Retroviral Vectors for Analysis of Viral Mutagenesis and Recombination


Abstract
:1. Introduction
2. Vectors for Examination of Retroviral Mutagenesis
2.1. Reporter Gene Vectors for Reversion Assays

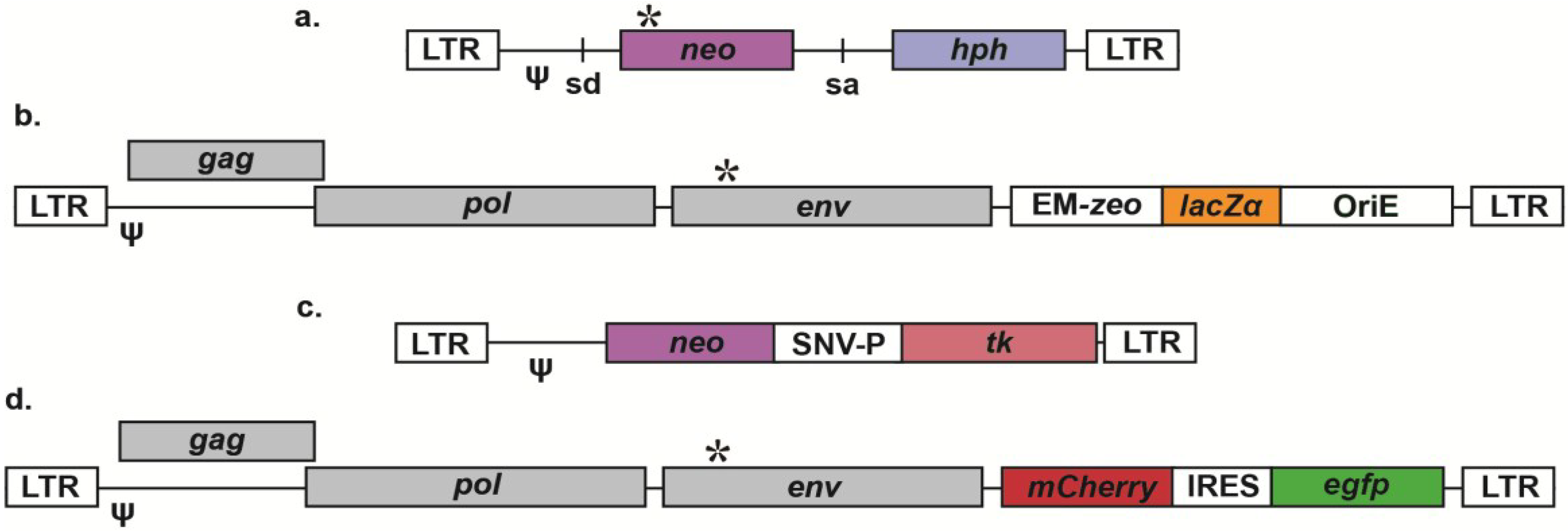{kind=link}
{kind=link}
| Target1 | Assay Type2 | Virus | References |
|---|---|---|---|
| neo | Reporter (R) | SNV, MLV | [90,91] |
| tk | Reporter (F) | MLV, HIV-1 | [8,97,98] |
| lacZα/lacZ | Reporter (F) | SNV, BLV, HTLV-1, HIV-1 | [4,5,6,7,27,28,29,31,32,33,34,35,99,100,101,102] |
| luc | Reporter (R) | HIV-1 | [30] |
| bsr | Reporter (R) | ALV, MLV, HIV-1 | [96] |
| hsa | Reporter (F) | HIV-1 | [103,104] |
| egfp | Reporter (F) | HIV-1 | [105,106,107,108] |
| Viral | HTA | RSV | [109] |
| RNase T1 | MLV | [110] | |
| SSCP | HIV-1 | [12] | |
| Sequencing (Sanger) | HIV-1 | [111,112] | |
| Sequencing (NGS) | HIV-1 | [40] |
2.2. Reporter Gene Vectors for Forward Mutational Assays
2.2.1. LacZα/LacZ (β-galactosidase)
2.2.2. Thymidine Kinase (TK)
2.2.3. Fluorescent Proteins and Cell Surface Markers
2.3. Detection of Mutations in Viral Genes
2.3.1. Biochemical and Sanger Sequencing-Based Approaches
2.3.2. Next-Generation Sequencing-Based Approaches
3. Vectors for Examination of Retroviral Recombination
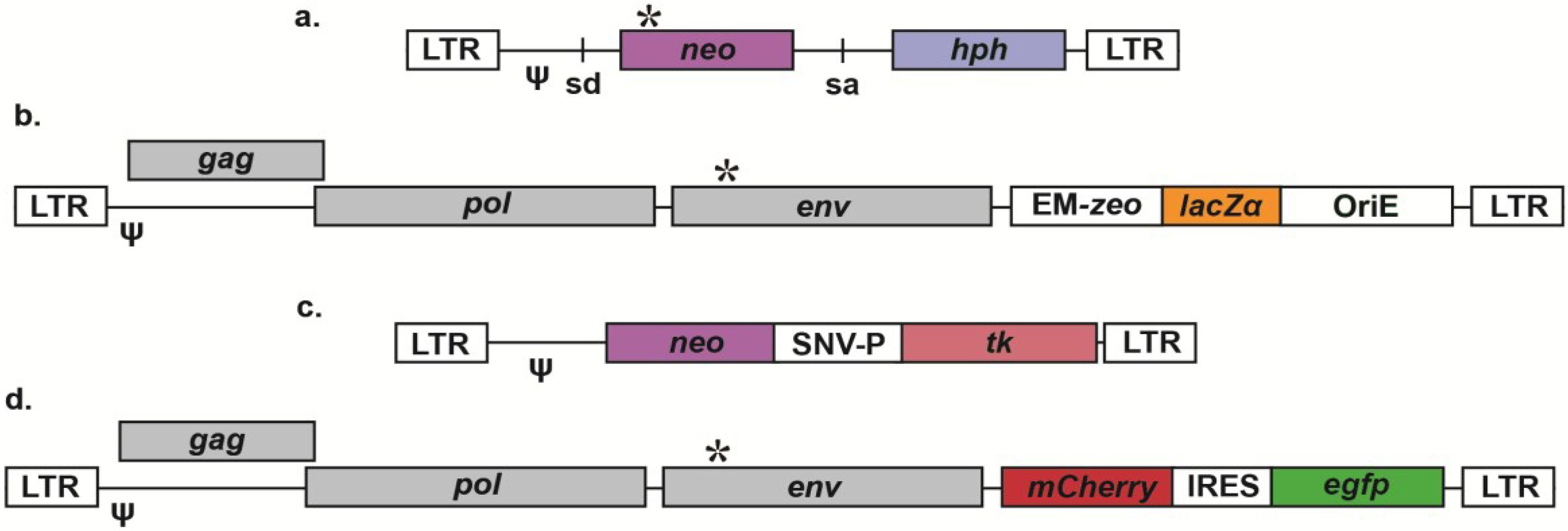
3.1. Reporter Gene Vectors
3.1.1. Antibiotic Resistance Genes
| Target1 | Assay Type2 | Virus | References |
|---|---|---|---|
| neo/hph | Reporter Gene | SNV, MLV, HIV-1 | [41,43,139,140,141,142,143] |
| tk | Reporter Gene | MLV | [144] |
| lacZ | Reporter Gene | MLV, HIV-1 | [42] |
| gfp | Reporter Gene | MLV, HIV-1, HIV-2, SIVagm | [38,49,68,69,70,71,145,146,147] |
| ecfp/eyfp | Reporter Gene | HIV-1 | [55] |
| viral | Gag Reconstitution | HIV-1 | [71] |
| Modified LacZ | HIV-1 | [77,148,149] | |
| PCR/qPCR | HIV-1 | [148,150,151,152] | |
| HTA | HIV-1 | [36,37,152] | |
| Restriction Mapping | HIV-1 | [37,45] | |
| Sequencing (Sanger) | HIV-1 | [37,39,45,151] | |
| Sequencing (NGS) | HIV-1 | [40,153] |

3.1.2. Fluorescent Proteins
3.1.3. LacZ (β-galactosidase)
3.1.4. Direct Repeat Reporter Vectors
3.2. Detection of Recombinants in Viral Genes
3.2.1. Gag Reconstitution Assay
3.2.2. Use of LacZ (β-galactosidase) to Detect Recombinants in Viral Genes
3.2.3. Heteroduplex Tracking, Quantitative PCR, and Restriction Mapping
3.2.4. Direct Sequencing of Viral Genes-Sanger
3.2.5. Direct Sequencing of Viral Genes-NGS
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Delviks-Frankenberry, K.; Galli, A.; Nikolaitchik, O.; Mens, H.; Pathak, V.K.; Hu, W.S. Mechanisms and factors that influence high frequency retroviral recombination. Viruses 2011, 3, 1650–1680. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.P.; Davenport, M.P.; Mak, J. The origin of genetic diversity in hiv-1. Virus Res. 2012, 169, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Hughes, S.H. Hiv-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [PubMed]
- Pathak, V.K.; Temin, H.M. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: Substitutions, frameshifts, and hypermutations. Proc. Natl. Acad. Sci. USA 1990, 87, 6019–6023. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. In vivo analysis of human t-cell leukemia virus type 1 reverse transcription accuracy. J. Virol. 2000, 74, 9525–9531. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Temin, H.M. Lower mutation rate of bovine leukemia virus relative to that of spleen necrosis virus. J. Virol. 1994, 68, 494–499. [Google Scholar] [PubMed]
- Varela-Echavarria, A.; Prorock, C.M.; Ron, Y.; Dougherty, J.P. High rate of genetic rearrangement during replication of a moloney murine leukemia virus-based vector. J. Virol. 1993, 67, 6357–6364. [Google Scholar] [PubMed]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S.D.; Kunkel, T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008, 18, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 2009, 1, 1137–1165. [Google Scholar] [CrossRef] [PubMed]
- O'Neil, P.K.; Sun, G.; Yu, H.; Ron, Y.; Dougherty, J.P.; Preston, B.D. Mutational analysis of hiv-1 long terminal repeats to explore the relative contribution of reverse transcriptase and rna polymerase ii to viral mutagenesis. J. Biol. Chem. 2002, 277, 38053–38061. [Google Scholar] [CrossRef] [PubMed]
- Refsland, E.W.; Harris, R.S. The apobec3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 2013, 371, 1–27. [Google Scholar] [PubMed]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple apobec3 restriction factors for hiv-1 and one vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed]
- Mulder, L.C.; Harari, A.; Simon, V. Cytidine deamination induced hiv-1 drug resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 5501–5506. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Bhattacharya, T.; Kunstman, K.; Swantek, P.; Koning, F.A.; Malim, M.H.; Wolinsky, S.M. Human apobec3g-mediated editing can promote hiv-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 2010, 84, 10402–10405. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Zennou, V.; Murray, D.; Huang, Y.; Ho, D.D.; Bieniasz, P.D. Natural variation in vif: Differential impact on apobec3g/3f and a potential role in hiv-1 diversification. PLoS Pathog. 2005, 1, e6. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, Y.; Kinomoto, M.; Tatsumi, M.; Fujita, H.; Shimura, M.; Tanaka, Y.; Ishizaka, Y.; Nolan, D.; Mallal, S.; Sata, T.; et al. Differential anti-apobec3g activity of hiv-1 vif proteins derived from different subtypes. J. Biol. Chem. 2010, 285, 35350–35358. [Google Scholar]
- Binka, M.; Ooms, M.; Steward, M.; Simon, V. The activity spectrum of vif from multiple hiv-1 subtypes against apobec3g, apobec3f, and apobec3h. J. Virol. 2012, 86, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Fourati, S.; Lambert-Niclot, S.; Soulie, C.; Wirden, M.; Malet, I.; Valantin, M.A.; Tubiana, R.; Simon, A.; Katlama, C.; Carcelain, G.; et al. Differential impact of apobec3-driven mutagenesis on hiv evolution in diverse anatomical compartments. AIDS 2014, 28, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Janini, M.; Rogers, M.; Birx, D.R.; McCutchan, F.E. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of cd4(+) t cells. J. Virol. 2001, 75, 7973–7986. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.L.; Kwon, P.; Nettles, R.E.; Han, Y.; Ray, S.C.; Siliciano, R.F. G-->a hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting cd4+ t cells in vivo. J. Virol. 2005, 79, 1975–1980. [Google Scholar] [CrossRef] [PubMed]
- Kijak, G.H.; Janini, L.M.; Tovanabutra, S.; Sanders-Buell, E.; Arroyo, M.A.; Robb, M.L.; Michael, N.L.; Birx, D.L.; McCutchan, F.E. Variable contexts and levels of hypermutation in hiv-1 proviral genomes recovered from primary peripheral blood mononuclear cells. Virology 2008, 376, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.; Keller, J.; Nolan, D.; James, I.; Gaudieri, S.; Moore, C.; Mallal, S. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with apobec3g and vif genetic variation. J. Virol. 2006, 80, 9259–9269. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Xing, H.; Hong, K.; Huang, H.; Tang, H.; Qin, G.; Shao, Y. Biased g-to-a hypermutation in hiv-1 proviral DNA from a long-term non-progressor. AIDS 2004, 18, 1863–1865. [Google Scholar] [CrossRef] [PubMed]
- Wood, N.; Bhattacharya, T.; Keele, B.F.; Giorgi, E.; Liu, M.; Gaschen, B.; Daniels, M.; Ferrari, G.; Haynes, B.F.; McMichael, A.; et al. Hiv evolution in early infection: Selection pressures, patterns of insertion and deletion, and the impact of apobec. PLoS Pathog. 2009, 5, e1000414. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Bernard, L.C. 3'-azido-3'-deoxythymidine (azt) and azt-resistant reverse transcriptase can increase the in vivo mutation rate of human immunodeficiency virus type 1. J. Virol. 2000, 74, 9532–9539. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; le Rouzic, E.; Benichou, S.; Gajary, L.C. Influence of reverse transcriptase variants, drugs, and vpr on human immunodeficiency virus type 1 mutant frequencies. J. Virol. 2003, 77, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.K.; Chen, R.; Skasko, M.; Reynolds, H.M.; Lee, K.; Bambara, R.A.; Mansky, L.M.; Kim, B. A role for dntp binding of human immunodeficiency virus type 1 reverse transcriptase in viral mutagenesis. Biochemistry 2004, 43, 4490–4500. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yokoyama, M.; Sato, H.; Reilly, C.; Mansky, L.M. Human immunodeficiency virus mutagenesis during antiviral therapy: Impact of drug-resistant reverse transcriptase and nucleoside and nonnucleoside reverse transcriptase inhibitors on human immunodeficiency virus type 1 mutation frequencies. J. Virol. 2005, 79, 12045–12057. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.E.; Ferris, A.L.; Das, K.; Quinones, O.; Shao, W.; Tuske, S.; Alvord, W.G.; Arnold, E.; Hughes, S.H. Mutations in hiv-1 reverse transcriptase affect the errors made in a single cycle of viral replication. J. Virol. 2014, 88, 7589–7601. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. Mutagenic outcome of combined antiviral drug treatment during human immunodeficiency virus type 1 replication. Virology 2003, 307, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology 1996, 222, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Preveral, S.; Selig, L.; Benarous, R.; Benichou, S. The interaction of vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 in vivo mutation rate. J. Virol. 2000, 74, 7039–7047. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; le Rouzic, E.; Kearney, J.A.; Mansky, L.M.; Benichou, S. Vpr-mediated incorporation of ung2 into hiv-1 particles is required to modulate the virus mutation rate and for replication in macrophages. J. Biol. Chem. 2004, 279, 28419–28425. [Google Scholar] [CrossRef] [PubMed]
- Jetzt, A.E.; Yu, H.; Klarmann, G.J.; Ron, Y.; Preston, B.D.; Dougherty, J.P. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 2000, 74, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Jetzt, A.E.; Sun, G.; Yu, H.; Klarmann, G.; Ron, Y.; Preston, B.D.; Dougherty, J.P. Human immunodeficiency virus type 1 recombination: Rate, fidelity, and putative hot spots. J. Virol. 2002, 76, 11273–11282. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, T.D.; Nikolaitchik, O.; Chen, J.; Powell, D.; Hu, W.S. Genetic recombination of human immunodeficiency virus type 1 in one round of viral replication: Effects of genetic distance, target cells, accessory genes, and lack of high negative interference in crossover events. J. Virol. 2005, 79, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- Schlub, T.E.; Smyth, R.P.; Grimm, A.J.; Mak, J.; Davenport, M.P. Accurately measuring recombination between closely related hiv-1 genomes. PLoS Comput. Biol. 2010, 6, e1000766. [Google Scholar] [CrossRef] [PubMed]
- Schlub, T.E.; Grimm, A.J.; Smyth, R.P.; Cromer, D.; Chopra, A.; Mallal, S.; Venturi, V.; Waugh, C.; Mak, J.; Davenport, M.P. Fifteen to twenty percent of hiv substitution mutations are associated with recombination. J. Virol. 2014, 88, 3837–3849. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, T.; Wargo, H.; Hu, W.S. High rates of human immunodeficiency virus type 1 recombination: Near-random segregation of markers one kilobase apart in one round of viral replication. J. Virol. 2003, 77, 11193–11200. [Google Scholar] [CrossRef] [PubMed]
- Onafuwa, A.; An, W.; Robson, N.D.; Telesnitsky, A. Human immunodeficiency virus type 1 genetic recombination is more frequent than that of moloney murine leukemia virus despite similar template switching rates. J. Virol. 2003, 77, 4577–4587. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Temin, H.M. Genetic consequences of packaging two rna genomes in one retroviral particle: Pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA 1990, 87, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Kellam, P.; Larder, B.A. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. J. Virol. 1995, 69, 669–674. [Google Scholar] [PubMed]
- Moutouh, L.; Corbeil, J.; Richman, D.D. Recombination leads to the rapid emergence of hiv-1 dually resistant mutants under selective drug pressure. Proc. Natl. Acad. Sci. USA 1996, 93, 6106–6111. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Busch, M.; Abel, K.; Fritts, L.; Bustamante, P.; Stanton, J.; Lu, D.; Wu, S.; Glowczwskie, J.; Rourke, T.; et al. Retroviral recombination in vivo: Viral replication patterns and genetic structure of simian immunodeficiency virus (siv) populations in rhesus macaques after simultaneous or sequential intravaginal inoculation with sivmac239deltavpx/deltavpr and sivmac239deltanef. J. Virol. 2005, 79, 4886–4895. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Liang, C.; Brenner, B.G.; Wainberg, M.A. Multidrug-resistant variants of hiv type 1 (hiv-1) can exist in cells as defective quasispecies and be rescued by superinfection with other defective hiv-1 variants. J. Infect. Dis. 2009, 200, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Xu, H.; Wainberg, M.A. Defective hiv-1 quasispecies in the form of multiply drug-resistant proviral DNA within cells can be rescued by superinfection with different subtype variants of hiv-1 and by hiv-2 and siv. J. Antimicrob. Chemother. 2014, 69, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Kearney, M.; Nikolaitchik, O.A.; Yu, S.; Chin, M.P.; Maldarelli, F.; Coffin, J.M.; Pathak, V.K.; Hu, W.S. Patterns of human immunodeficiency virus type 1 recombination ex vivo provide evidence for coadaptation of distant sites, resulting in purifying selection for intersubtype recombinants during replication. J. Virol. 2010, 84, 7651–7661. [Google Scholar] [CrossRef] [PubMed]
- Los alamos national laboratory hiv databases. Available online: http://www.hiv.lanl.gov/ (accessed on 14 August 2014).
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global trends in molecular epidemiology of hiv-1 during 2000–2007. AIDS 2011, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Dang, Q.; Unutmaz, D.; Pathak, V.K.; Maldarelli, F.; Powell, D.; Hu, W.S. Mechanisms of nonrandom human immunodeficiency virus type 1 infection and double infection: Preference in virus entry is important but is not the sole factor. J. Virol. 2005, 79, 4140–4149. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.; Chen, J.; Unutmaz, D.; Coffin, J.M.; Pathak, V.K.; Powell, D.; KewalRamani, V.N.; Maldarelli, F.; Hu, W.S. Nonrandom hiv-1 infection and double infection via direct and cell-mediated pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Del Portillo, A.; Tripodi, J.; Najfeld, V.; Wodarz, D.; Levy, D.N.; Chen, B.K. Multiploid inheritance of hiv-1 during cell-to-cell infection. J. Virol. 2011, 85, 7169–7176. [Google Scholar]
- Levy, D.N.; Aldrovandi, G.M.; Kutsch, O.; Shaw, G.M. Dynamics of hiv-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4204–4209. [Google Scholar] [CrossRef] [PubMed]
- Bregnard, C.; Pacini, G.; Danos, O.; Basmaciogullari, S. Suboptimal provirus expression explains apparent nonrandom cell coinfection with hiv-1. J. Virol. 2012, 86, 8810–8820. [Google Scholar] [CrossRef] [PubMed]
- Nethe, M.; Berkhout, B.; van der Kuyl, A.C. Retroviral superinfection resistance. Retrovirology 2005, 2, 52. [Google Scholar] [CrossRef] [PubMed]
- Chohan, B.; Lavreys, L.; Rainwater, S.M.; Overbaugh, J. Evidence for frequent reinfection with human immunodeficiency virus type 1 of a different subtype. J. Virol. 2005, 79, 10701–10708. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, A.; Chohan, B.; Chohan, V.; McClelland, R.S.; Overbaugh, J. Chronic hiv-1 infection frequently fails to protect against superinfection. PLoS Pathog. 2007, 3, e177. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, A.; Ngayo, M.O.; Chohan, B.; Overbaugh, J. Examination of a second region of the hiv type 1 genome reveals additional cases of superinfection. AIDS Res. Hum. Retrovir. 2008, 24, 1221. [Google Scholar] [CrossRef] [PubMed]
- Redd, A.D.; Mullis, C.E.; Serwadda, D.; Kong, X.; Martens, C.; Ricklefs, S.M.; Tobian, A.A.; Xiao, C.; Grabowski, M.K.; Nalugoda, F.; et al. The rates of hiv superinfection and primary hiv incidence in a general population in rakai, uganda. J. Infect. Dis. 2012, 206, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Ronen, K.; McCoy, C.O.; Matsen, F.A.; Boyd, D.F.; Emery, S.; Odem-Davis, K.; Jaoko, W.; Mandaliya, K.; McClelland, R.S.; Richardson, B.A.; et al. Hiv-1 superinfection occurs less frequently than initial infection in a cohort of high-risk kenyan women. PLoS Pathog. 2013, 9, e1003593. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Wong, J.K.; Hightower, G.K.; Ignacio, C.C.; Koelsch, K.K.; Daar, E.S.; Richman, D.D.; Little, S.J. Incidence of hiv superinfection following primary infection. JAMA: J. Am. Med. Assoc. 2004, 292, 1177–1178. [Google Scholar]
- Gratton, S.; Cheynier, R.; Dumaurier, M.J.; Oksenhendler, E.; Wain-Hobson, S. Highly restricted spread of hiv-1 and multiply infected cells within splenic germinal centers. Proc. Natl. Acad. Sci. USA 2000, 97, 14566–14571. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.; Maier, R.; Vartanian, J.P.; Bocharov, G.; Jung, V.; Fischer, U.; Meese, E.; Wain-Hobson, S.; Meyerhans, A. Recombination: Multiply infected spleen cells in hiv patients. Nature 2002, 418, 144. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Sopper, S.; Sauermann, U.; Meyerhans, A.; Suspene, R. Stable multi-infection of splenocytes during siv infection--the basis for continuous recombination. Retrovirology 2012, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, L.; King, M.S.; Makitalo, B.; Brannstrom, J.; Shao, W.; Maldarelli, F.; Kearney, M.F.; Hu, W.S.; Chen, J.; Gaines, H.; et al. Majority of cd4+ t cells from peripheral blood of hiv-1-infected individuals contain only one hiv DNA molecule. Proc. Natl. Acad. Sci. USA 2011, 108, 11199–11204. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Rhodes, T.D.; Chen, J.; Fu, W.; Hu, W.S. Identification of a major restriction in hiv-1 intersubtype recombination. Proc. Natl. Acad. Sci. USA 2005, 102, 9002–9007. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Chen, J.; Nikolaitchik, O.A.; Hu, W.S. Molecular determinants of hiv-1 intersubtype recombination potential. Virology 2007, 363, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Lee, S.K.; Chen, J.; Nikolaitchik, O.A.; Powell, D.A.; Fivash, M.J., Jr.; Hu, W.S. Long-range recombination gradient between hiv-1 subtypes b and c variants caused by sequence differences in the dimerization initiation signal region. J. Mol. Biol. 2008, 377, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Nikolaitchik, O.A.; Galli, A.; Moore, M.D.; Pathak, V.K.; Hu, W.S. Multiple barriers to recombination between divergent hiv-1 variants revealed by a dual-marker recombination assay. J. Mol. Biol. 2011, 407, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Svarovskaia, E.S.; Delviks, K.A.; Hwang, C.K.; Pathak, V.K. Structural determinants of murine leukemia virus reverse transcriptase that affect the frequency of template switching. J. Virol. 2000, 74, 7171–7178. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.K.; Svarovskaia, E.S.; Pathak, V.K. Dynamic copy choice: Steady state between murine leukemia virus polymerase and polymerase-dependent rnase h activity determines frequency of in vivo template switching. Proc. Natl. Acad. Sci. USA 2001, 98, 12209–12214. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Topping, R.S.; Shin, N.H.; Telesnitsky, A. Altering the intracellular environment increases the frequency of tandem repeat deletion during moloney murine leukemia virus reverse transcription. J. Virol. 1999, 73, 8441–8447. [Google Scholar] [PubMed]
- Nikolenko, G.N.; Svarovskaia, E.S.; Delviks, K.A.; Pathak, V.K. Antiretroviral drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase increase template-switching frequency. J. Virol. 2004, 78, 8761–8770. [Google Scholar] [CrossRef] [PubMed]
- Operario, D.J.; Balakrishnan, M.; Bambara, R.A.; Kim, B. Reduced dntp interaction of human immunodeficiency virus type 1 reverse transcriptase promotes strand transfer. J. Biol. Chem. 2006, 281, 32113–32121. [Google Scholar] [CrossRef] [PubMed]
- Galetto, R.; Moumen, A.; Giacomoni, V.; Veron, M.; Charneau, P.; Negroni, M. The structure of hiv-1 genomic rna in the gp120 gene determines a recombination hot spot in vivo. J. Biol. Chem. 2004, 279, 36625–36632. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, J.J.; Mallaber, L.M.; Rodriguez-Rodriguez, L.; Fay, P.J.; Bambara, R.A. Requirements for strand transfer between internal regions of heteropolymer templates by human immunodeficiency virus reverse transcriptase. J. Virol. 1992, 66, 6370–6378. [Google Scholar] [PubMed]
- Wu, W.; Blumberg, B.M.; Fay, P.J.; Bambara, R.A. Strand transfer mediated by human immunodeficiency virus reverse transcriptase in vitro is promoted by pausing and results in misincorporation. J. Biol. Chem. 1995, 270, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Brincat, J.L.; Pfeiffer, J.K.; Telesnitsky, A. Rnase h activity is required for high-frequency repeat deletion during moloney murine leukemia virus replication. J. Virol. 2002, 76, 88–95. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Telesnitsky, A. Effects of varying sequence similarity on the frequency of repeat deletion during reverse transcription of a human immunodeficiency virus type 1 vector. J. Virol. 2002, 76, 7897–7902. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Nickle, D.C.; Justement, J.S.; Meyers, J.H.; Roby, G.; Hallahan, C.W.; Kottilil, S.; Moir, S.; Mican, J.M.; Mullins, J.I.; et al. Persistence of hiv in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J. Infect. Dis. 2008, 197, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Gras, G.; Kaul, M. Molecular mechanisms of neuroinvasion by monocytes-macrophages in hiv-1 infection. Retrovirology 2010, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An integrated overview of hiv-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Pursuing Later Treatment Options II (PLATO II) project team; Collaboration of Observational HIV Epidemiological Research Europe (COHERE) Group; Nakagawa, F.; Lodwick, R.; Costagliola, D.; van Sighem, A.; Torti, C.; Podzamczer, D.; Mocroft, A.; Ledergerber, B.; et al. Calendar time trends in the incidence and prevalence of triple-class virologic failure in antiretroviral drug-experienced people with hiv in europe. J. Acquir. Immune Defic. Syndr. 2012, 59, 294–299. [Google Scholar]
- Galli, A.; Bukh, J. Comparative analysis of the molecular mechanisms of recombination in hepatitis c virus. Trends Microbiol. 2014, 22, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Yang, Z.W.; Iang, S.B.; Chao, M. Reduced genetic distance and high replication levels increase the rna recombination rate of hepatitis delta virus. Virus Res. 2014. [Google Scholar] [CrossRef]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length 'imprecise' intermediates. PLoS pathogens 2014, 10, e1004191. [Google Scholar] [CrossRef] [PubMed]
- Diamond, T.L.; Roshal, M.; Jamburuthugoda, V.K.; Reynolds, H.M.; Merriam, A.R.; Lee, K.Y.; Balakrishnan, M.; Bambara, R.A.; Planelles, V.; Dewhurst, S.; et al. Macrophage tropism of hiv-1 depends on efficient cellular dntp utilization by reverse transcriptase. J. Biol. Chem. 2004, 279, 51545–51553. [Google Scholar]
- Dougherty, J.P.; Temin, H.M. Determination of the rate of base-pair substitution and insertion mutations in retrovirus replication. J. Virol. 1988, 62, 2817–2822. [Google Scholar] [PubMed]
- Varela-Echavarria, A.; Garvey, N.; Preston, B.D.; Dougherty, J.P. Comparison of moloney murine leukemia virus mutation rate with the fidelity of its reverse transcriptase in vitro. J. Biol. Chem. 1992, 267, 24681–24688. [Google Scholar] [PubMed]
- Bebenek, K.; Abbotts, J.; Roberts, J.D.; Wilson, S.H.; Kunkel, T.A. Specificity and mechanism of error-prone replication by human immunodeficiency virus-1 reverse transcriptase. J. Biol. Chem. 1989, 264, 16948–16956. [Google Scholar] [PubMed]
- Boyer, J.C.; Bebenek, K.; Kunkel, T.A. Unequal human immunodeficiency virus type 1 reverse transcriptase error rates with rna and DNA templates. Proc. Natl. Acad. Sci. USA 1992, 89, 6919–6923. [Google Scholar] [CrossRef] [PubMed]
- Eckert, K.A.; Kunkel, T.A. Fidelity of DNA synthesis catalyzed by human DNA polymerase alpha and hiv-1 reverse transcriptase: Effect of reaction ph. Nucleic Acids Res. 1993, 21, 5212–5220. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Loeb, L.A. Fidelity of hiv-1 reverse transcriptase copying a hypervariable region of the hiv-1 env gene. Virology 1994, 199, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M.M.; Sutton, R.E. Replicative fidelity of lentiviral vectors produced by transient transfection. Virology 2006, 348, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathi, S.; Varela-Echavarria, A.; Ron, Y.; Preston, B.D.; Dougherty, J.P. Genetic rearrangements occurring during a single cycle of murine leukemia virus vector replication: Characterization and implications. J. Virol. 1995, 69, 7991–8000. [Google Scholar] [PubMed]
- Huang, K.J.; Wooley, D.P. A new cell-based assay for measuring the forward mutation rate of hiv-1. J. Virol. Methods 2005, 124, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Mudry, R.A., Jr.; Rexrode, C.A., 2nd; Pathak, V.K. Retroviral mutation rates and a-to-g hypermutations during different stages of retroviral replication. J. Virol. 1996, 70, 7594–7602. [Google Scholar]
- Mansky, L.M. Forward mutation rate of human immunodeficiency virus type 1 in a t lymphoid cell line. AIDS Res. Hum. Retrovir. 1996, 12, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, position, and frequency of mutations made in a single cycle of hiv-1 replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef] [PubMed]
- Pathak, V.K.; Temin, H.M. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: Deletions and deletions with insertions. Proc. Natl. Acad. Sci. USA 1990, 87, 6024–6028. [Google Scholar] [CrossRef] [PubMed]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. Apobec3g contributes to hiv-1 variation through sublethal mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar] [CrossRef] [PubMed]
- Holtz, C.M.; Mansky, L.M. Variation of hiv-1 mutation spectra among cell types. J. Virol. 2013, 87, 5296–5299. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.J.; Clouser, C.L.; Patterson, S.; Mansky, L.M. 5-azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 2009, 83, 11950–11958. [Google Scholar] [CrossRef] [PubMed]
- Clouser, C.L.; Patterson, S.E.; Mansky, L.M. Exploiting drug repositioning for discovery of a novel hiv combination therapy. J. Virol. 2010, 84, 9301–9309. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.M.; Heineman, R.H.; Beach, L.B.; Martin, J.L.; Schnettler, E.K.; Dapp, M.J.; Patterson, S.E.; Mansky, L.M. 5,6-dihydro-5-aza-2'-deoxycytidine potentiates the anti-hiv-1 activity of ribonucleotide reductase inhibitors. Bioorganic Med. Chem. 2013, 21, 7222–7228. [Google Scholar] [CrossRef]
- Dapp, M.J.; Holtz, C.M.; Mansky, L.M. Concomitant lethal mutagenesis of human immunodeficiency virus type 1. J. Mol. Biol. 2012, 419, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Leider, J.M.; Palese, P.; Smith, F.I. Determination of the mutation rate of a retrovirus. J. Virol. 1988, 62, 3084–3091. [Google Scholar] [PubMed]
- Monk, R.J.; Malik, F.G.; Stokesberry, D.; Evans, L.H. Direct determination of the point mutation rate of a murine retrovirus. J. Virol. 1992, 66, 3683–3689. [Google Scholar] [PubMed]
- Gao, F.; Chen, Y.; Levy, D.N.; Conway, J.A.; Kepler, T.B.; Hui, H. Unselected mutations in the human immunodeficiency virus type 1 genome are mostly nonsynonymous and often deleterious. J. Virol. 2004, 78, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.J.; Bonnac, L.; Patterson, S.E.; Mansky, L.M. Discovery of novel ribonucleoside analogs with activity against human immunodeficiency virus type 1. J. Virol. 2014, 88, 354–363. [Google Scholar] [CrossRef]
- Bebenek, K.; Kunkel, T.A. Analyzing fidelity of DNA polymerases. Methods Enzymol. 1995, 262, 217–232. [Google Scholar] [PubMed]
- Boyer, J.C.; Bebenek, K.; Kunkel, T.A. Analyzing the fidelity of reverse transcription and transcription. Methods Enzymol. 1996, 275, 523–537. [Google Scholar] [PubMed]
- Beach, L.B.; Rawson, J.M.; Kim, B.; Patterson, S.E.; Mansky, L.M. Novel inhibitors of human immunodeficiency virus type 2 infectivity. J. Gen. Virol. 2014. [Google Scholar] [CrossRef]
- Clouser, C.L.; Chauhan, J.; Bess, M.A.; van Oploo, J.L.; Zhou, D.; Dimick-Gray, S.; Mansky, L.M.; Patterson, S.E. Anti-hiv-1 activity of resveratrol derivatives and synergistic inhibition of hiv-1 by the combination of resveratrol and decitabine. Bioorganic Med. Chem. Lett. 2012, 22, 6642–6646. [Google Scholar] [CrossRef]
- Dapp, M.J.; Heineman, R.H.; Mansky, L.M. Interrelationship between hiv-1 fitness and mutation rate. J. Mol. Biol. 2013, 425, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Essigmann, J.M.; Kazazi, F.; Zhang, J.; Rose, K.D.; Mullins, J.I. Lethal mutagenesis of hiv with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA 1999, 96, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Brabant, W.; Styrchak, S.; Gall, A.; Daifuku, R. Kp-1212/1461, a nucleoside designed for the treatment of hiv by viral mutagenesis. Antivir. Res. 2005, 67, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Swenson, L.C.; Mo, T.; Dong, W.W.; Zhong, X.; Woods, C.K.; Jensen, M.A.; Thielen, A.; Chapman, D.; Lewis, M.; James, I.; et al. Deep sequencing to infer hiv-1 co-receptor usage: Application to three clinical trials of maraviroc in treatment-experienced patients. J. Infect. Dis. 2011, 203, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, I.; van Marck, H.; Mostmans, W.; van Eygen, V.; Rondelez, E.; Thys, K.; van Baelen, K.; Fransen, K.; Vaira, D.; Kabeya, K.; et al. Hiv-1 v3 envelope deep sequencing for clinical plasma specimens failing in phenotypic tropism assays. AIDS Res. Ther. 2010, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Ganusov, V.V.; Giorgi, E.E.; Hraber, P.T.; Keele, B.F.; Leitner, T.; Han, C.S.; Gleasner, C.D.; Green, L.; Lo, C.C.; et al. Transmission of single hiv-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 2010, 5, e12303. [Google Scholar] [CrossRef] [PubMed]
- Simen, B.B.; Simons, J.F.; Hullsiek, K.H.; Novak, R.M.; Macarthur, R.D.; Baxter, J.D.; Huang, C.; Lubeski, C.; Turenchalk, G.S.; Braverman, M.S.; et al. Low-abundance drug-resistant viral variants in chronically hiv-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 2009, 199, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, C.; Mild, M.; Jernberg, J.; Sherwood, E.; Bratt, G.; Leitner, T.; Lundeberg, J.; Andersson, B.; Albert, J. Dynamics of hiv-1 quasispecies during antiviral treatment dissected using ultra-deep pyrosequencing. PLoS One 2010, 5, e11345. [Google Scholar] [CrossRef] [PubMed]
- Jabara, C.B.; Jones, C.D.; Roach, J.; Anderson, J.A.; Swanstrom, R. Accurate sampling and deep sequencing of the hiv-1 protease gene using a primer id. Proc. Natl. Acad. Sci. USA 2011, 108, 20166–20171. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Chapman, B.; Charlebois, P.; Hofmann, O.; Weiner, B.; Porter, A.J.; Samuel, R.; Vardhanabhuti, S.; Zheng, L.; Eron, J.; et al. Comparison of illumina and 454 deep sequencing in participants failing raltegravir-based antiretroviral therapy. PLoS One 2014, 9, e90485. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Oshima, T.; Morimoto, T.; Ikeda, S.; Yoshikawa, H.; Shiwa, Y.; Ishikawa, S.; Linak, M.C.; Hirai, A.; Takahashi, H.; et al. Sequence-specific error profile of illumina sequencers. Nucleic Acids Res. 2011, 39, e90. [Google Scholar] [PubMed]
- Gilles, A.; Meglecz, E.; Pech, N.; Ferreira, S.; Malausa, T.; Martin, J.F. Accuracy and quality assessment of 454 gs-flx titanium pyrosequencing. BMC Genomics 2011, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, I.; van Marck, H.; Verhasselt, P.; Thys, K.; Mostmans, W.; Dumont, S.; van Eygen, V.; Coen, K.; Tuefferd, M.; Aerssens, J. Minor variant detection in amplicons using 454 massive parallel pyrosequencing: Experiences and considerations for successful applications. BioTechniques 2011, 51, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Minoche, A.E.; Dohm, J.C.; Himmelbauer, H. Evaluation of genomic high-throughput sequencing data generated on illumina hiseq and genome analyzer systems. Genome Biol. 2011, 12, R112. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The long-term stability of the human gut microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.I.; Hussmann, J.A.; McBee, R.M.; Acevedo, A.; Andino, R.; Press, W.H.; Sawyer, S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 19872–19877. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; Andino, R. Library preparation for highly accurate population sequencing of rna viruses. Nat. Protoc. 2014, 9, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; Brodsky, L.; Andino, R. Mutational and fitness landscapes of an rna virus revealed through population sequencing. Nature 2014, 505, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.A.; Bowman, E.H.; Hu, W.S. Retroviral recombination rates do not increase linearly with marker distance and are limited by the size of the recombining subpopulation. J. Virol. 1998, 72, 1195–1202. [Google Scholar] [PubMed]
- Anderson, J.A.; Pathak, V.K.; Hu, W.S. Effect of the murine leukemia virus extended packaging signal on the rates and locations of retroviral recombination. J. Virol. 2000, 74, 6953–6963. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Bowman, E.H.; Delviks, K.A.; Pathak, V.K. Homologous recombination occurs in a distinct retroviral subpopulation and exhibits high negative interference. J. Virol. 1997, 71, 6028–6036. [Google Scholar] [PubMed]
- Bircher, L.A.; Rigano, J.C.; Ponferrada, V.G.; Wooley, D.P. High fidelity of homologous retroviral recombination in cell culture. Arch. Virol. 2002, 147, 1665–1683. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.A.; Teufel, R.J., 2nd; Yin, P.D.; Hu, W.S. Correlated template-switching events during minus-strand DNA synthesis: A mechanism for high negative interference during retroviral recombination. J. Virol. 1998, 72, 1186–1194. [Google Scholar]
- Delviks, K.A.; Pathak, V.K. Effect of distance between homologous sequences and 3' homology on the frequency of retroviral reverse transcriptase template switching. J. Virol. 1999, 73, 7923–7932. [Google Scholar] [PubMed]
- Chen, J.; Rhodes, T.D.; Hu, W.S. Comparison of the genetic recombination rates of human immunodeficiency virus type 1 in macrophages and t cells. J. Virol. 2005, 79, 9337–9340. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Powell, D.; Hu, W.S. High frequency of genetic recombination is a common feature of primate lentivirus replication. J. Virol. 2006, 80, 9651–9658. [Google Scholar] [CrossRef] [PubMed]
- Motomura, K.; Chen, J.; Hu, W.S. Genetic recombination between human immunodeficiency virus type 1 (hiv-1) and hiv-2, two distinct human lentiviruses. J. Virol. 2008, 82, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Baird, H.A.; Gao, Y.; Galetto, R.; Lalonde, M.; Anthony, R.M.; Giacomoni, V.; Abreha, M.; Destefano, J.J.; Negroni, M.; Arts, E.J. Influence of sequence identity and unique breakpoints on the frequency of intersubtype hiv-1 recombination. Retrovirology 2006, 3, 91. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Galetto, R.; Hamoudi, M.; Archer, J.; Lefeuvre, P.; Martin, D.P.; Robertson, D.L.; Negroni, M. Molecular mechanisms of recombination restriction in the envelope gene of the human immunodeficiency virus. PLoS Pathog. 2009, 5, e1000418. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, T.; Miyazaki, Y.; Igarashi, T.; Takehisa, J.; Hayami, M. The rapid spread of recombinants during a natural in vitro infection with two human immunodeficiency virus type 1 strains. J. Virol. 1997, 71, 7088–7091. [Google Scholar] [PubMed]
- Iglesias-Sanchez, M.J.; Lopez-Galindez, C. Analysis, quantification, and evolutionary consequences of hiv-1 in vitro recombination. Virology 2002, 304, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Quinones-Mateu, M.E.; Gao, Y.; Ball, S.C.; Marozsan, A.J.; Abraha, A.; Arts, E.J. In vitro intersubtype recombinants of human immunodeficiency virus type 1: Comparison to recent and circulating in vivo recombinant forms. J. Virol. 2002, 76, 9600–9613. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.P.; Schlub, T.E.; Grimm, A.J.; Waugh, C.; Ellenberg, P.; Chopra, A.; Mallal, S.; Cromer, D.; Mak, J.; Davenport, M.P. Identifying recombination hot spots in the hiv-1 genome. J. Virol. 2014, 88, 2891–2902. [Google Scholar] [CrossRef] [PubMed]
- Czernilofsky, A.P.; Levinson, A.D.; Varmus, H.E.; Bishop, J.M.; Tischer, E.; Goodman, H.M. Nucleotide sequence of an avian sarcoma virus oncogene (src) and proposed amino acid sequence for gene product. Nature 1980, 287, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Kosik, E. Mutagenesis of the region between env and src of the sr-a strain of rous sarcoma virus for the purpose of constructing helper-independent vectors. Virology 1984, 136, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Omer, C.A.; Pogue-Geile, K.; Guntaka, R.; Staskus, K.A.; Faras, A.J. Involvement of directly repeated sequences in the generation of deletions of the avian sarcoma virus src gene. J. Virol. 1983, 47, 380–382. [Google Scholar] [PubMed]
- Rhode, B.W.; Emerman, M.; Temin, H.M. Instability of large direct repeats in retrovirus vectors. J. Virol. 1987, 61, 925–927. [Google Scholar] [PubMed]
- Lovmand, S.; Kjeldgaard, N.O.; Jorgensen, P.; Pedersen, F.S. Enhancer functions in u3 of akv virus: A role for cooperativity of a tandem repeat unit and its flanking DNA sequences. J. Virol. 1990, 64, 3185–3191. [Google Scholar] [PubMed]
- Trainor, C.D.; Scott, M.L.; Josephs, S.F.; Fry, K.E.; Reitz, M.S., Jr. Nucleotide sequence of the large terminal repeat of two different strains of gibbon ape leukemia virus. Virology 1984, 137, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Georgiadis, M.M.; Telesnitsky, A. Structure-based moloney murine leukemia virus reverse transcriptase mutants with altered intracellular direct-repeat deletion frequencies. J. Virol. 2000, 74, 9629–9636. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Telesnitsky, A. Frequency of direct repeat deletion in a human immunodeficiency virus type 1 vector during reverse transcription in human cells. Virology 2001, 286, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.A.; Kim, D.H.; Daly, M.B.; Allan, K.C.; Kim, B. Host samhd1 protein promotes hiv-1 recombination in macrophages. J. Biol. Chem. 2014, 289, 16642. [Google Scholar] [CrossRef]
- Clavel, F.; Hoggan, M.D.; Willey, R.L.; Strebel, K.; Martin, M.A.; Repaske, R. Genetic recombination of human immunodeficiency virus. J. Virol. 1989, 63, 1455–1459. [Google Scholar] [PubMed]
- St Louis, D.C.; Gotte, D.; Sanders-Buell, E.; Ritchey, D.W.; Salminen, M.O.; Carr, J.K.; McCutchan, F.E. Infectious molecular clones with the nonhomologous dimer initiation sequences found in different subtypes of human immunodeficiency virus type 1 can recombine and initiate a spreading infection in vitro. J. Virol. 1998, 72, 3991–3998. [Google Scholar] [PubMed]
- Delwart, E.L.; Shpaer, E.G.; Louwagie, J.; McCutchan, F.E.; Grez, M.; Rubsamen-Waigmann, H.; Mullins, J.I. Genetic relationships determined by a DNA heteroduplex mobility assay: Analysis of hiv-1 env genes. Science 1993, 262, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Wang, N.; Carr, A.; Nam, D.S.; Moor-Jankowski, R.; Cooper, D.A.; Ho, D.D. Genetic characterization of human immunodeficiency virus type 1 in blood and genital secretions: Evidence for viral compartmentalization and selection during sexual transmission. J. Virol. 1996, 70, 3098–3107. [Google Scholar] [PubMed]
- Smyth, R.P.; Schlub, T.E.; Grimm, A.; Venturi, V.; Chopra, A.; Mallal, S.; Davenport, M.P.; Mak, J. Reducing chimera formation during pcr amplification to ensure accurate genotyping. Gene 2010, 469, 45–51. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rawson, J.M.O.; Mansky, L.M. Retroviral Vectors for Analysis of Viral Mutagenesis and Recombination. Viruses 2014, 6, 3612-3642. https://doi.org/10.3390/v6093612
Rawson JMO, Mansky LM. Retroviral Vectors for Analysis of Viral Mutagenesis and Recombination. Viruses. 2014; 6(9):3612-3642. https://doi.org/10.3390/v6093612
Chicago/Turabian StyleRawson, Jonathan M.O., and Louis M. Mansky. 2014. "Retroviral Vectors for Analysis of Viral Mutagenesis and Recombination" Viruses 6, no. 9: 3612-3642. https://doi.org/10.3390/v6093612
APA StyleRawson, J. M. O., & Mansky, L. M. (2014). Retroviral Vectors for Analysis of Viral Mutagenesis and Recombination. Viruses, 6(9), 3612-3642. https://doi.org/10.3390/v6093612