
The Complete Sequence of the First Spodoptera frugiperda Betabaculovirus Genome: A Natural Multiple Recombinant Virus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Insect’s Source, Rearing and Virus Production
2.1.1. Larvae of S. frugiperda
2.1.2. Occlusion Body Purification
2.2. SfGV VG008 Genome Sequencing
2.3. Phylogenetic Inference for SfGV VG008
2.3.1. ORF Identification
2.3.2. Phylogeny

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baculovirus | Acc. Number | Abbreviation |
|---|---|---|
| Antheraea pernyi MNPV Isolate L2 | EF207986 | AnpeMNPV |
| Antheraea pernyi NPV Isolate Z | NC_008035 | AnpeNPV |
| Anticarsia gemmatalis MNPV | NC_008520 | AgMNPV |
| Autographa californica MNPV Clone C6 | NC_001623 | AcMNPV |
| Bombyx mandarina NPV S1 | NC_012672 | BomaNPV S1 |
| Bombyx mandarina NPV S2 | JQ071499 | BomaNPV S2 |
| Bombyx mori NPV Isolate T3 | NC_001962 | BmNPV |
| Choristoneura fumiferana MNPV | NC_004778 | CfMNPV |
| Choristoneura fumiferana Defective MNPV | NC_005137 | CfDEFMNPV |
| Choristoneura murinana NPV Strain Darmstadt | NC_023177 | ChmuNPV |
| Choristoneura occidentalis NPV Isolate BC1 | NC_021925 | ChocNPV |
| Choristoneura rosaceana NPV Isolate NB1 | NC_021924 | ChroNPV |
| Epiphyas postvittana NPV | NC_003083 | EppoNPV |
| Hyphantria cunea NPV | NC_007767 | HycuNPV |
| Maruca vitrata NPV | NC_008725 | MaviNPV |
| Orgyia pseudotsugata MNPV | NC_001875 | OpMNPV |
| Philosamia cynthia ricini NPV | JX404026 | PhcyNPV |
| Plutella xylostella MNPV Isolate CL3 | NC_008349 | PlxyMNPV |
| Rachiplusia ou MNPV | NC_004323 | RoMNPV |
| Thysanoplusia orichalcea NPV P2 | NC_019945 | ThorNPV P2 |
| Adoxophyes honmai NPV | NC_004690 | AdhoNPV |
| Adoxophyes orana NPV | NC_011423 | AdorNPV |
| Agrotis ipsilon MNPV | NC_011345 | AgipMNPV |
| Agrotis segetum NPV | NC_007921 | AgseNPV |
| Apocheima cinerarium NPV | NC_018504 | ApciNPV |
| Buzura suppressaria NPV Isolate Hubei | NC_023442 | BusuNPV |
| Chrysodeixis chalcites NPV | NC_007151 | ChchNPV |
| Clanis bilineata NPV Isolate DZ1 | NC_008293 | ClbiNPV |
| Ectropis obliqua NPV Strain A1 | NC_008586 | EcobNPV |
| Euproctis pseudoconspersa NPV | NC_012639 | EupsNPV |
| Helicoverpa armigera MNPV | NC_011615 | HaMNPV |
| Helicoverpa armigera NPV Isolate Australia | JN584482 | HaNPV Aus |
| Helicoverpa armigera NPV Strain C1 | NC_003094 | HaNPV C1 |
| Helicoverpa armigera NPV Strain G4 | NC_002654 | HaNPV G4 |
| Helicoverpa armigera SNPV Strain NNg1 | NC_011354 | HaSNPV |
| Helicoverpa zea NPV | NC_003349 | HezeNPV |
| Hemileuca sp. NPV | NC_021923 | HespNPV |
| Leucania separata NPV Strain AH1 | NC_008348 | LeseNPV |
| Lymantria dispar MNPV | NC_001973 | LdMNPV |
| Lymantria xylina MNPV | NC_013953 | LyxyMNPV |
| Mamestra brassicae MNPV Isolate Chb1 | JX138237 | MabrMNPV Chb1 |
| Mamestra brassicae MNPV Isolate K1 | NC_023681 | MabrMNPV K1 |
| Mamestra configurata NPV Strain 90-2 | NC_003529 | MacoNPV 90 2 |
| Mamestra configurata NPV Strain A90-4 | AF539999 | MacoNPV A90 4 |
| Mamestra configurata NPV Strain B | NC_004117 | MacoNPV B |
| Orgyia leucostigma NPV IsolateCFS77 | NC_010276 | OrleNPV |
| Spodoptera exigua MNPV | NC_002169 | SeMNPV |
| Spodoptera frugiperda MNPV Isolate 3AP2 | NC_009011 | SfMNPV 3AP2 |
| Spodoptera frugiperda MNPV Isolate Nicaraguan | HM595733 | SfMNPV Nic |
| Spodoptera frugiperda MNPV Isolate Nicaraguan DefG | JF899325 | SfMNPV NicG |
| Spodoptera frugiperda MNPV Strain 19 | EU258200 | SfMNPV 19 |
| Spodoptera littoralis NPV Isolate AN1956 | JX454574 | SpltNPV AN1956 |
| Spodoptera litura II MNPV | NC_011616 | SpliMNPV II |
| Spodoptera litura MNPV Strain G2 | NC_003102 | SpliMNPV G2 |
| Trichoplusia ni SNPV | NC_007383 | TnSNPV |
| Adoxophyes orana GV | NC_005038 | AdorGV |
| Agrotis segetum GV | NC_005839 | AgseGV |
| Choristoneura occidentalis GV | NC_008168 | ChocGV |
| Clostera anastomosis GV Strain Henan | NC_022646 | CalGV |
| Cryptophlebia leucotreta GV | NC_005068 | CrleGV |
| Cydia pomonella GV | NC_002816 | CpGV |
| Epinotia aporema GV | NC_018875 | EpapGV |
| Helicoverpa armigera GV | NC_010240 | HearGV |
| Phthorimaea operculella GV | NC_004062 | PhopGV |
| Pseudaletia unipuncta GV Strain Hawaiin | NC_013772 | PsunGV |
| Pieris rapae GV | NC_013797 | PiraGV |
| Plutella xylostella GV | NC_002593 | PlxyGV |
| Spodoptera litura GV Strain K1 | NC_009503 | SpliGV |
| Xestia c-nigrum GV | NC_002331 | XecnGV |
| Neodiprion abietis NPV | DQ317692 | NeabNPV |
| Neodiprion lecontei NPV | NC_005906 | NeleNPV |
| Neodiprion sertifer NPV | NC_005905 | NeseNPV |
| Culex nigripalpus NPV | NC_003084 | CuniNPV |
2.4. Protein Synteny
2.5. Non-Coding Region Analyses
2.5.1. Homologous Regions (hrs)
2.5.2. A + T-Rich Regions
2.6. Analyses of Genes Putatively Derived from Horizontal Transfer
2.6.1. BlastP Relationships
2.6.2. Recombination Analyses
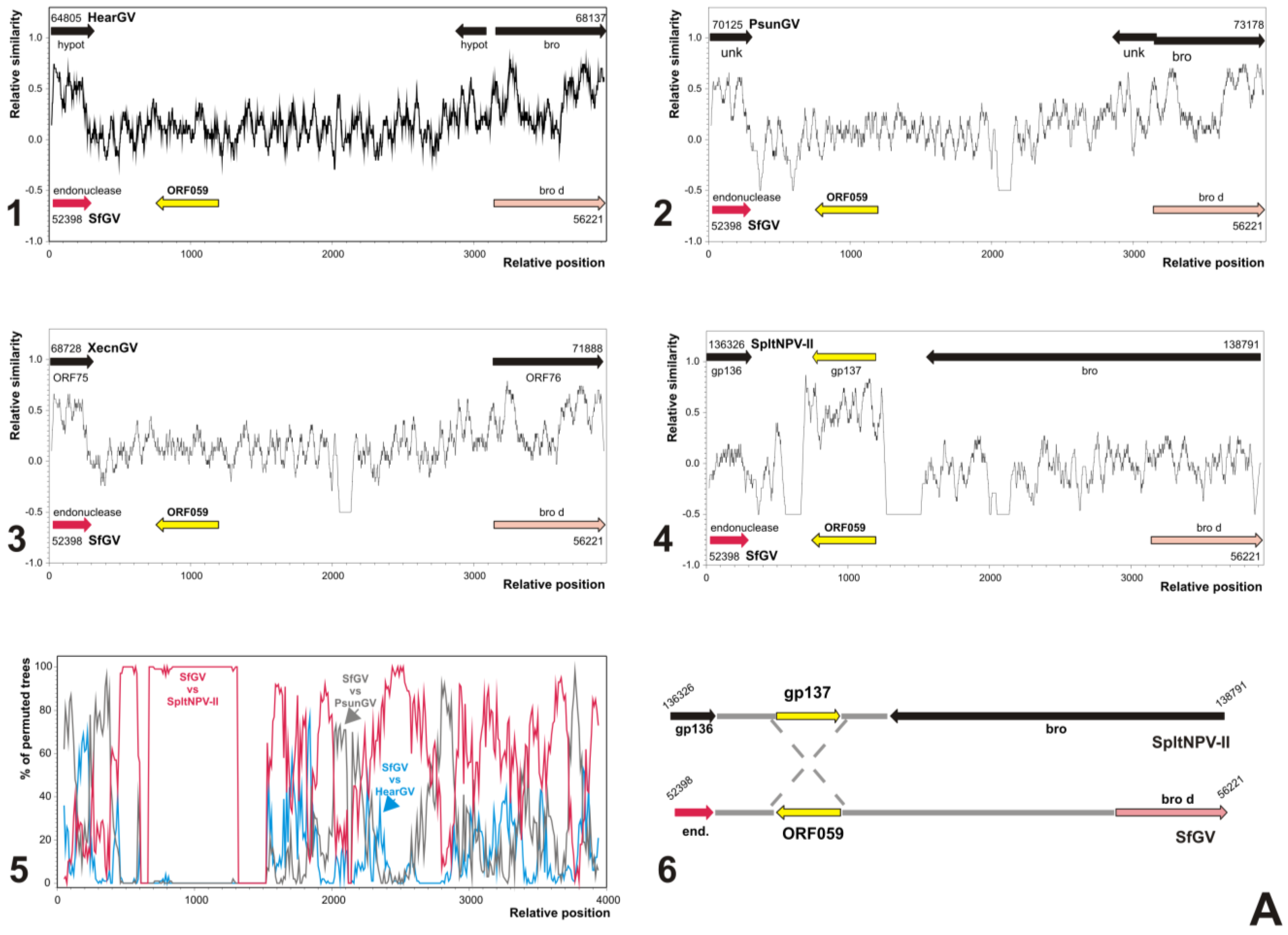
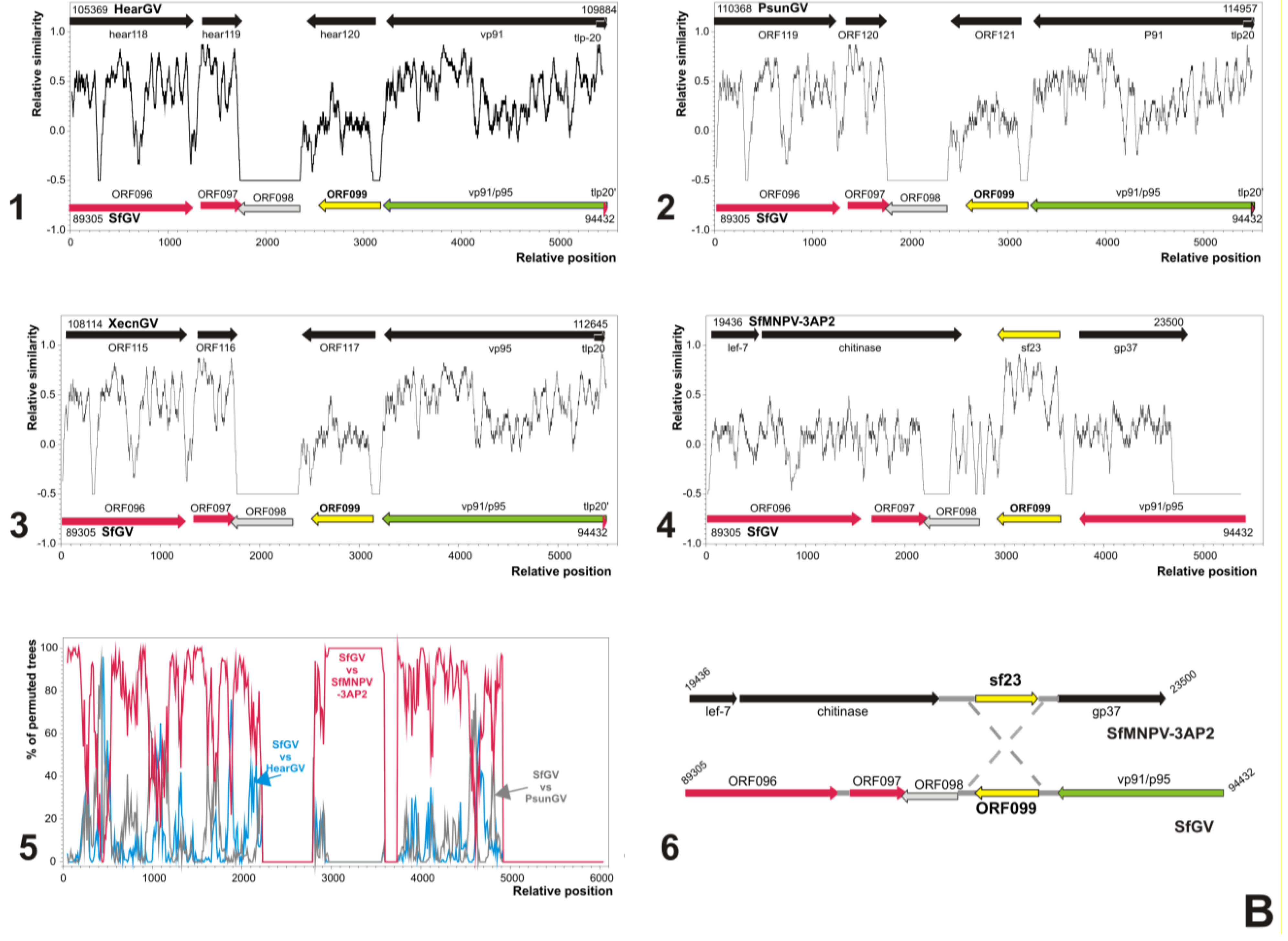
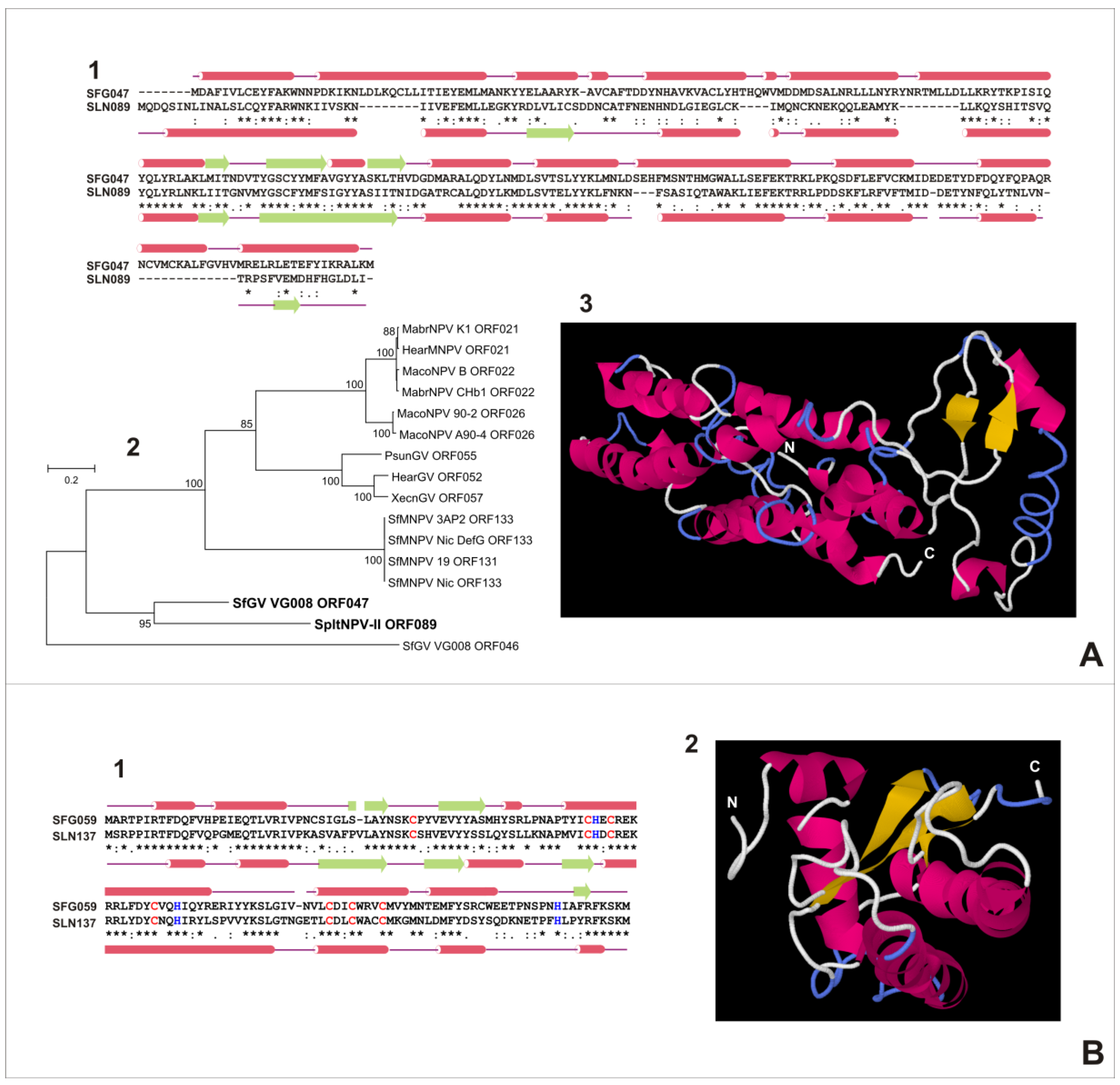
2.7. Characterization of SfGV VG008 ORFs 047/059/089/099
3. Results and Discussion
3.1. Genome of SfGV VG008 and Gene Content
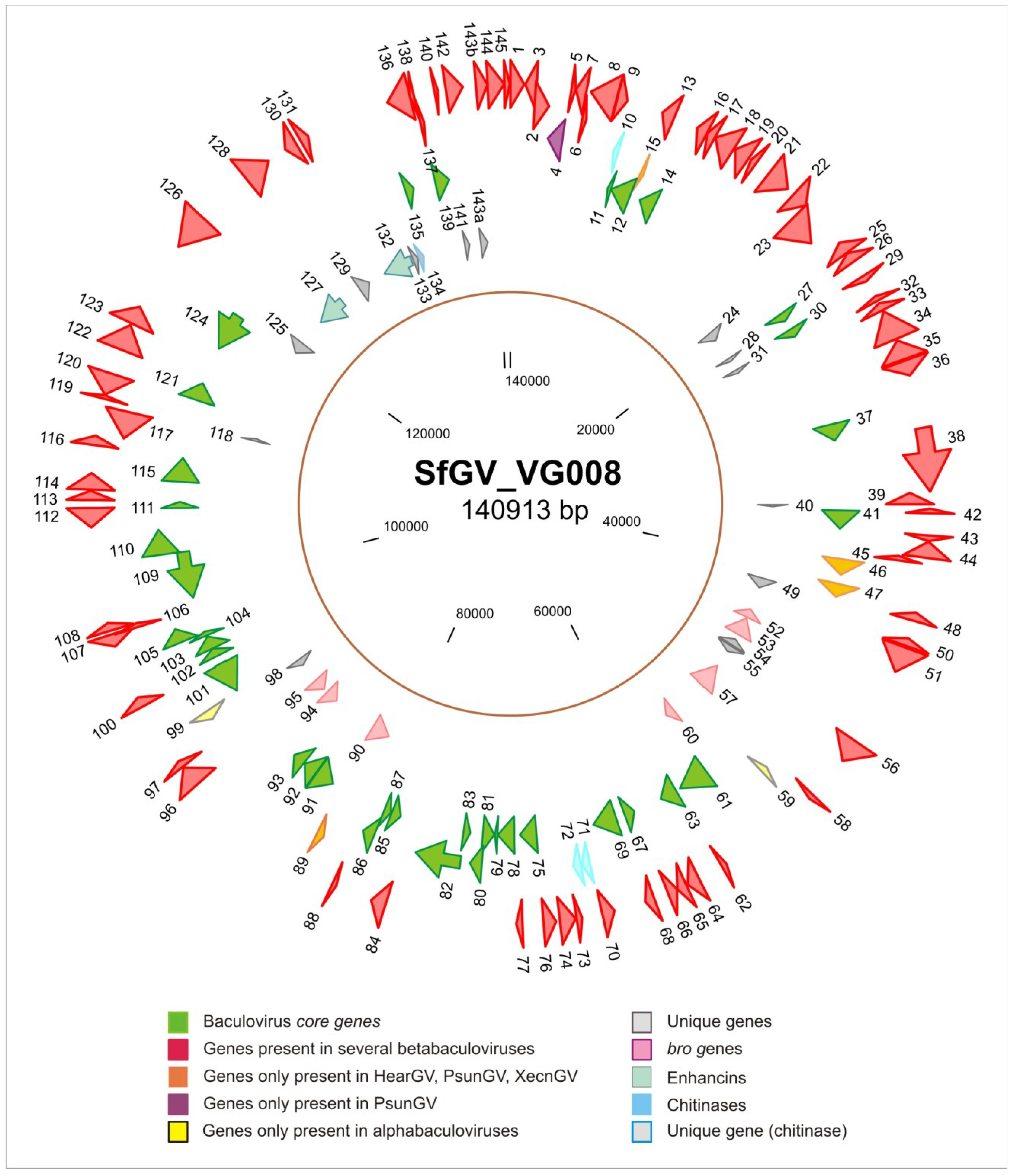
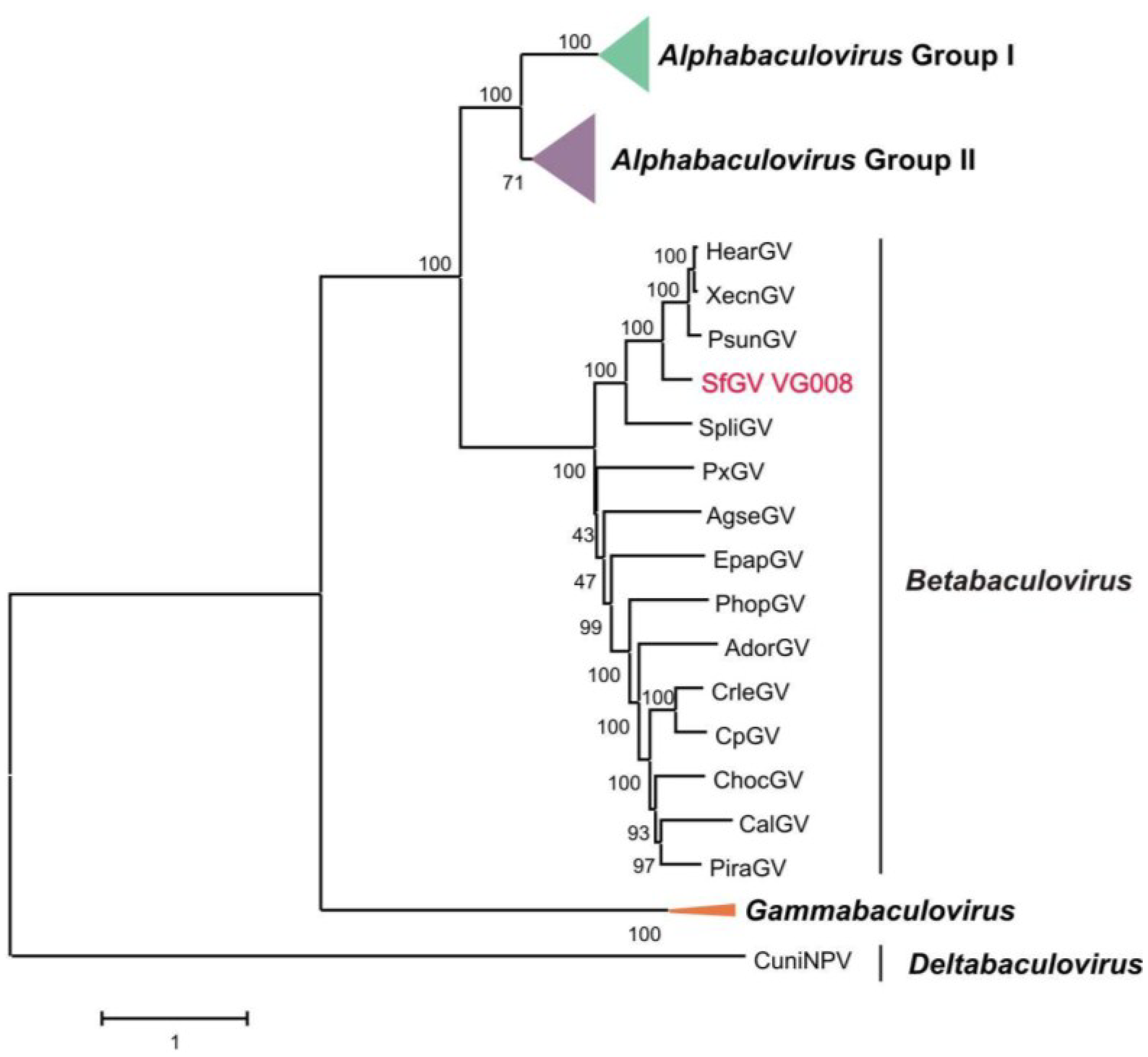
3.2. Phylogenetic Inference for SfGV VG008

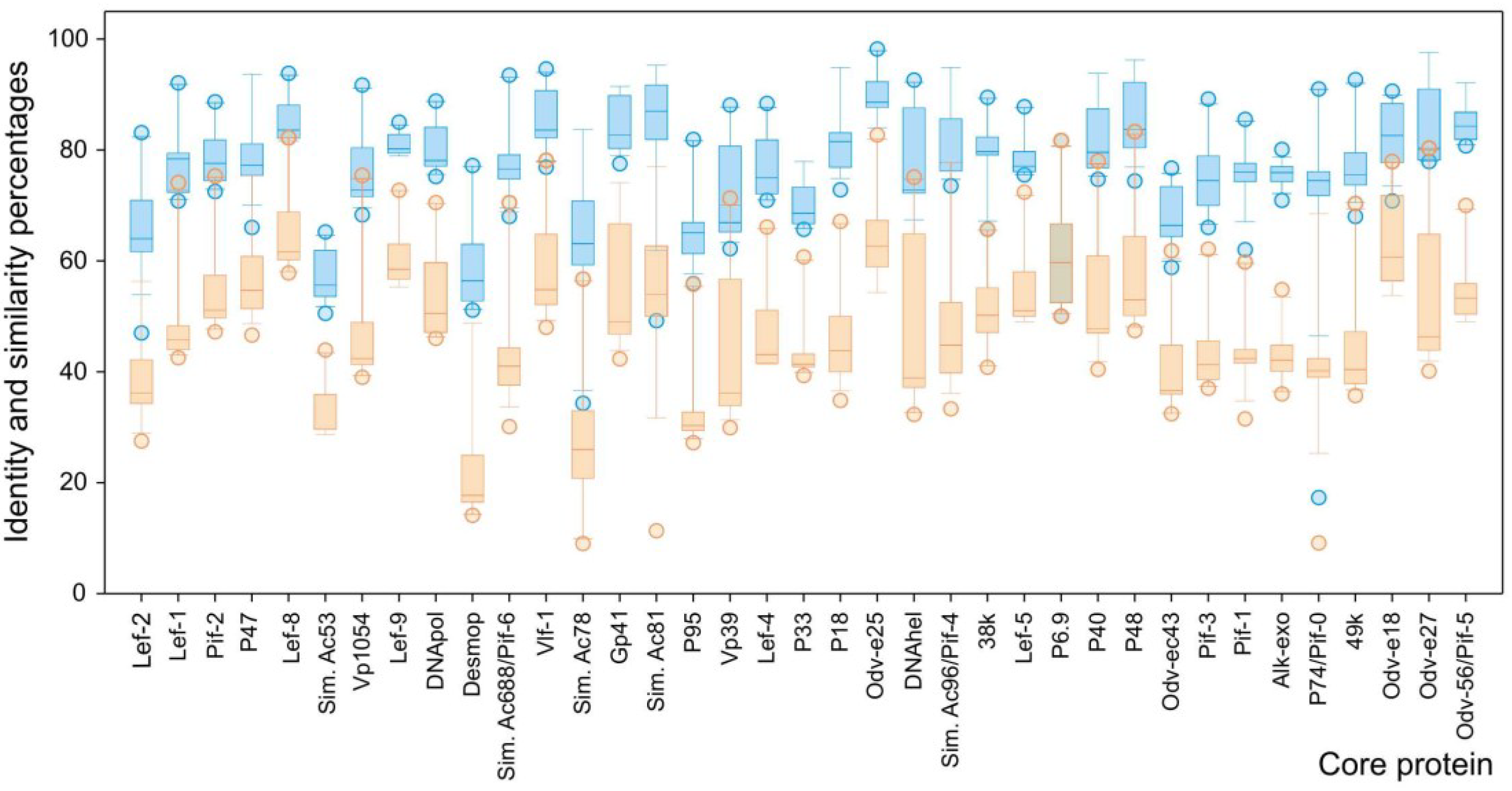
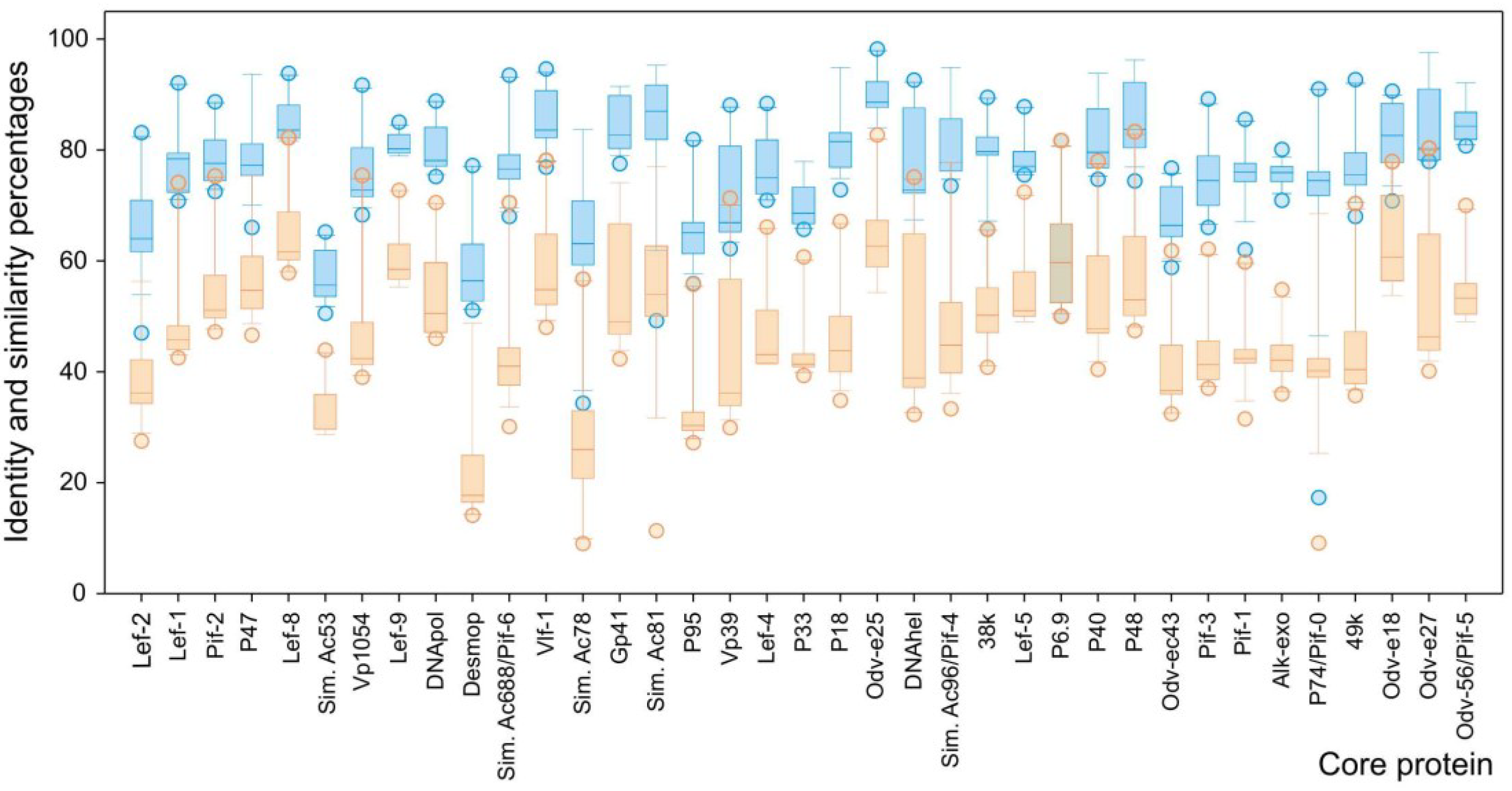
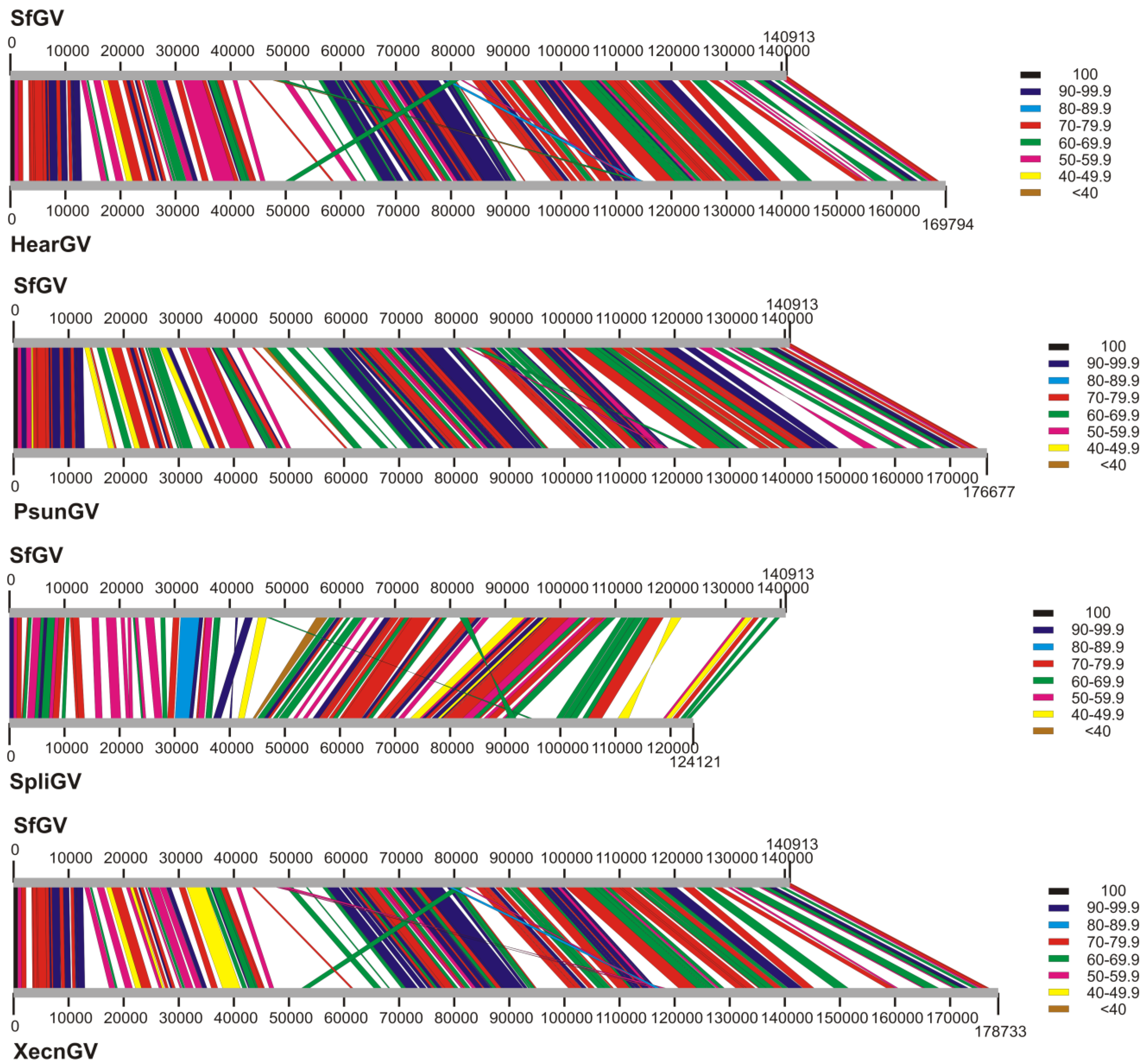
3.3. Genome Collinearity Analysis
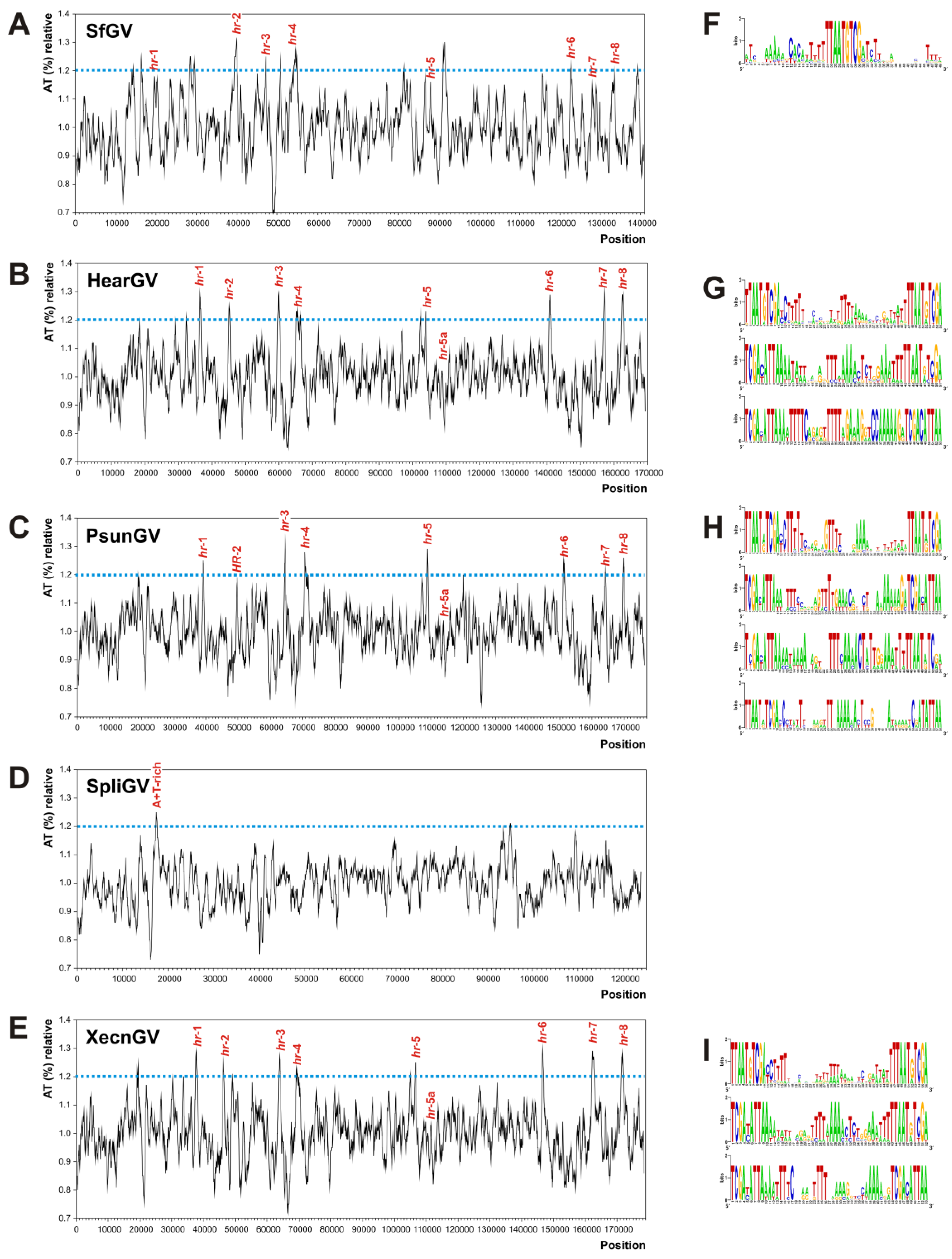
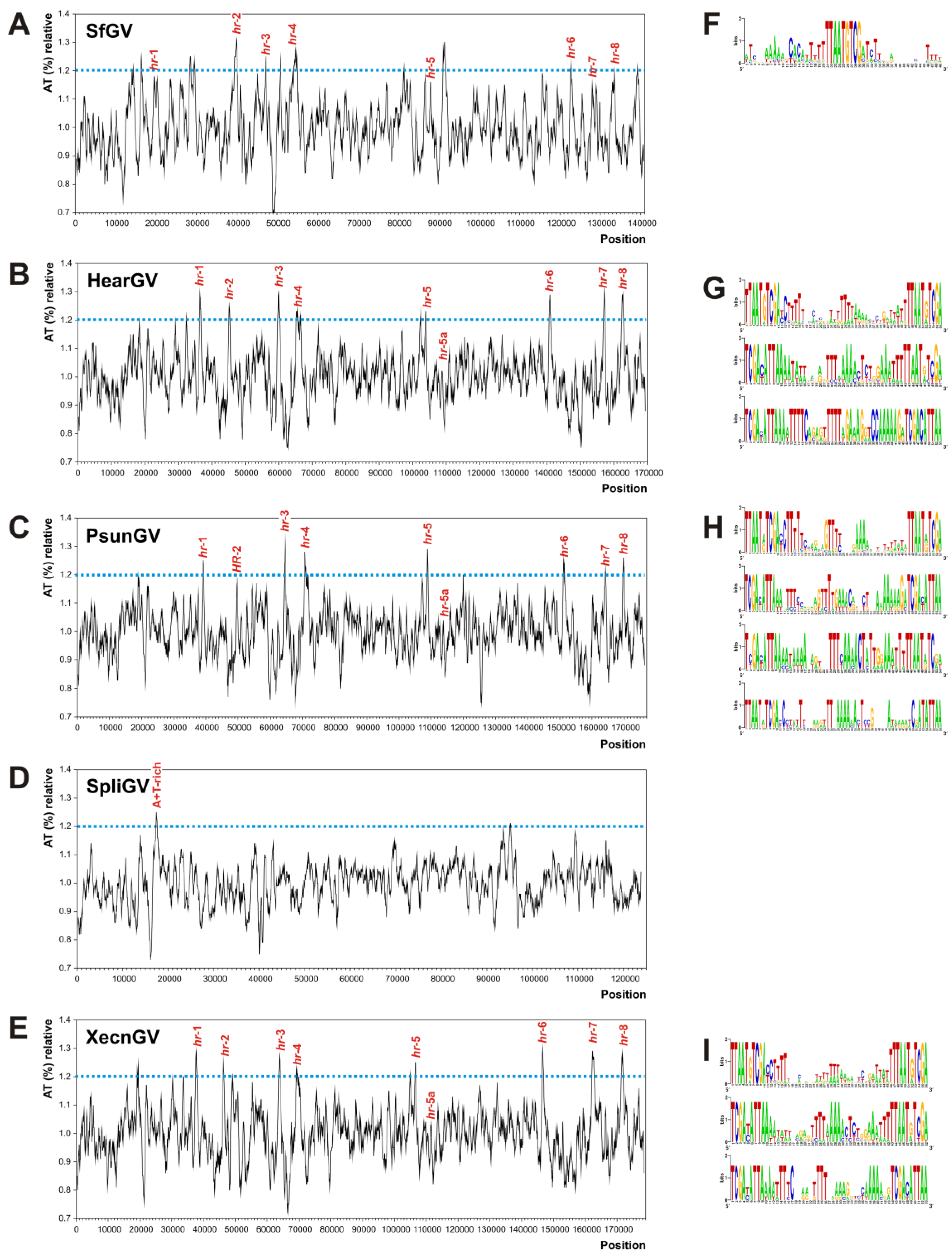
3.4. Homologous Regions (hrs) and A + T-Rich Regions
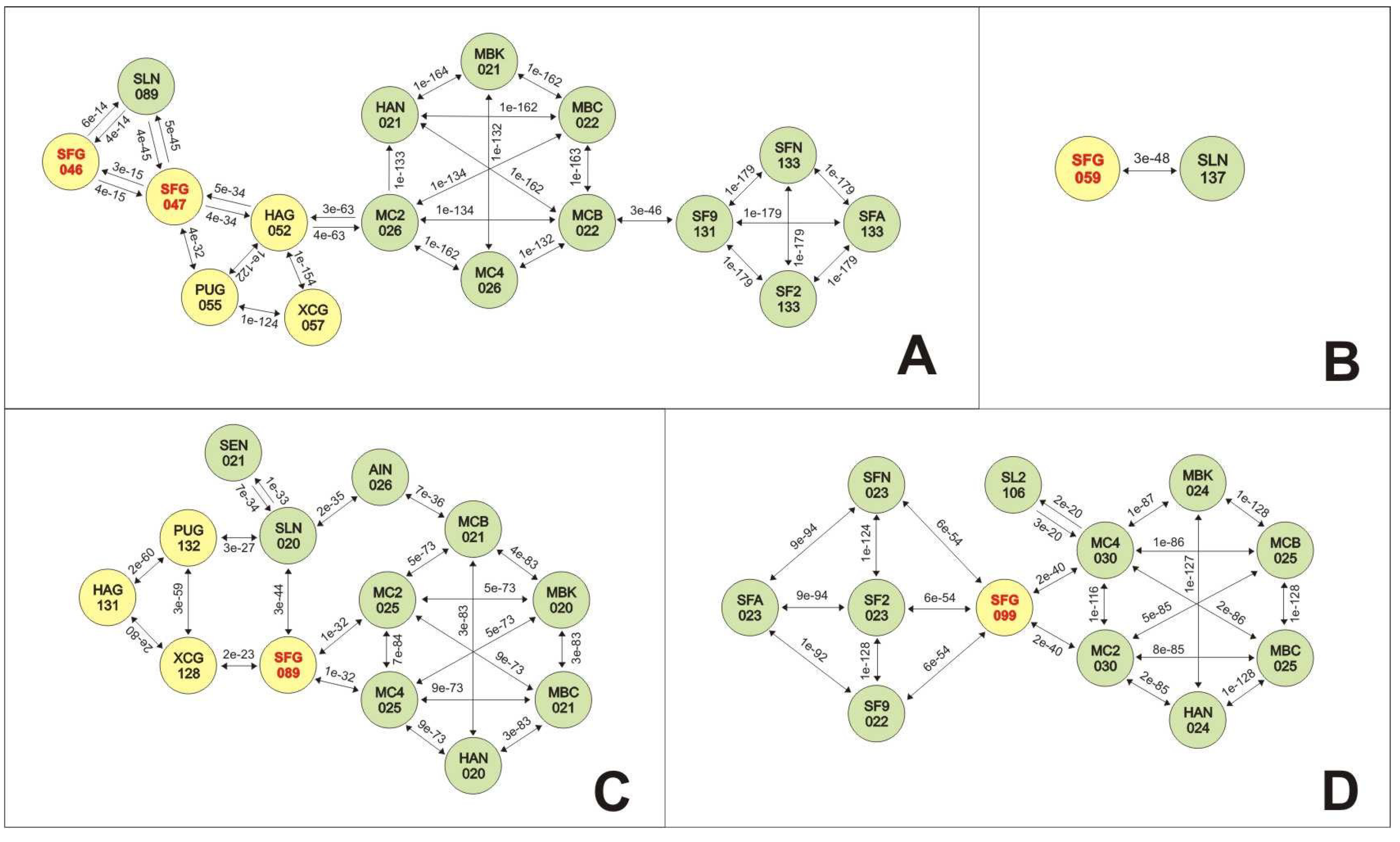
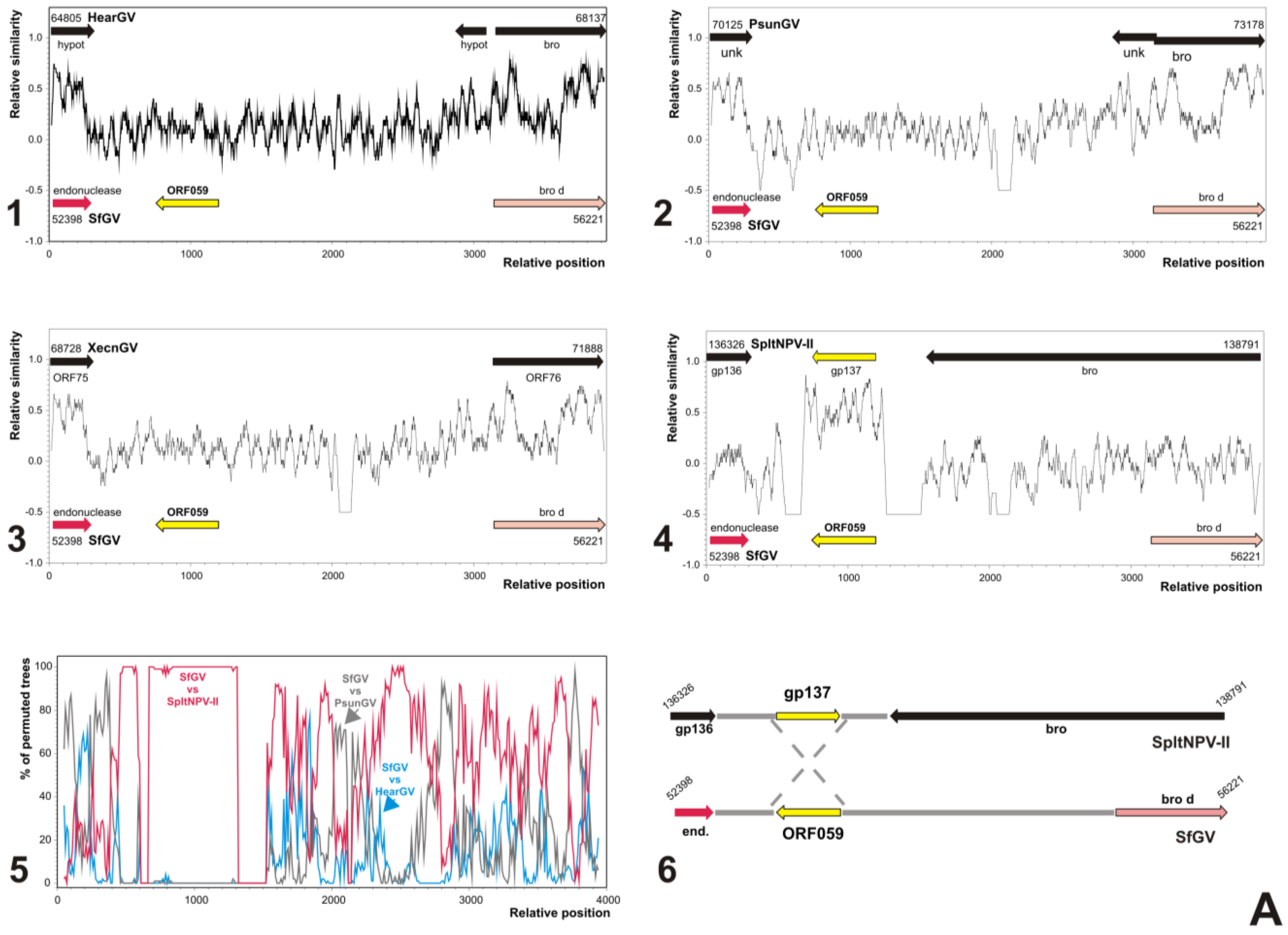
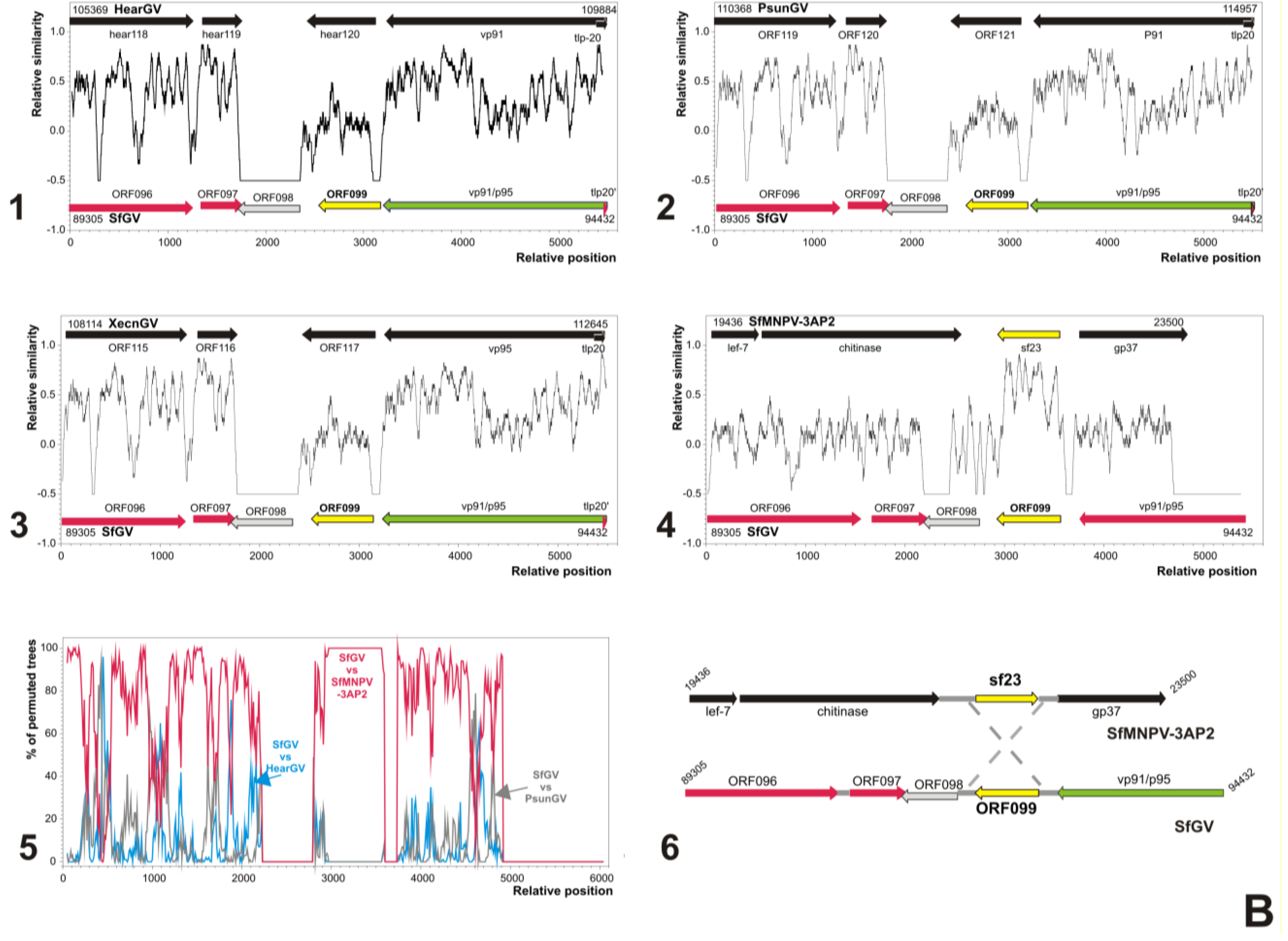
3.5. Analyses of Genes Putatively Derived from Horizontal Transfer
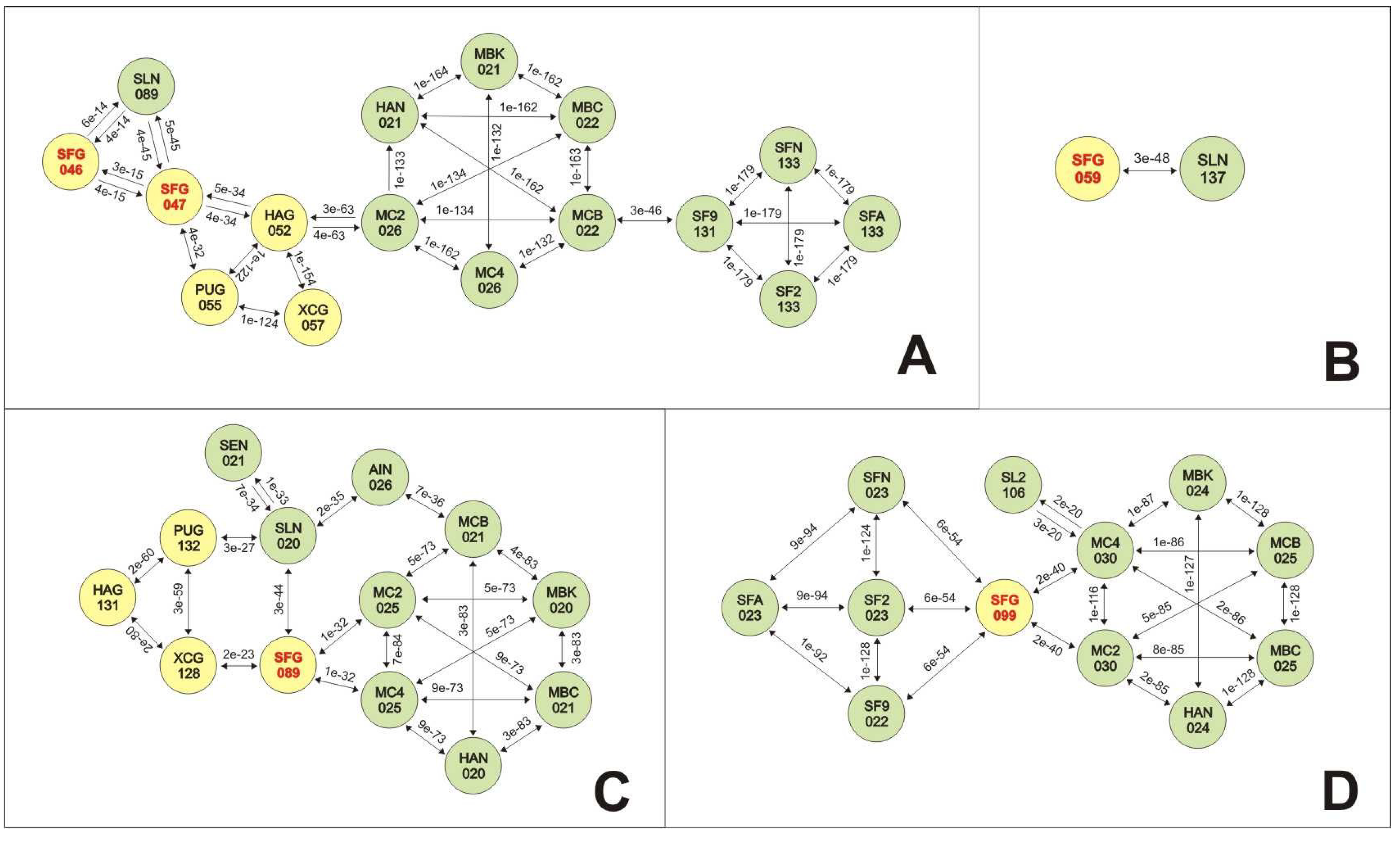
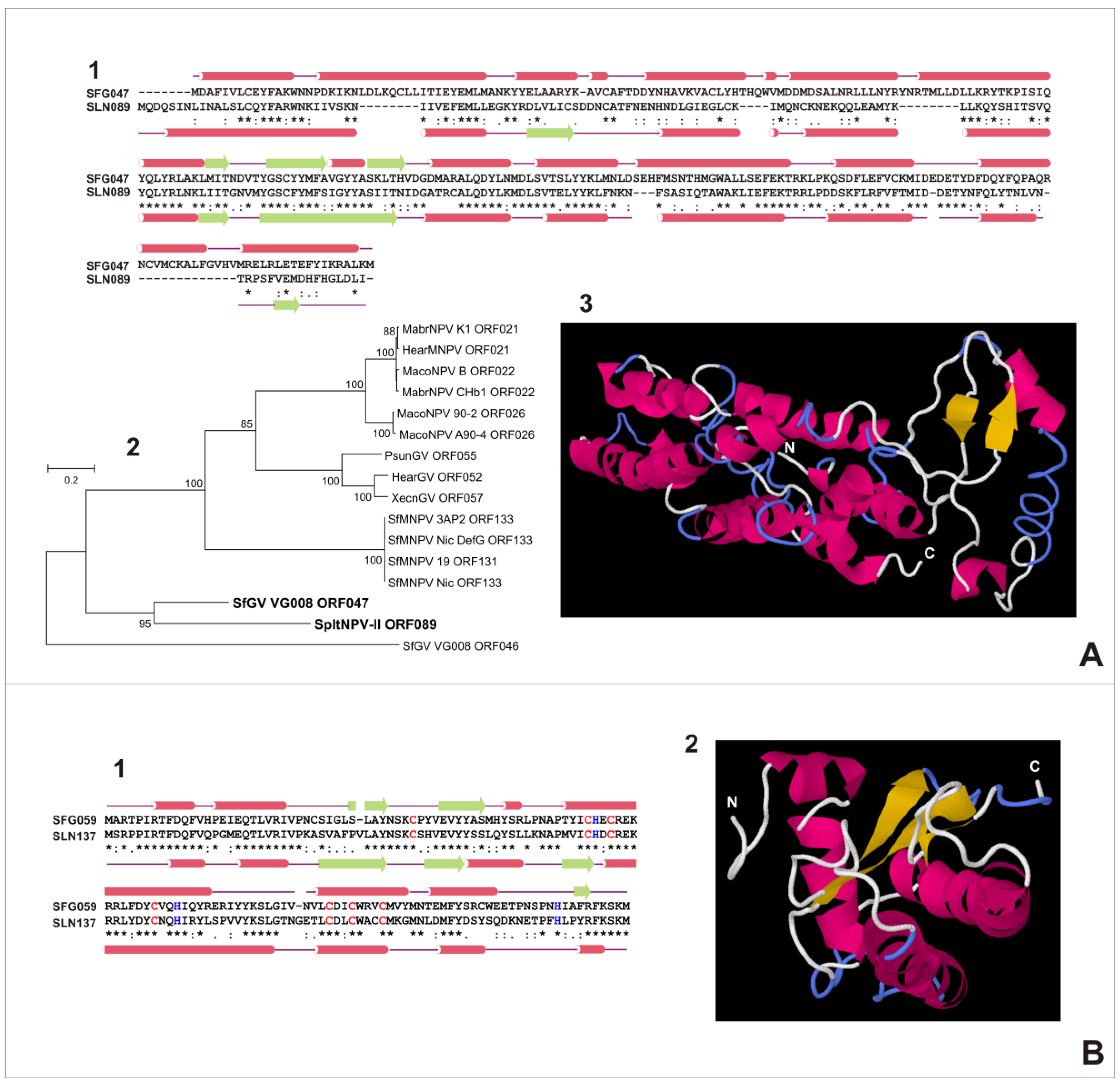
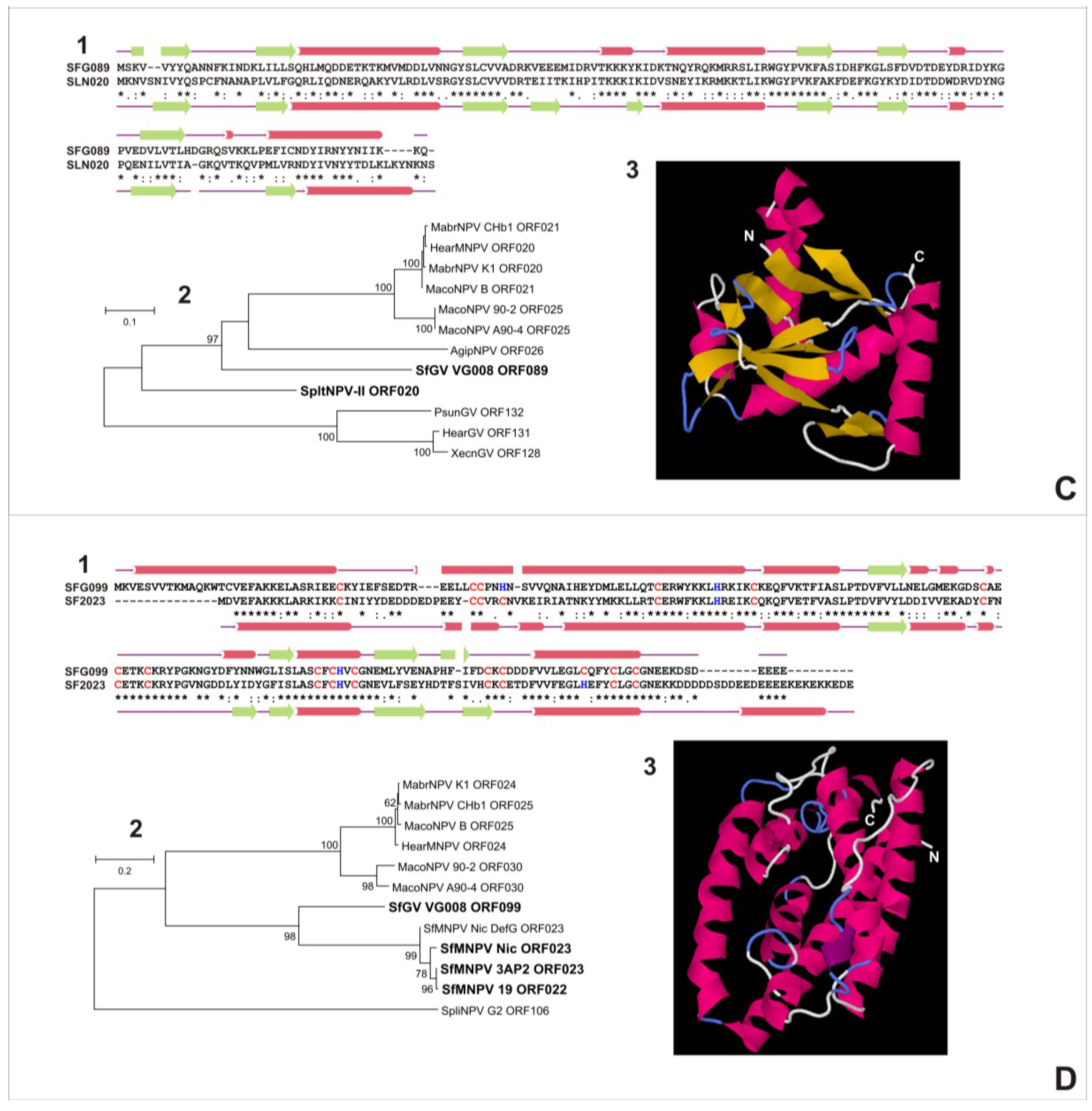
3.5.1. SfGV VG008 ORFs 046/047/089
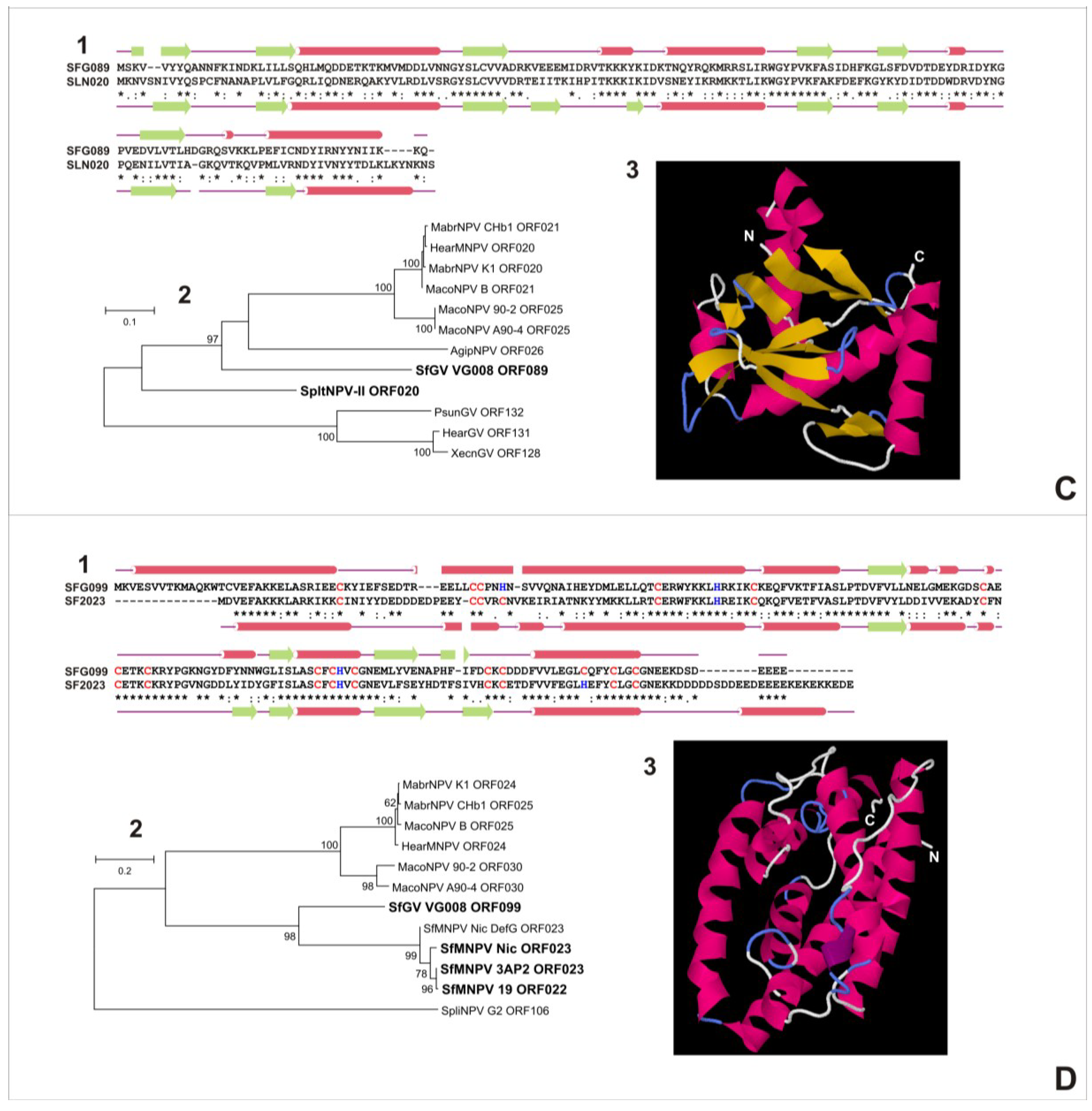
3.5.2. SfGV VG008 ORFs 059/099
3.6. Characterization of Proteins Encoded by SfGV VG008 ORFs 047/059/089/099
4. Concluding Remarks
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clark, P.L.; Molina-Ochoa, J.; Martinelli, S.; Skoda, S.R.; Isenhour, D.J.; Lee, D.J.; Krumm, J.T.; Foster, J.E. Population variation of the fall armyworm, Spodoptera frugiperda, in the western hemisphere. J. Insect Sci. 2007, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Goulson, D.; Caballero, P.; Cisneros, J.; Martínez, A.M.; Chapman, J.W.; Roman, D.X.; Cave, R.D. Evaluation of a baculovirus bioinsecticide for small-scale maize growers in latin america. Biol. Control 1999, 14, 67–75. [Google Scholar] [CrossRef]
- Flores, F. Manejo de Plagas en el Cultivo de Maíz; Estación Experimental Agropecuaria: INTA, Russia, 2010; p. 7. [Google Scholar]
- Barrera, G.; Simón, O.; Villamizar, L.; Williams, T.; Caballero, P. Spodoptera frugiperda multiple nucleopolyhedrovirus as a potential biological insecticide: Genetic and phenotypic comparison of field isolates from colombia. Biol. Control 2011, 58, 113–120. [Google Scholar] [CrossRef]
- Lapied, B.; Pennetier, C.; Apaire-Marchais, V.; Licznar, P.; Corbel, V. Innovative applications for insect viruses: Towards insecticide sensitization. Trends Biotechnol. 2009, 27, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Moscardi, F.; Souza, M.; Castro, M.; Moscardi, M.; Szewczyk, B. Baculovirus pesticides: Present state and future perspectives. In Microbes and Microbial Technology; Ahmad, I., Ahmad, F., Pichtel, J., Eds.; Springer: New York, NY, USA, 2011; pp. 415–445. [Google Scholar]
- Herniou, E.; Arif, B.M.; Becnel, J.; Blissard, G.W.; Bonning, B.C.; Harrison, R.L.; Jehle, J.A.; Theilmann, D.; Vlak, J.M. Family baculoviridae. In Virus Taxonomy—Classification and Nomenclature of Viruses: Ninth Report of the Internationalcommittee on Taxonomy of Viruses; King, A.M., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012. [Google Scholar]
- Jehle, J.A.; Lange, M.; Wang, H.L.; Hu, Z.H.; Wang, Y.J.; Hauschild, W. Molecular identification and phylogenetic analysis of baculoviruses from lepidoptera. Virology 2006, 346, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G.F. Baculovirus Molecular Biology. Available online: http://www.ncbi.nlm.nih.gov/books/NBK114593/ (accessed on 1 October 2014).
- Ferrelli, M.L.; Berretta, M.F.; Belaich, M.N.; Ghiringhelli, P.D.; Sciocco-Cap, A.; Romanowski, V. The baculoviral genome. In Viral Genomes—Molecular Structure, Diversity, Gene Expression Mechanisms and Host-Virus Interactions; Garcia, M.L., Romanowsky, V., Eds.; InTech: Croatia, Yugoslavia, 2012; pp. 1–32. [Google Scholar]
- Slack, J.; Arif, B.M. The baculoviruses occlusion-derived virus: Virion structure and function. Adv. Virus Res. 2007, 69, 99–165. [Google Scholar] [PubMed]
- Ji, X.; Sutton, G.; Evans, G.; Axford, D.; Owen, R.; Stuart, D.I. How baculovirus polyhedra fit square pegs into round holes to robustly package viruses. EMBO J. 2010, 29, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Jackes, R.P. Stability of insect viruses in the environment. In Viral Insecticides for Biological Control; Maromorosch, E., Sherman, K., Eds.; Academic Press Inc.: Orlando, FL, USA, 1985. [Google Scholar]
- Williams, G.V.; Faulkner, P. Cytological changes and viral morphogenesis during baculovirus infection. In The Baculoviruses; Miller, L., Ed.; Plenum Press Inc.: New York, NY, USA, 1997; pp. 61–107. [Google Scholar]
- Hoover, K.; Humphries, M.A.; Gendron, A.R.; Slavicek, J.M. Impact of viral enhancin genes on potency of lymantria dispar multiple nucleopolyhedrovirus in L. dispar following disruption of the peritrophic matrix. J. Invertebr. Pathol. 2010, 104, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Mukawa, S.; Goto, C. Enhancing effect of proteins derived from Xestia c-nigrum granulovirus on Mamestra brassicae nucleopolyhedrovirus infection in larvae of Autographa nigrisigna (lepidoptera: Noctuidae) on cabbage. Appl. Entomol. Zool. 2011, 46, 55–63. [Google Scholar] [CrossRef]
- Gómez, J.A.; Barrera, G.; Guevara, J.; Villamizar, L. Aislamiento y evaluación de un nucleopoliedrovirus colombiano de Spodoptera frugiperda para su control. Mem. XXXIII Congr. Nac. Control Biol. 2010, 1, 49–53. [Google Scholar]
- Cuartas, P.; Barrera, G.; Barreto, E.; Villamizar, L. Characterisation of a colombian granulovirus (Baculoviridae: Betabaculovirus) isolated from Spodoptera frugiperda (lepidoptera: Noctuidae) larvae. Biocontrol. Sci. Technol. 2014, 24, 1265–1285. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov (accessed on 1 October 2014).
- Escasa, S.R.; Lauzon, H.A.; Mathur, A.C.; Krell, P.J.; Arif, B.M. Sequence analysis of the Choristoneura occidentalis granulovirus genome. J. Gen. Virol. 2006, 87, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Ferrelli, M.L.; Salvador, R.; Biedma, M.E.; Berretta, M.F.; Haase, S.; Sciocco-Cap, A.; Ghiringhelli, P.D.; Romanowski, V. Genome of Epinotia aporema granulovirus (epapgv), a polyorganotropic fast killing betabaculovirus with a novel thymidylate kinase gene. BMC Genomics 2012, 13, e548. [Google Scholar] [CrossRef]
- Harrison, R.L.; Popham, H.J.R. Genomic sequence analysis of a granulovirus isolated from the old world bollworm, Helicoverpa armigera. Virus Genes 2008, 36, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Hayakawa, T.; Ueno, Y.; Fujita, T.; Sano, Y.; Matsumoto, T. Sequence analysis of the Plutella xylostella granulovirus genome. Virology 2000, 275, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Ko, R.; Okano, K.; Seong, S.-I.; Goto, C.; Maeda, S. Sequence analysis of the Xestia c-nigrum granulovirus genome. Virology 1999, 262, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Jehle, J.A. The genome of the Cryptophlebia leucotreta granulovirus. Virology 2003, 317, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhang, X.; Yin, X.; Cao, S.; Xu, F. Genomic sequencing and analysis of Clostera anachoreta granulovirus. Arch. Virol. 2011, 156, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhang, X.; Yin, X.; Song, X.; Shao, X.; Wang, L. Comparative analysis of the genomes of Clostera anastomosis (l) granulovirus and Clostera anachoreta granulovirus. Arch. Virol. 2013, 158, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Luque, T.; Finch, R.; Crook, N.; O’Reilly, D.R.; Winstanley, D. The complete sequence of the Cydia pomonella granulovirus genome. J. Gen. Virol. 2001, 82, 2531–2547. [Google Scholar] [PubMed]
- Wang, Y.; Choi, J.Y.; Roh, J.Y.; Liu, Q.; Tao, X.Y.; Park, J.B.; Kim, J.S.; Je, Y.H. Genomic sequence analysis of granulovirus isolated from the tobacco cutworm, Spodoptera litura. PLOS ONE 2011, 6, e28163. [Google Scholar] [CrossRef] [PubMed]
- Wormleaton, S.; Kuzio, J.; Winstanley, D. The complete sequence of the Adoxophyes orana granulovirus genome. Virology 2003, 311, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-Q.; Cheng, R.-L.; Wang, X.-F.; Zhang, C.-X. The genome of Pieris rapae granulovirus. J. Virol. 2012, 86, 9544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liang, Z.; Yin, X.; Wang, J.; Shao, X. Complete genome sequence of Agrotis segetum granulovirus shanghai strain. Arch. Virol. 2014, 1–4. [Google Scholar]
- Greene, G.L.; Leppla, N.C.; Dickerson, W.A. Velvetbean caterpillar (Lepidoptera, noctuidae) rearing procedure and artificial medium. J. Econ. Entomol. 1976, 69, 487–488. [Google Scholar]
- Hughes, P.R.; Wood, H.A. In vivo and in vitro bioassay methods for baculoviruses. In The Biology of Baculoviruses; Granados, R.R., Federici, B.B., Eds.; CRC Press: Boca Raton, FL, USA, 1986; Volume II, pp. 1–30. [Google Scholar]
- Espinel-Correal, C.; Léry, X.; Villamizar, L.; Gómez, J.; Zeddam, J.L.; Cotes, A.M.; López-Ferber, M. Genetic and biological analysis of colombian Phthorimaea operculella granulovirus isolated from tecia solanivora (lepidoptera: Gelechiidae). Appl. Environ. Microbiol. 2010, 76, 7617–7625. [Google Scholar] [CrossRef] [PubMed]
- Caballero, P.; Zuidema, D.; Santiago-Alvarez, C.; Vlak, J.M. Biochemical and biological characterization of four isolates of Spodoptera exigua nuclear polyhedrosis virus. Biocontrol Sci. Technol. 1992, 2, 145–157. [Google Scholar] [CrossRef]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.-A. Artemis and act: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [PubMed]
- Fickett, J.W. Recognition of protein coding regions in DNA sequences. Nucleic Acids Res. 1982, 10, 5303–5318. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.; Blackshields, G.; Brown, N.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal w and clustal x version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Higgins, D.; Gibson, T. Clustal w: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Garavaglia, M.J.; Miele, S.A.B.; Iserte, J.A.; Belaich, M.N.; Ghiringhelli, P.D. The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. J. Virol. 2012, 86, 12069–12079. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov (accessed on 1 May 2014).
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Gonnet, G.H.; Cohen, M.A.; Benner, S.A. Exhaustive matching of the entire protein sequence database. Science 1992, 256, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M.; Mathews, D.H.; Turner, D.H. Algorithms and thermodynamics for RNA secondary structure prediction: A practical guide. In RNA Biochemistry and Biotechnology; Springer: New York, NY, USA, 1999; pp. 11–43. [Google Scholar]
- Matzura, O.; Wennborg, A. Rnadraw: An integrated program for RNA secondary structure calculation and analysis under 32-bit microsoft windows. Comput. Appl. Biosci. 1996, 12, 247–249. [Google Scholar] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The clustal_x windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype c-infected seroconverters in india, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [PubMed]
- Salminen, M.O.; Koch, C.; Sanders-Buell, E.; Ehrenberg, P.K.; Michael, N.L.; Carr, J.K.; Burke, D.S.; McCutchan, F.E. Recovery of virtually full-length HIV-1 provirus of diverse subtypes from primary virus cultures using the polymerase chain reaction. Virology 1995, 213, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Cornette, J.L.; Cease, K.B.; Margalit, H.; Spouge, J.L.; Berzofsky, J.A.; DeLisi, C. Hydrophobicity scales and computational techniques for detecting amphipathic structures in proteins. J. Mol. Biol. 1987, 195, 659–685. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA 1984, 81, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signalp 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Söding, J. Protein homology detection by hmm–hmm comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y. Lomets: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-tasser: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge- based force field. Proteins Struct. Funct. Bioinform. 2012, 80, 1715–1735. [Google Scholar] [CrossRef]
- Friesen, P.D. Regulation of baculovirus early gene expression. In The Baculoviruses; Miller, L.K., Ed.; Plenum Press: New York, NY, USA, 1997; p. 141. [Google Scholar]
- Lu, A.; Miller, L.K.; Krell, P.; Vlak, J.M.; Rohrmann, G.F. Baculovirus DNA replication. In The Baculoviruses; Miller, L.K., Ed.; Plenum Press: New York, NY, USA, 1997. [Google Scholar]
- Van Oers, M.M.; Vlak, J.M. Baculovirus genomics. Curr. Drug Targets 2007, 8, 1051–1068. [Google Scholar]
- Miele, S.A.B.; Garavaglia, M.J.; Belaich, M.N.; Ghiringhelli, P.D. Baculovirus: Molecular insights on their diversity and conservation. Int. J. Evol. Biol. 2011, 211, 379424. [Google Scholar]
- Herniou, E.A.; Jehle, J.A. Baculovirus phylogeny and evolution. Curr. Drug Targets 2007, 8, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Berretta, M.; Romanowski, V. Baculovirus homologous regions (hrs): Pleiotropic functional cis elements in viral genomes and insect and mammalian cells. Curr. Top. Virol. 2008, 7, 47–56. [Google Scholar]
- Hilton, S.; Winstanley, D. The origins of replication of granuloviruses. Arch. Virol. 2008, 153, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Choi, J.Y.; Roh, J.Y.; Woo, S.D.; Jin, B.R.; Je, Y.H. Molecular and phylogenetic characterization of Spodoptera litura granulovirus. J. Microbiol. 2008, 46, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yin, F.; Zhu, Z.; Hou, D.; Wang, J.; Zhang, L.; Wang, M.; Wang, H.; Hu, Z.; Deng, F. Genomic sequencing and analysis of Sucra jujuba nucleopolyhedrovirus. PLOS ONE 2014, 9, e110023. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L. Structural divergence among genomes of closely related baculoviruses and its implications for baculovirus evolution. J. Invertebr. Pathol. 2009, 101, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Olszewski, J.A.; Cory, J.S.; O’Reilly, D.R. The genome sequence and evolution of baculoviruses. Ann. Rev. Entomol. 2003, 48, 211–234. [Google Scholar] [CrossRef]
- Maeda, S.; Kamita, S.G.; Kondo, A. Host range expansion of autographa californica nuclear polyhedrosis virus (npv) following recombination of a 0.6-kilobase-pair DNA fragment originating from Bombyx mori npv. J. Virol. 1993, 67, 6234–6238. [Google Scholar] [PubMed]
- Xu, Y.-P.; Gu, L.-Z.; Lou, Y.-H.; Cheng, R.-L.; Xu, H.-J.; Wang, W.-B.; Zhang, C.-X. A baculovirus isolated from wild silkworm encompasses the host ranges of Bombyx mori nucleopolyhedrosis virus and Autographa californica multiple nucleopolyhedrovirus in cultured cells. J. Gen. Virol. 2012, 93, 2480–2489. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Fritsch, E.; Huber, J.; Backhaus, H. Intra-specific and inter-specific recombination of tortricid-specific granuloviruses during co-infection in insect larvae. Arch. Virol. 2003, 148, 1317–1333. [Google Scholar] [CrossRef] [PubMed]
- Croizier, G.; Croizier, L.; Quiot, J.M.; Lereclus, D. Recombination of Autographa californica and Rachiplusia ou nuclear polyhedrosis viruses in galleria mellonella l. J. Gen. Virol. 1988, 69, 177–185. [Google Scholar] [CrossRef]
- Meijer, M.; Karimi-Busheri, F.; Huang, T.Y.; Weinfeld, M.; Young, D. DNA: Replication, repair, and recombination-pnk1, a DNA kinase/phosphatase required for normal response to DNA damage by g-radiation or camptothecin in Schizosaccharomyces pombe. J. Biol. Chem. 2002, 277, 4050–4055. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuartas, P.E.; Barrera, G.P.; Belaich, M.N.; Barreto, E.; Ghiringhelli, P.D.; Villamizar, L.F. The Complete Sequence of the First Spodoptera frugiperda Betabaculovirus Genome: A Natural Multiple Recombinant Virus. Viruses 2015, 7, 394-421. https://doi.org/10.3390/v7010394
Cuartas PE, Barrera GP, Belaich MN, Barreto E, Ghiringhelli PD, Villamizar LF. The Complete Sequence of the First Spodoptera frugiperda Betabaculovirus Genome: A Natural Multiple Recombinant Virus. Viruses. 2015; 7(1):394-421. https://doi.org/10.3390/v7010394
Chicago/Turabian StyleCuartas, Paola E., Gloria P. Barrera, Mariano N. Belaich, Emiliano Barreto, Pablo D. Ghiringhelli, and Laura F. Villamizar. 2015. "The Complete Sequence of the First Spodoptera frugiperda Betabaculovirus Genome: A Natural Multiple Recombinant Virus" Viruses 7, no. 1: 394-421. https://doi.org/10.3390/v7010394
APA StyleCuartas, P. E., Barrera, G. P., Belaich, M. N., Barreto, E., Ghiringhelli, P. D., & Villamizar, L. F. (2015). The Complete Sequence of the First Spodoptera frugiperda Betabaculovirus Genome: A Natural Multiple Recombinant Virus. Viruses, 7(1), 394-421. https://doi.org/10.3390/v7010394