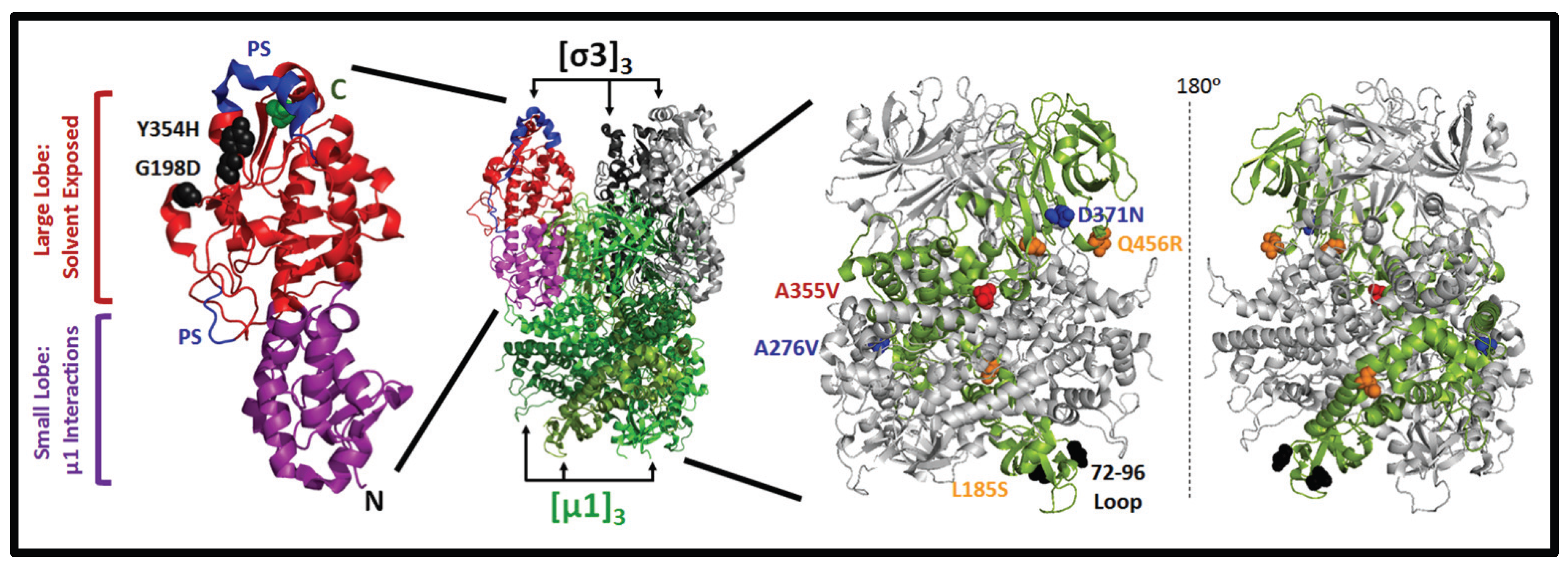
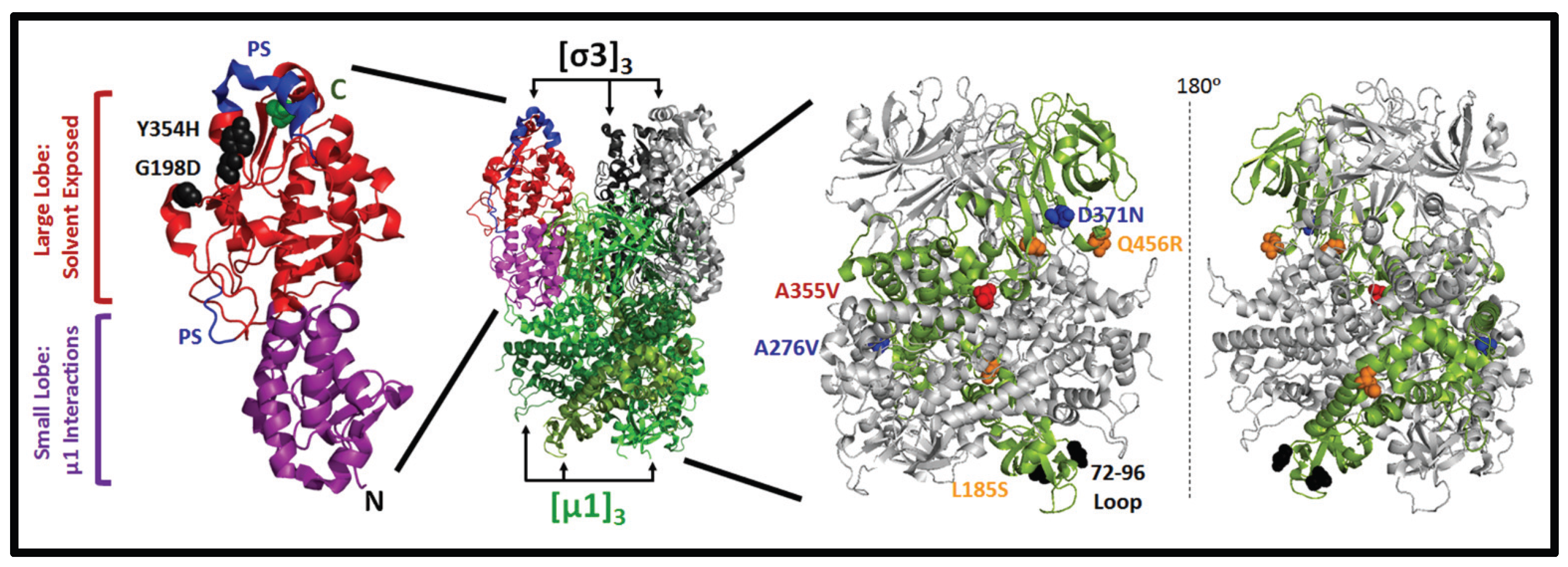
3.3. Mutations in µ1 Modulate Reovirus Uncoating
3.4. Reduced Virion-Associated σ1 Promotes Reovirus Oncolysis
4. Macromolecular Synthesis, Progeny Virus Production and Virus Release
5. Improving Reovirus Specificity and Safety
5.1. Modifying Reovirus-JAM-A Binding
5.2. Increasing Interferon Sensitivity
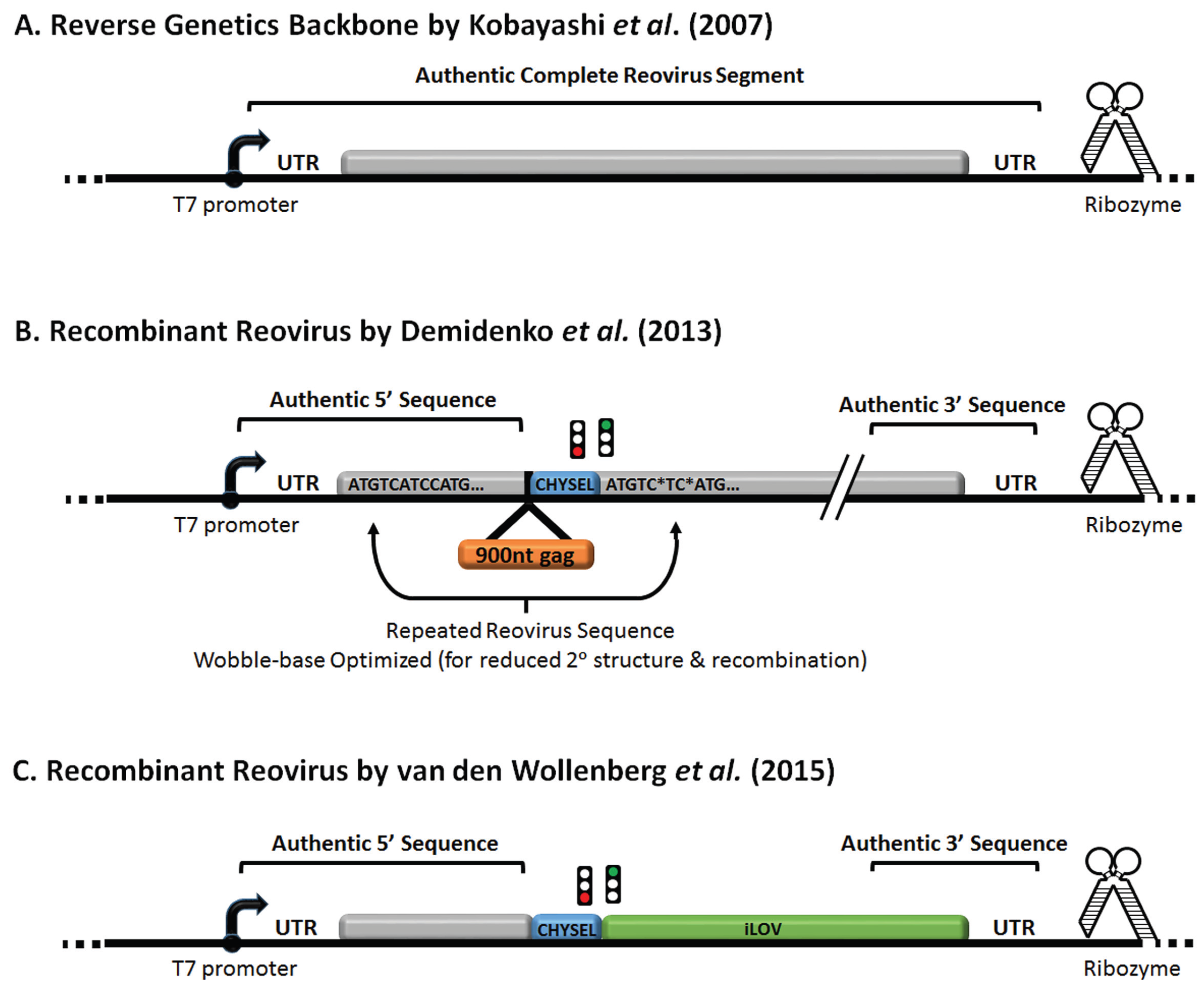
6. Reovirus Reverse Engineering and Expression of Exogenous Genes
7. Future Directions
Acknowledgments
Conflicts of Interest
References
- Rosen, L. Serologic grouping of reoviruses by hemagglutination-inhibition. Am. J. Epidemiol. 1960, 71, 242–249. [Google Scholar]
- Drayna, D.; Fields, B.N. Activation and characterization of the reovirus transcriptase: Genetic analysis. J. Virol. 1982, 41, 110–118. [Google Scholar] [PubMed]
- Adams, D.J.; Ridinger, D.N.; Spendlove, R.S.; Barnett, B.B. Protamine precipitation of two reovirus particle types from polluted waters. Appl. Environ. Microbiol. 1982, 44, 589–596. [Google Scholar] [PubMed]
- Hrdy, D.B.; Rosen, L.; Fields, B.N. Polymorphism of the migration of double-stranded RNA genome segments of reovirus isolates from humans, cattle, and mice. J. Virol. 1979, 31, 104–111. [Google Scholar] [PubMed]
- Dermody, T.S.; Nibert, M.L.; Bassel-Duby, R.; Fields, B.N. Sequence diversity in S1 genes and S1 translation products of 11 serotype 3 reovirus strains. J. Virol. 1990, 64, 4842–4850. [Google Scholar] [PubMed]
- Irving, L.G.; Smith, F.A. One-year survey of enteroviruses, adenoviruses, and reoviruses isolated from effluent at an activated-sludge purification plant. Appl. Environ. Microbiol. 1981, 41, 51–59. [Google Scholar] [PubMed]
- Ridinger, D.N.; Spendlove, R.S.; Barnett, B.B.; George, D.B.; Roth, J.C. Evaluation of cell lines and immunofluorescence and plaque assay procedures for quantifying reoviruses in sewage. Appl. Environ. Microbiol. 1982, 43, 740–746. [Google Scholar] [PubMed]
- Sedmak, G.; Bina, D.; Macdonald, J.; Couillard, L. Nine-year study of the occurrence of culturable viruses in source water for two drinking water treatment plants and the influent and effluent of a Wastewater Treatment Plant in Milwaukee, Wisconsin (August 1994 through July 2003). Appl. Environ. Microbiol. 2005, 71, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Minuk, G.Y.; Paul, R.W.; Lee, P.W. The prevalence of antibodies to reovirus type 3 in adults with idiopathic cholestatic liver disease. J. Med. Virol. 1985, 16, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.; Evans, H.E.; Spickard, A. Reovirus infections in human volunteers. Am. J. Hyg. 1963, 77, 29–37. [Google Scholar] [PubMed]
- Tai, J.H.; Williams, J.V.; Edwards, K.M.; Wright, P.F.; Crowe, J.E., Jr.; Dermody, T.S. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J. Infect. Dis. 2005, 191, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Amerongen, H.M.; Wilson, G.A.; Fields, B.N.; Neutra, M.R. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J. Virol. 1994, 68, 8428–8432. [Google Scholar] [PubMed]
- Helander, A.; Silvey, K.J.; Mantis, N.J.; Hutchings, A.B.; Chandran, K.; Lucas, W.T.; Nibert, M.L.; Neutra, M.R. The viral σ1 protein and glycoconjugates containing α2-3-linked sialic acid are involved in type 1 reovirus adherence to M cell apical surfaces. J. Virol. 2003, 77, 7964–7977. [Google Scholar] [CrossRef] [PubMed]
- Organ, E.L.; Rubin, D.H. Pathogenesis of reovirus gastrointestinal and hepatobiliary disease. Curr. Top. Microbiol. Immunol. 1998, 233, 67–83. [Google Scholar] [PubMed]
- Wolf, J.L.; Rubin, D.H.; Finberg, R.; Kauffman, R.S.; Sharpe, A.H.; Trier, J.S.; Fields, B.N. Intestinal M cells: A pathway for entry of reovirus into the host. Science 1981, 212, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.H.; Kornstein, M.J.; Anderson, A.O. Reovirus serotype 1 intestinal infection: A novel replicative cycle with ileal disease. J. Virol. 1985, 53, 391–398. [Google Scholar] [PubMed]
- Rubin, D.H. Reovirus serotype 1 binds to the basolateral membrane of intestinal epithelial cells. Microb. Pathog. 1987, 3, 215–219. [Google Scholar] [CrossRef]
- Barton, E.S.; Forrest, J.C.; Connolly, J.L.; Chappell, J.D.; Liu, Y.; Schnell, F.J.; Nusrat, A.; Parkos, C.A.; Dermody, T.S. Junction adhesion molecule is a receptor for reovirus. Cell 2001, 104, 441–451. [Google Scholar] [CrossRef]
- Nibert, M.L.; Chappell, J.D.; Dermody, T.S. Infectious subvirion particles of reovirus type 3 Dearing exhibit a loss in infectivity and contain a cleaved σ 1 protein. J. Virol. 1995, 69, 5057–5067. [Google Scholar] [PubMed]
- Cleveland, D.R.; Zarbl, H.; Millward, S. Reovirus guanylyltransferase is L2 gene product lambda 2. J. Virol. 1986, 60, 307–311. [Google Scholar] [PubMed]
- Kim, J.; Parker, J.S.; Murray, K.E.; Nibert, M.L. Nucleoside and RNA triphosphatase activities of orthoreovirus transcriptase cofactor μ2. J. Biol. Chem. 2004, 279, 4394–4403. [Google Scholar] [CrossRef] [PubMed]
- Bisaillon, M.; Lemay, G. Characterization of the reovirus λ1 protein RNA 5′-triphosphatase activity. J. Biol. Chem. 1997, 272, 29954–29957. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Hashiro, G.; Loh, P.C.; Yau, J.T. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch. Virol. 1977, 54, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.R.; Stanish, S.M.; Cox, D.C. Differential sensitivity of normal and transformed human cells to reovirus infection. J. Virol. 1978, 28, 444–449. [Google Scholar] [PubMed]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [PubMed]
- Marcato, P.; Shmulevitz, M.; Pan, D.; Stoltz, D.; Lee, P.W. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 2007, 15, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Alain, T.; Kim, T.S.; Lun, X.; Liacini, A.; Schiff, L.A.; Senger, D.L.; Forsyth, P.A. Proteolytic disassembly is a critical determinant for reovirus oncolysis. Mol. Ther. 2007, 15, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; van den Wollenberg, D.J.; Borel, R.I.; Hoeben, R.C.; Kranenburg, O. Sensitization to apoptosis underlies KrasD12-dependent oncolysis of murine C26 colorectal carcinoma cells by reovirus T3D. J. Virol. 2005, 79, 14981–14985. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Pan, L.Z.; Garant, K.; Pan, D.; Lee, P.W. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010, 70, 4912–4921. [Google Scholar] [CrossRef] [PubMed]
- Rudd, P.; Lemay, G. Correlation between interferon sensitivity of reovirus isolates and ability to discriminate between normal and Ras-transformed cells. J. Gen. Virol. 2005, 86, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Errington, F.; White, C.L.; Twigger, K.R.; Rose, A.; Scott, K.; Steele, L.; Ilett, L.J.; Prestwich, R.; Pandha, H.S.; Coffey, M.; et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther. 2008, 15, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Himeno, Y.; Matsumoto, T.; Aramaki, M.; Kawano, K.; Nishizono, A.; Kitano, S. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin. Cancer Res. 2003, 9, 1218–1223. [Google Scholar] [PubMed]
- Hirasawa, K.; Nishikawa, S.G.; Norman, K.L.; Alain, T.; Kossakowska, A.; Lee, P.W. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002, 62, 1696–1701. [Google Scholar] [PubMed]
- Ikeda, Y.; Nishimura, G.; Yanoma, S.; Kubota, A.; Furukawa, M.; Tsukuda, M. Reovirus oncolysis in human head and neck squamous carcinoma cells. Auris Nasus Larynx 2004, 31, 407–412. [Google Scholar] [CrossRef]
- Norman, K.L.; Coffey, M.C.; Hirasawa, K.; Demetrick, D.J.; Nishikawa, S.G.; DiFrancesco, L.M.; Strong, J.E.; Lee, P.W. Reovirus oncolysis of human breast cancer. Hum. Gene Ther. 2002, 13, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Comins, C.; Spicer, J.; Protheroe, A.; Roulstone, V.; Twigger, K.; White, C.M.; Vile, R.; Melcher, A.; Coffey, M.C.; Mettinger, K.L.; et al. REO-10, a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin. Cancer Res. 2010, 16, 5564–5572. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, P.; Roldan, G.; George, D.; Wallace, C.; Palmer, C.A.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol. Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Karapanagiotou, E.M.; Roulstone, V.; Twigger, K.R.; White, C.L.; Vidal, L.; Beirne, D.; Prestwich, R.; Newbold, K.; Ahmed, M.; et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin. Cancer Res. 2010, 16, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Karapanagiotou, E.M.; Roulstone, V.; Twigger, K.; Ball, M.; Tanay, M.; Nutting, C.; Newbold, K.; Gore, M.E.; Larkin, J.; Syrigos, K.N.; et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin. Cancer Res. 2012, 18, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Lolkema, M.P.; Arkenau, H.T.; Harrington, K.; Roxburgh, P.; Morrison, R.; Roulstone, V.; Twigger, K.; Coffey, M.; Mettinger, K.; Gill, G.; et al. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2011, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Vidal, L.; Pandha, H.S.; Yap, T.A.; White, C.L.; Twigger, K.; Vile, R.G.; Melcher, A.; Coffey, M.; Harrington, K.J.; Debono, J.S. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin. Cancer Res. 2008, 14, 7127–7137. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Twigger, K.R.; Vidal, L.; de Bono, J.S.; Coffey, M.; Heinemann, L.; Morgan, R.; Merrick, A.; Errington, F.; Vile, R.G.; et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 2008, 15, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Gollamudi, R.; Ghalib, M.H.; Desai, K.K.; Chaudhary, I.; Wong, B.; Einstein, M.; Coffey, M.; Gill, G.M.; Mettinger, K.; Mariadason, J.M.; et al. Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest. New Drugs 2010, 28, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Phase 3 Trial Examining REOLYSIN® in Combination with Paclitaxel and Carboplatin in Patients with Platinum-Refractory Head and Neck Cancers. Oncolytics Biotech Inc., 2011. Available online: http://www.oncolyticsbiotech.com/clinical_pr.html (accessed on 26 November 2015).
- Heinemann, L.; Simpson, G.R.; Boxall, A.; Kottke, T.; Relph, K.L.; Vile, R.; Melcher, A.; Prestwich, R.; Harrington, K.J.; Morgan, R.; et al. Synergistic effects of oncolytic reovirus and docetaxel chemotherapy in prostate cancer. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Melcher, A.; Vassaux, G.; Pandha, H.S.; Vile, R.G. Exploiting synergies between radiation and oncolytic viruses. Curr. Opin. Mol. Ther. 2008, 10, 362–370. [Google Scholar] [PubMed]
- Harrington, K.J.; Vile, R.G.; Melcher, A.; Chester, J.; Pandha, H.S. Clinical trials with oncolytic reovirus: Moving beyond phase I into combinations with standard therapeutics. Cytokine Growth Factor Rev. 2010, 21, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.M.; Pandha, H.; Harrington, K.; et al. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D.M.; Grin, I.; Dunin-Horkawicz, S.; Deiss, S.; Linke, D.; Lupas, A.N.; Hernandez, A.B. Complete fiber structures of complex trimeric autotransporter adhesins conserved in enterobacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 20907–20912. [Google Scholar] [CrossRef] [PubMed]
- Reiter, D.M.; Frierson, J.M.; Halvorson, E.E.; Kobayashi, T.; Dermody, T.S.; Stehle, T. Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides. PLoS. Pathog. 2011, 7, e1002166. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, E.; Guglielmi, K.M.; Strauss, H.M.; Dermody, T.S.; Stehle, T. Structure of reovirus σ1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog. 2008, 4, e1000235. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; Mah, D.C.; Lee, P.W. The incorporation of reovirus cell attachment protein σ 1 into virions requires the N-terminal hydrophobic tail and the adjacent heptad repeat region. Virology 1991, 182, 346–350. [Google Scholar] [CrossRef]
- Leone, G.; Duncan, R.; Mah, D.C.; Price, A.; Cashdollar, L.W.; Lee, P.W. The N-terminal heptad repeat region of reovirus cell attachment protein σ 1 is responsible for σ 1 oligomer stability and possesses intrinsic oligomerization function. Virology 1991, 182, 336–345. [Google Scholar] [CrossRef]
- Barton, E.S.; Connolly, J.L.; Forrest, J.C.; Chappell, J.D.; Dermody, T.S. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 2001, 276, 2200–2211. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.D.; Barton, E.S.; Smith, T.H.; Baer, G.S.; Duong, D.T.; Nibert, M.L.; Dermody, T.S. Cleavage susceptibility of reovirus attachment protein σ1 during proteolytic disassembly of virions is determined by a sequence polymorphism in the σ1 neck. J. Virol. 1998, 72, 8205–8213. [Google Scholar] [PubMed]
- Campbell, J.A.; Schelling, P.; Wetzel, J.D.; Johnson, E.M.; Forrest, J.C.; Wilson, G.A.; Aurrand-Lions, M.; Imhof, B.A.; Stehle, T.; Dermody, T.S. Junctional adhesion molecule a serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J. Virol. 2005, 79, 7967–7978. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Campbell, J.A.; Schelling, P.; Forrest, J.C.; Watson, M.J.; Peters, T.R.; Aurrand-Lions, M.; Imhof, B.A.; Dermody, T.S.; Stehle, T. Crystal structure of human junctional adhesion molecule 1: Implications for reovirus binding. Proc. Natl. Acad. Sci. USA 2003, 100, 5366–5371. [Google Scholar] [CrossRef] [PubMed]
- Konopka-Anstadt, J.L.; Mainou, B.A.; Sutherland, D.M.; Sekine, Y.; Strittmatter, S.M.; Dermody, T.S. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe 2014, 15, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S.; Forrest, J.C.; Kopecky-Bromberg, S.A.; Dickeson, S.K.; Santoro, S.A.; Zutter, M.M.; Nemerow, G.R.; Bergelson, J.M.; Dermody, T.S. β1 integrin mediates internalization of mammalian reovirus. J. Virol. 2006, 80, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S.; Mainou, B.A.; Derdowski, A.; Johnson, E.M.; Zent, R.; Dermody, T.S. NPXY motifs in the β1 integrin cytoplasmic tail are required for functional reovirus entry. J. Virol. 2008, 82, 3181–3191. [Google Scholar] [CrossRef] [PubMed]
- Stencel-Baerenwald, J.E.; Reiss, K.; Reiter, D.M.; Stehle, T.; Dermody, T.S. The sweet spot: Defining virus-sialic acid interactions. Nat. Rev. Microbiol. 2014, 12, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Gentsch, J.R.; Pacitti, A.F. Differential interaction of reovirus type 3 with sialylated receptor components on animal cells. Virology 1987, 161, 245–248. [Google Scholar] [CrossRef]
- Gentsch, J.R.; Pacitti, A.F. Effect of neuraminidase treatment of cells and effect of soluble glycoproteins on type 3 reovirus attachment to murine L cells. J. Virol. 1985, 56, 356–364. [Google Scholar] [PubMed]
- Paul, R.W.; Choi, A.H.; Lee, P.W. The alpha-anomeric form of sialic acid is the minimal receptor determinant recognized by reovirus. Virology 1989, 172, 382–385. [Google Scholar] [CrossRef]
- Chappell, J.D.; Gunn, V.L.; Wetzel, J.D.; Baer, G.S.; Dermody, T.S. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein σ1. J. Virol. 1997, 71, 1834–1841. [Google Scholar] [PubMed]
- Chappell, J.D.; Duong, J.L.; Wright, B.W.; Dermody, T.S. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J. Virol. 2000, 74, 8472–8479. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Stencel, J.E.; Liu, Y.; Blaum, B.S.; Reiter, D.M.; Feizi, T.; Dermody, T.S.; Stehle, T. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS. Pathog. 2012, 8, e1003078. [Google Scholar] [CrossRef] [PubMed]
- Barton, E.S.; Youree, B.E.; Ebert, D.H.; Forrest, J.C.; Connolly, J.L.; Valyi-Nagy, T.; Washington, K.; Wetzel, J.D.; Dermody, T.S. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J. Clin. Invest. 2003, 111, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Frierson, J.M.; Pruijssers, A.J.; Konopka, J.L.; Reiter, D.M.; Abel, T.W.; Stehle, T.; Dermody, T.S. Utilization of sialylated glycans as coreceptors enhances the neurovirulence of serotype 3 reovirus. J. Virol. 2012, 86, 13164–13173. [Google Scholar] [CrossRef] [PubMed]
- Boligan, K.F.; Mesa, C.; Fernandez, L.E.; von Gunten, S. Cancer intelligence acquired (CIA): Tumor glycosylation and sialylation codes dismantling antitumor defense. Cell Mol. Life Sci. 2015, 72, 1231–1248. [Google Scholar] [CrossRef] [PubMed]
- Padler-Karavani, V.; Hurtado-Ziola, N.; Pu, M.; Yu, H.; Huang, S.; Muthana, S.; Chokhawala, H.A.; Cao, H.; Secrest, P.; Friedmann-Morvinski, D.; et al. Human xeno-autoantibodies against a non-human sialic acid serve as novel serum biomarkers and immunotherapeutics in cancer. Cancer Res. 2011, 71, 3352–3363. [Google Scholar] [CrossRef] [PubMed]
- Sandekian, V.; Lemay, G. Amino acids substitutions in σ1 and μ1 outer capsid proteins of a Vero cell-adapted mammalian orthoreovirus are required for optimal virus binding and disassembly. Virus Res. 2015, 196, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jabre, R.; Sandekian, V.; Lemay, G. Amino acid substitutions in σ1 and μ1 outer capsid proteins are selected during mammalian reovirus adaptation to Vero cells. Virus Res. 2013, 176, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K.; Suzuki, A.; Ohno, S.; Vestweber, D. Junctional adhesion molecules (JAMs): More molecules with dual functions? J. Cell Sci. 2004, 117, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Ikeo, K.; Oshima, T.; Shan, J.; Matsui, H.; Tomita, T.; Fukui, H.; Watari, J.; Miwa, H. Junctional adhesion molecule-A promotes proliferation and inhibits apoptosis of gastric cancer. Hepatogastroenterology 2015, 62, 540–545. [Google Scholar] [PubMed]
- Tian, Y.; Tian, Y.; Zhang, W.; Wei, F.; Yang, J.; Luo, X.; Zhou, T.; Hou, B.; Qian, S.; Deng, X.; et al. Junctional adhesion molecule-A, an epithelial-mesenchymal transition inducer, correlates with metastasis and poor prognosis in human nasopharyngeal cancer. Carcinogenesis 2015, 36, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Lu, F.; Chen, H.; Zhao, X.; Sun, J.; Chen, H. Dysregulation of JAM-A plays an important role in human tumor progression. Int. J. Clin. Exp. Pathol. 2014, 7, 7242–7248. [Google Scholar] [PubMed]
- Lathia, J.D.; Li, M.; Sinyuk, M.; Alvarado, A.G.; Flavahan, W.A.; Stoltz, K.; Rosager, A.M.; Hale, J.; Hitomi, M.; Gallagher, J.; et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 2014, 6, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Luo, W.; Huang, B.; Liu, Z.; Sun, L.; Zhang, Q.; Qiu, X.; Xu, K.; Wang, E. Overexpression of JAM-A in non-small cell lung cancer correlates with tumor progression. PLoS. ONE 2013, 8, e79173. [Google Scholar] [CrossRef] [PubMed]
- Goetsch, L.; Haeuw, J.F.; Beau-Larvor, C.; Gonzalez, A.; Zanna, L.; Malissard, M.; Lepecquet, A.M.; Robert, A.; Bailly, C.; Broussas, M.; et al. A novel role for junctional adhesion molecule-A in tumor proliferation: Modulation by an anti-JAM-A monoclonal antibody. Int. J. Cancer 2013, 132, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Giampietro, C.; Giannotta, M.; Corada, M.; Torselli, I.; Orsenigo, F.; Cocito, A.; d’Ario, G.; Mazzarol, G.; Confalonieri, S.; et al. Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PLoS ONE 2011, 6, e21242. [Google Scholar] [CrossRef] [PubMed]
- Ponten, F.; Jirstrom, K.; Uhlen, M. The Human Protein Atlas—A tool for pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Berglund, L.; Bjorling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Szigyarto, C.A.; Persson, A.; Ottosson, J.; Wernerus, H.; Nilsson, P.; et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell. Proteom. 2008, 7, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Terasawa, Y.; Hotani, T.; Katayama, Y.; Tachibana, M.; Mizuguchi, H.; Sakurai, F. Activity levels of cathepsins B and L in tumor cells are a biomarker for efficacy of reovirus-mediated tumor cell killing. Cancer Gene Ther. 2015, 22, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, W.J.; Smakman, N.; van den Wollenberg, D.J.; Emmink, B.L.; Veenendaal, L.M.; van Diest, P.J.; Hoeben, R.C.; Borel, R.I.; Kranenburg, O. Transient infection of freshly isolated human colorectal tumor cells by reovirus T3D intermediate subviral particles. Cancer Gene Ther. 2008, 15, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Van den Wollenberg, D.J.; Dautzenberg, I.J.; van den Hengel, S.K.; Cramer, S.J.; de Groot, R.J.; Hoeben, R.C. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-A. PLoS ONE 2012, 7, e48064. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.D.; Prota, A.E.; Dermody, T.S.; Stehle, T. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 2002, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mercier, G.T.; Campbell, J.A.; Chappell, J.D.; Stehle, T.; Dermody, T.S.; Barry, M.A. A chimeric adenovirus vector encoding reovirus attachment protein σ1 targets cells expressing junctional adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2004, 101, 6188–6193. [Google Scholar] [CrossRef] [PubMed]
- Schagen, F.H.; Graat, H.C.; Carette, J.E.; Vellinga, J.; van Geer, M.A.; Hoeben, R.C.; Dermody, T.S.; van Beusechem, V.W. Replacement of native adenovirus receptor-binding sites with a new attachment moiety diminishes hepatic tropism and enhances bioavailability in mice. Hum. Gene Ther. 2008, 19, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, Y.; Pereboeva, L.; Glasgow, J.N.; Luongo, C.L.; Komarova, S.; Kawakami, Y.; Curiel, D.T. Reovirus σ1 fiber incorporated into adenovirus serotype 5 enhances infectivity via a CAR-independent pathway. Biochem. Biophys. Res. Commun. 2005, 335, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, Y.; Pereboeva, L.; Glasgow, J.N.; Rein, D.T.; Kawakami, Y.; Alvarez, R.D.; Rocconi, R.P.; Siegal, G.P.; Dent, P.; Fisher, P.B.; et al. A mosaic fiber adenovirus serotype 5 vector containing reovirus σ 1 and adenovirus serotype 3 knob fibers increases transduction in an ovarian cancer ex vivo system via a coxsackie and adenovirus receptor-independent pathway. Clin. Cancer Res. 2007, 13, 2777–2783. [Google Scholar] [CrossRef] [PubMed]
- Van den Wollenberg, D.J.; van den Hengel, S.K.; Dautzenberg, I.J.; Cramer, S.J.; Kranenburg, O.; Hoeben, R.C. A strategy for genetic modification of the spike-encoding segment of human reovirus T3D for reovirus targeting. Gene Ther. 2008, 15, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- Bass, D.M.; Bodkin, D.; Dambrauskas, R.; Trier, J.S.; Fields, B.N.; Wolf, J.L. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J. Virol. 1990, 64, 1830–1833. [Google Scholar] [PubMed]
- Bodkin, D.K.; Nibert, M.L.; Fields, B.N. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J. Virol. 1989, 63, 4676–4681. [Google Scholar] [PubMed]
- Jane-Valbuena, J.; Breun, L.A.; Schiff, L.A.; Nibert, M.L. Sites and determinants of early cleavages in the proteolytic processing pathway of reovirus surface protein σ3. J. Virol. 2002, 76, 5184–5197. [Google Scholar] [CrossRef] [PubMed]
- Borsa, J.; Sargent, M.D.; Ewing, D.D.; Einspenner, M. Perturbation of the switch-on of transcriptase activity in intermediate subviral particles from reovirus. J. Cell. Physiol. 1982, 112, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Odegard, A.L.; Agosto, M.A.; Chandran, K.; Schiff, L.A. Putative autocleavage of reovirus mu1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J. Mol. Biol. 2005, 345, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Odegard, A.L.; Chandran, K.; Zhang, X.; Parker, J.S.; Baker, T.S.; Nibert, M.L. Putative autocleavage of outer capsid protein micro1, allowing release of myristoylated peptide micro1N during particle uncoating, is critical for cell entry by reovirus. J. Virol. 2004, 78, 8732–8745. [Google Scholar] [CrossRef] [PubMed]
- Borsa, J.; Sargent, M.D.; Lievaart, P.A.; Copps, T.P. Reovirus: Evidence for a second step in the intracellular uncoating and transcriptase activation process. Virology 1981, 111, 191–200. [Google Scholar] [CrossRef]
- Chandran, K.; Farsetta, D.L.; Nibert, M.L. Strategy for nonenveloped virus entry: A hydrophobic conformer of the reovirus membrane penetration protein micro 1 mediates membrane disruption. J. Virol. 2002, 76, 9920–9933. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Parker, J.S.; Ehrlich, M.; Kirchhausen, T.; Nibert, M.L. The delta region of outer-capsid protein micro 1 undergoes conformational change and release from reovirus particles during cell entry. J. Virol. 2003, 77, 13361–13375. [Google Scholar] [CrossRef] [PubMed]
- Dryden, K.A.; Wang, G.; Yeager, M.; Nibert, M.L.; Coombs, K.M.; Furlong, D.B.; Fields, B.N.; Baker, T.S. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: Analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J. Cell Biol. 1993, 122, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Danthi, P. Determinants of strain-specific differences in efficiency of reovirus entry. J. Virol. 2010, 84, 12723–12732. [Google Scholar] [CrossRef] [PubMed]
- Agosto, M.A.; Myers, K.S.; Ivanovic, T.; Nibert, M.L. A positive-feedback mechanism promotes reovirus particle conversion to the intermediate associated with membrane penetration. Proc. Natl. Acad. Sci. USA 2008, 105, 10571–10576. [Google Scholar] [CrossRef] [PubMed]
- Agosto, M.A.; Ivanovic, T.; Nibert, M.L. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc. Natl. Acad. Sci. USA 2006, 103, 16496–16501. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P.; Guglielmi, K.M.; Kirchner, E.; Mainou, B.; Stehle, T.; Dermody, T.S. From touchdown to transcription: The reovirus cell entry pathway. Curr. Top. Microbiol. Immunol. 2010, 343, 91–119. [Google Scholar] [PubMed]
- Danthi, P.; Holm, G.H.; Stehle, T.; Dermody, T.S. Reovirus receptors, cell entry, and proapoptotic signaling. Adv. Exp. Med. Biol. 2013, 790, 42–71. [Google Scholar] [PubMed]
- Mainou, B.A.; Dermody, T.S. In search of cathepsins: How reovirus enters host cells. DNA Cell Biol. 2012, 31, 1646–1649. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.H.; Deussing, J.; Peters, C.; Dermody, T.S. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 2002, 277, 24609–24617. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.M.; Doyle, J.D.; Wetzel, J.D.; McClung, R.P.; Katunuma, N.; Chappell, J.D.; Washington, M.K.; Dermody, T.S. Genetic and pharmacologic alteration of cathepsin expression influences reovirus pathogenesis. J. Virol. 2009, 83, 9630–9640. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.W.; Schiff, L.A. Neutrophil elastase, an acid-independent serine protease, facilitates reovirus uncoating and infection in U937 promonocyte cells. Virol. J. 2005, 2. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.W.; Bahe, J.A.; Lucas, W.T.; Nibert, M.L.; Schiff, L.A. Cathepsin S supports acid-independent infection by some reoviruses. J. Biol. Chem. 2004, 279, 8547–8557. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, R.M.; Golden, J.W.; Schiff, L.A. Impact of host proteases on reovirus infection in the respiratory tract. J. Virol. 2012, 86, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Tang, C.; Lai, L. Specificity of trypsin and chymotrypsin: Loop-motion-controlled dynamic correlation as a determinant. Biophys. J. 2005, 89, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.E. The lysosomal cysteine proteases. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Vajda, T.; Szabo, T. Specificity of trypsin and alpha-chymotrypsin towards neutral substrates. Acta Biochim. Biophys. Acad. Sci. Hung. 1976, 11, 287–294. [Google Scholar] [PubMed]
- Golden, J.W.; Linke, J.; Schmechel, S.; Thoemke, K.; Schiff, L.A. Addition of exogenous protease facilitates reovirus infection in many restrictive cells. J. Virol. 2002, 76, 7430–7443. [Google Scholar] [CrossRef] [PubMed]
- Dautzenberg, I.J.; van den Wollenberg, D.J.; van den Hengel, S.K.; Limpens, R.W.; Barcena, M.; Koster, A.J.; Hoeben, R.C. Mammalian orthoreovirus T3D infects U-118 MG cell spheroids independent of junction adhesion molecule-A. Gene Ther. 2014, 21, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.K.; Severson, T.F.; Chandran, K.; Gillian, A.L.; Yin, J.; Nibert, M.L. Thermostability of reovirus disassembly intermediates (ISVPs) correlates with genetic, biochemical, and thermodynamic properties of major surface protein μ1. J. Virol. 2002, 76, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Liemann, S.; Chandran, K.; Baker, T.S.; Nibert, M.L.; Harrison, S.C. Structure of the reovirus membrane-penetration protein, M1, in a complex with is protector protein, σ3. Cell 2002, 108, 283–295. [Google Scholar] [CrossRef]
- Metcalf, P.; Cyrklaff, M.; Adrian, M. The three-dimensional structure of reovirus obtained by cryo-electron microscopy. EMBO J. 1991, 10, 3129–3136. [Google Scholar] [PubMed]
- Olland, A.M.; Jane-Valbuena, J.; Schiff, L.A.; Nibert, M.L.; Harrison, S.C. Structure of the reovirus outer capsid and dsRNA-binding protein σ3 at 1.8 A resolution. EMBO J. 2001, 20, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Shepard, D.A.; Ehnstrom, J.G.; Schiff, L.A. Association of reovirus outer capsid proteins σ3 and μ1 causes a conformational change that renders σ 3 protease sensitive. J. Virol. 1995, 69, 8180–8184. [Google Scholar] [PubMed]
- Clark, K.M.; Wetzel, J.D.; Gu, Y.; Ebert, D.H.; McAbee, S.A.; Stoneman, E.K.; Baer, G.S.; Zhu, Y.; Wilson, G.J.; Prasad, B.V.; et al. Reovirus variants selected for resistance to ammonium chloride have mutations in viral outer-capsid protein σ3. J. Virol. 2006, 80, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, J.D.; Wilson, G.J.; Baer, G.S.; Dunnigan, L.R.; Wright, J.P.; Tang, D.S.; Dermody, T.S. Reovirus variants selected during persistent infections of L cells contain mutations in the viral S1 and S4 genes and are altered in viral disassembly. J. Virol. 1997, 71, 1362–1369. [Google Scholar] [PubMed]
- Wilson, G.J.; Nason, E.L.; Hardy, C.S.; Ebert, D.H.; Wetzel, J.D.; Prasad, B.V.V.; Dermody, T.S. A single mutation in the carboxy terminus of reovirus outer-capsid protein σ 3 confers enhanced kinetics of σ 3 proteolysis, resistance to inhibitors of viral disassembly, and alterations in σ 3 structure. J. Virol. 2002, 76, 9832–9843. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.D.; Danthi, P.; Kendall, E.A.; Ooms, L.S.; Wetzel, J.D.; Dermody, T.S. Molecular determinants of proteolytic disassembly of the reovirus outer capsid. J. Biol. Chem. 2012, 287, 8029–8038. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.J.; Wetzel, J.D.; Puryear, W.; Bassel-Duby, R.; Dermody, T.S. Persistent reovirus infections of L cells select mutations in viral attachment protein σ1 that alter oligomer stability. J. Virol. 1996, 70, 6598–6606. [Google Scholar] [PubMed]
- Doyle, J.D.; Stencel-Baerenwald, J.E.; Copeland, C.A.; Rhoads, J.P.; Brown, J.J.; Boyd, K.L.; Atkinson, J.B.; Dermody, T.S. Diminished reovirus capsid stability alters disease pathogenesis and littermate transmission. PLoS. Pathog. 2015, 11, e1004693. [Google Scholar] [CrossRef] [PubMed]
- Denzler, K.L.; Jacobs, B.L. Site-directed mutagenic analysis of reovirus σ3 protein binding to dsRNA. Virology 1994, 204, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Shatkin, A.J. Double-stranded RNA-dependent protein kinase (PKR) is regulated by reovirus structural proteins. Virology 1997, 234, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Madren, J.A.; Sarkar, P.; Danthi, P. Cell entry-associated conformational changes in reovirus particles are controlled by host protease activity. J. Virol. 2012, 86, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Danthi, P. The μ1 72–96 loop controls conformational transitions during reovirus cell entry. J. Virol. 2013, 87, 13532–13542. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Gujar, S.A.; Ahn, D.G.; Mohamed, A.; Lee, P.W. Reovirus variants with mutations in S1 and L2 genome segments exhibit enhanced virion infectivity and superior oncolysis. J. Virol. 2012, 86, 7403–7413. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Walker, S.B.; Chen, Y.; Contreras, C.M.; Schiff, L.A.; Baker, T.S.; Nibert, M.L. In vitro recoating of reovirus cores with baculovirus-expressed outer-capsid proteins μ1 and σ3. J. Virol. 1999, 73, 3941–3950. [Google Scholar] [PubMed]
- Mendez, I.I.; Weiner, S.G.; She, Y.M.; Yeager, M.; Coombs, K.M. Conformational changes accompany activation of reovirus RNA-dependent RNA transcription. J. Struct. Biol. 2008, 162, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, K.M.; Nibert, M.L.; Harrison, S.C. Structure of the reovirus core at 3.6 A resolution. Nature 2000, 404, 960–967. [Google Scholar] [PubMed]
- Tao, Y.; Farsetta, D.L.; Nibert, M.L.; Harrison, S.C. RNA synthesis in a cage—Structural studies of reovirus polymerase λ3. Cell 2002, 111, 733–745. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, L.; Fang, Q.; Hui, W.H.; Zhou, Z.H. 3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell 2010, 141, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Walker, S.B.; Chipman, P.R.; Nibert, M.L.; Baker, T.S. Reovirus polymerase λ3 localized by cryo-electron microscopy of virions at a resolution of 7.6 A. Nat. Struct. Biol. 2003, 10, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Teicher, C.; Haefliger, S.; Shmulevitz, M. Reduction of virion-associated σ1 fibers on oncolytic reovirus variants promotes adaptation toward tumorigenic cells. J. Virol. 2015, 89, 4319–4334. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Antczak, J.B.; Joklik, W.K. Reovirus exists in the form of 13 particle species that differ in their content of protein σ1. Virology 1994, 201, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Garant, K.A.; zur Nieden, N.I.; Alain, T.; Loken, S.D.; Urbanski, S.J.; Forsyth, P.A.; Rancourt, D.E.; Lee, P.W.; Johnston, R.N. Attenuated reovirus displays oncolysis with reduced host toxicity. Br. J. Cancer 2011, 104, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Egan, C.; Alain, T.; Urbanski, S.J.; Lee, P.W.; Forsyth, P.A.; Johnston, R.N. Acquired resistance to reoviral oncolysis in Ras-transformed fibrosarcoma cells. Oncogene 2007, 26, 4124–4134. [Google Scholar] [CrossRef] [PubMed]
- Antar, A.A.; Konopka, J.L.; Campbell, J.A.; Henry, R.A.; Perdigoto, A.L.; Carter, B.D.; Pozzi, A.; Abel, T.W.; Dermody, T.S. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 2009, 5, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.L.; Barton, E.S.; Dermody, T.S. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J. Virol. 2001, 75, 4029–4039. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.; Kauffman, E.B.; Martinez, H.; Perkus, M.E.; Jacobs, B.L.; Paoletti, E.; Tartaglia, J. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes 1996, 12, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Imani, F.; Jacobs, B.L. Inhibitory activity for the interferon-induced protein kinase is associated with the reovirus serotype 1 σ3 protein. Proc. Natl. Acad. Sci. USA 1988, 85, 7887–7891. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; Langland, J.O. Reovirus σ3 protein: dsRNA binding and inhibition of RNA-activated protein kinase. Curr. Top. Microbiol. Immunol. 1998, 233, 185–196. [Google Scholar] [PubMed]
- Mabrouk, T.; Danis, C.; Lemay, G. Two basic motifs of reovirus σ3 protein are involved in double-stranded RNA binding. Biochem. Cell Biol. 1995, 73, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Irvin, S.C.; Zurney, J.; Ooms, L.S.; Chappell, J.D.; Dermody, T.S.; Sherry, B. A single amino acid polymorphism in reovirus protein M2 determines repression of interferon signaling and modulates myocarditis. J. Virol. 2012, 86, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Zurney, J.; Kobayashi, T.; Holm, G.H.; Dermody, T.S.; Sherry, B. Reovirus μ2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J. Virol. 2009, 83, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Sherry, B.; Torres, J.; Blum, M.A. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J. Virol. 1998, 72, 1314–1323. [Google Scholar] [PubMed]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Yang, K.; He, B. Dephosphorylation of eIF-2alpha mediated by the gamma(1)34.5 protein of herpes simplex virus type 1 is required for viral response to interferon but is not sufficient for efficient viral replication. J. Virol. 2003, 77, 10154–10161. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, C.; Chen, X.; Yu, J.; Wang, Y.; Yang, Y.; Du, M.; Jin, H.; Ma, Y.; He, B.; et al. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J. Biol. Chem. 2011, 286, 24785–24792. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Jin, H.; Valyi-Nagy, T.; Cao, Y.; Yan, Z.; He, B. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J. Virol. 2012, 86, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Grekova, S.; Zawatzky, R.; Horlein, R.; Cziepluch, C.; Mincberg, M.; Davis, C.; Rommelaere, J.; Daeffler, L. Activation of an antiviral response in normal but not transformed mouse cells: A new determinant of minute virus of mice oncotropism. J. Virol. 2010, 84, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [PubMed]
- Krishnamurthy, S.; Takimoto, T.; Scroggs, R.A.; Portner, A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J. Virol. 2006, 80, 5145–5155. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.E.; Samuel, C.E. Proteolytic cleavage of the reovirus σ 3 protein results in enhanced double-stranded RNA-binding activity: Identification of a repeated basic amino acid motif within the C-terminal binding region. J. Virol. 1992, 66, 5347–5356. [Google Scholar] [PubMed]
- Schiff, L.A. Reovirus capsid proteins σ 3 and μ 1: Interactions that influence viral entry, assembly, and translational control. Curr. Top. Microbiol. Immunol. 1998, 233, 167–183. [Google Scholar] [PubMed]
- Bergeron, J.; Mabrouk, T.; Garzon, S.; Lemay, G. Characterization of the thermosensitive ts453 reovirus mutant: Increased dsRNA binding of σ3 protein correlates with interferon resistance. Virology 1998, 246, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Zhang, X.; Olson, N.H.; Walker, S.B.; Chappell, J.D.; Dermody, T.S.; Baker, T.S.; Nibert, M.L. Complete in vitro assembly of the reovirus outer capsid produces highly infectious particles suitable for genetic studies of the receptor-binding protein. J. Virol. 2001, 75, 5335–5342. [Google Scholar] [CrossRef] [PubMed]
- Roner, M.R.; Joklik, W.K. Reovirus reverse genetics: Incorporation of the CAT gene into the reovirus genome. Proc. Natl. Acad. Sci. USA 2001, 98, 8036–8041. [Google Scholar] [CrossRef] [PubMed]
- Roner, M.R.; Bassett, K.; Roehr, J. Identification of the 5' sequences required for incorporation of an engineered ssRNA into the Reovirus genome. Virology 2004, 329, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Roner, M.R.; Roehr, J. The 3' sequences required for incorporation of an engineered ssRNA into the Reovirus genome. Virol. J. 2006, 3. [Google Scholar] [CrossRef] [PubMed]
- Roner, M.R.; Steele, B.G. Features of the mammalian orthoreovirus 3 Dearing l1 single-stranded RNA that direct packaging and serotype restriction. J. Gen. Virol. 2007, 88, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Roner, M.R.; Steele, B.G. Localizing the reovirus packaging signals using an engineered m1 and s2 ssRNA. Virology 2007, 358, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Baric, R.S.; Sims, A.C. A Reverse genetics system for dsRNA viruses. Cell Host Microbe 2007, 1, 90–91. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Antar, A.A.; Boehme, K.W.; Danthi, P.; Eby, E.A.; Guglielmi, K.M.; Holm, G.H.; Johnson, E.M.; Maginnis, M.S.; Naik, S.; et al. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 2007, 1, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ooms, L.S.; Ikizler, M.; Chappell, J.D.; Dermody, T.S. An improved reverse genetics system for mammalian orthoreoviruses. Virology 2010, 398, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Kawagishi, T.; Kobayashi, T.; Ikizler, M.; Iskarpatyoti, J.; Dermody, T.S.; Taniguchi, K. A plasmid-based reverse genetics system for mammalian orthoreoviruses driven by a plasmid-encoded T7 RNA polymerase. J. Virol. Methods 2014, 196, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, A.A.; Blattman, J.N.; Blattman, N.N.; Greenberg, P.D.; Nibert, M.L. Engineering recombinant reoviruses with tandem repeats and a tetravirus 2A-like element for exogenous polypeptide expression. Proc. Natl. Acad. Sci. USA 2013, 110, E1867–E1876. [Google Scholar] [CrossRef] [PubMed]
- Van den Wollenberg, D.J.; Dautzenberg, I.J.; Ros, W.; Lipinska, A.D.; van den Hengel, S.K.; Hoeben, R.C. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene Ther. 2015, 22, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Brochu-Lafontaine, V.; Lemay, G. Addition of exogenous polypeptides on the mammalian reovirus outer capsid using reverse genetics. J. Virol. Methods 2012, 179, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Rouault, E.; Lemay, G. Incorporation of epitope-tagged viral σ3 proteins to reovirus virions. Can. J. Microbiol. 2003, 49, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Adair, R.A.; Scott, K.J.; Fraser, S.; Errington-Mais, F.; Pandha, H.; Coffey, M.; Selby, P.; Cook, G.P.; Vile, R.; Harrington, K.J.; et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int. J. Cancer 2013, 132, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Adair, R.A.; Roulstone, V.; Scott, K.J.; Morgan, R.; Nuovo, G.J.; Fuller, M.; Beirne, D.; West, E.J.; Jennings, V.A.; Rose, A.; et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.J.; Prestwich, R.J.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Vile, R.G.; et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Clements, D.R.; Sterea, A.M.; Kim, Y.; Helson, E.; Dean, C.A.; Nunokawa, A.; Coyle, K.M.; Sharif, T.; Marcato, P.; Gujar, S.A.; et al. Newly recruited CD11b+, GR-1+, Ly6C(high) myeloid cells augment tumor-associated immunosuppression immediately following the therapeutic administration of oncolytic reovirus. J. Immunol. 2015, 194, 4397–4412. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Dielschneider, R.; Clements, D.; Helson, E.; Shmulevitz, M.; Marcato, P.; Pan, D.; Pan, L.Z.; Ahn, D.G.; Alawadhi, A.; et al. Multifaceted therapeutic targeting of ovarian peritoneal carcinomatosis through virus-induced immunomodulation. Mol. Ther. 2013, 21, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Marcato, P.; Pan, D.; Lee, P.W. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol. Cancer Ther. 2010, 9, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Pan, D.A.; Marcato, P.; Garant, K.A.; Lee, P.W. Oncolytic virus-initiated protective immunity against prostate cancer. Mol. Ther. 2011, 19, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Lee, P.W. Oncolytic virus-mediated reversal of impaired tumor antigen presentation. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Ilett, E.J.; Morgan, R.S.; Scott, K.J.; Kottke, T.; Thompson, J.; Morrison, E.E.; Harrington, K.J.; Pandha, H.S.; et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin. Cancer Res. 2008, 14, 7358–7366. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Hatfield, P.; Merrick, A.E.; Ilett, E.J.; Selby, P.J.; Melcher, A.A. The immune system—Is it relevant to cancer development, progression and treatment? Clin. Oncol. 2008, 20, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Steele, L.P.; Ilett, E.J.; Morgan, R.S.; Harrington, K.J.; Pandha, H.S.; Selby, P.J.; Vile, R.G.; Melcher, A.A. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J. Immunol. 2009, 183, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Ilett, E.J.; Errington, F.; Diaz, R.M.; Steele, L.P.; Kottke, T.; Thompson, J.; Galivo, F.; Harrington, K.J.; Pandha, H.S.; et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin. Cancer Res. 2009, 15, 4374–4381. [Google Scholar] [CrossRef] [PubMed]
- Steele, L.; Errington, F.; Prestwich, R.; Ilett, E.; Harrington, K.; Pandha, H.; Coffey, M.; Selby, P.; Vile, R.; Melcher, A. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Grekova, S.P.; Heller, A.; Kuhlmann, O.; Soyka, E.; Giese, T.; Aprahamian, M.; Bour, G.; Ruffer, S.; Cziepluch, C.; et al. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J. Virol. 2014, 88, 5263–5276. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, O.G.; Errington-Mais, F.; Steele, L.; Hadac, E.; Jennings, V.; Scott, K.; Peach, H.; Phillips, R.M.; Bond, J.; Pandha, H.; et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 2013, 20, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fueyo, J. Healing after death: Antitumor immunity induced by oncolytic adenoviral therapy. Oncoimmunology 2014, 3, e947872. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; de Vleeschouwer, S.; Agostinis, P.; Graf, N.; van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2015, 136, E313–E325. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Inoue, H.; Nakamura, T.; Yamada, M.; Sakamoto, C.; Urata, Y.; Okazaki, T.; Marumoto, T.; Takahashi, A.; Takayama, K.; et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012, 72, 2609–2621. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Pol, J.G.; Lichty, B.D.; Cummings, D.T.; Mossman, K.L. Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. Cancer Immunol. Res. 2013, 1, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Rewiring cancer cell death to enhance oncolytic viro-immunotherapy. Oncoimmunology 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Ketzer, P.; Kaufmann, J.K.; Engelhardt, S.; Bossow, S.; von Kalle, C.; Hartig, J.S.; Ungerechts, G.; Nettelbeck, D.M. Artificial riboswitches for gene expression and replication control of DNA and RNA viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E554–E562. [Google Scholar] [CrossRef] [PubMed]
- Strobel, B.; Klauser, B.; Hartig, J.S.; Lamla, T.; Gantner, F.; Kreuz, S. Riboswitch-mediated attenuation of transgene cytotoxicity increases adeno-associated virus vector yields in HEK-293 cells. Mol. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, A.; Johnston, R.N.; Shmulevitz, M. Potential for Improving Potency and Specificity of Reovirus Oncolysis with Next-Generation Reovirus Variants. Viruses 2015, 7, 6251-6278. https://doi.org/10.3390/v7122936
Mohamed A, Johnston RN, Shmulevitz M. Potential for Improving Potency and Specificity of Reovirus Oncolysis with Next-Generation Reovirus Variants. Viruses. 2015; 7(12):6251-6278. https://doi.org/10.3390/v7122936
Chicago/Turabian StyleMohamed, Adil, Randal N. Johnston, and Maya Shmulevitz. 2015. "Potential for Improving Potency and Specificity of Reovirus Oncolysis with Next-Generation Reovirus Variants" Viruses 7, no. 12: 6251-6278. https://doi.org/10.3390/v7122936
APA StyleMohamed, A., Johnston, R. N., & Shmulevitz, M. (2015). Potential for Improving Potency and Specificity of Reovirus Oncolysis with Next-Generation Reovirus Variants. Viruses, 7(12), 6251-6278. https://doi.org/10.3390/v7122936