The Cellular Bromodomain Protein Brd4 has Multiple Functions in E2-Mediated Papillomavirus Transcription Activation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
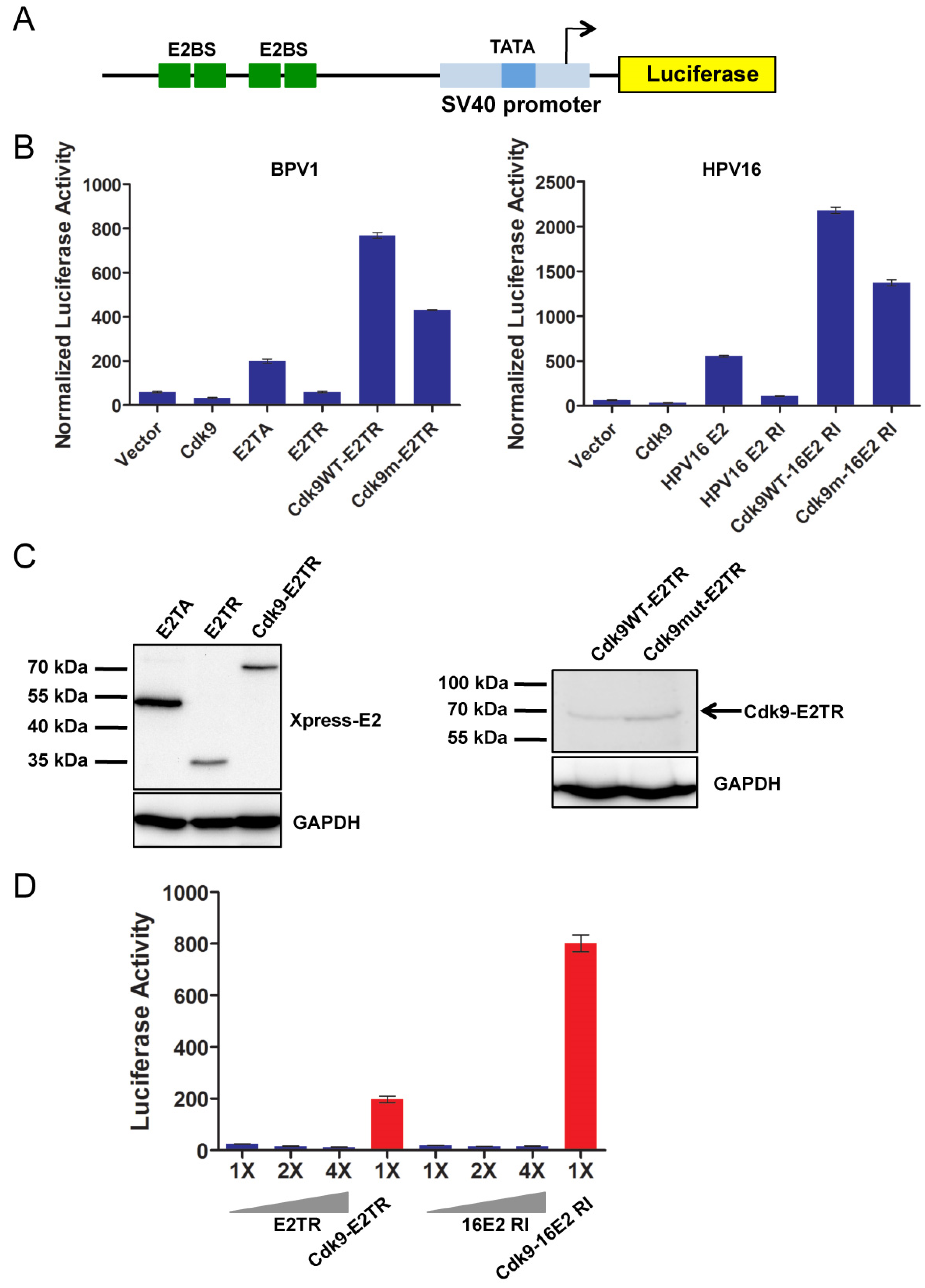
2.1. P-TEFb Is Important for Papillomavirus E2-Mediated Transcription Activation
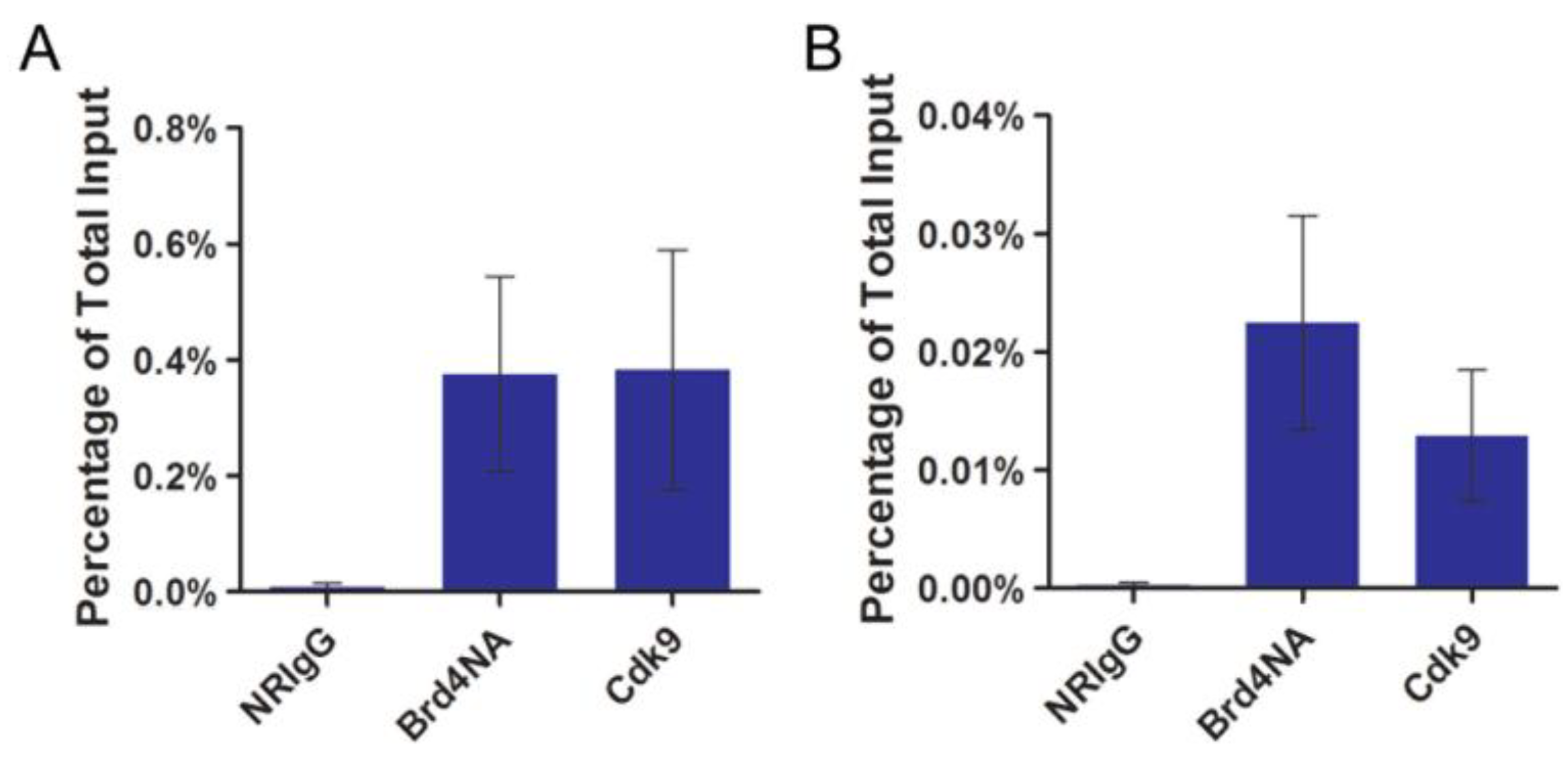
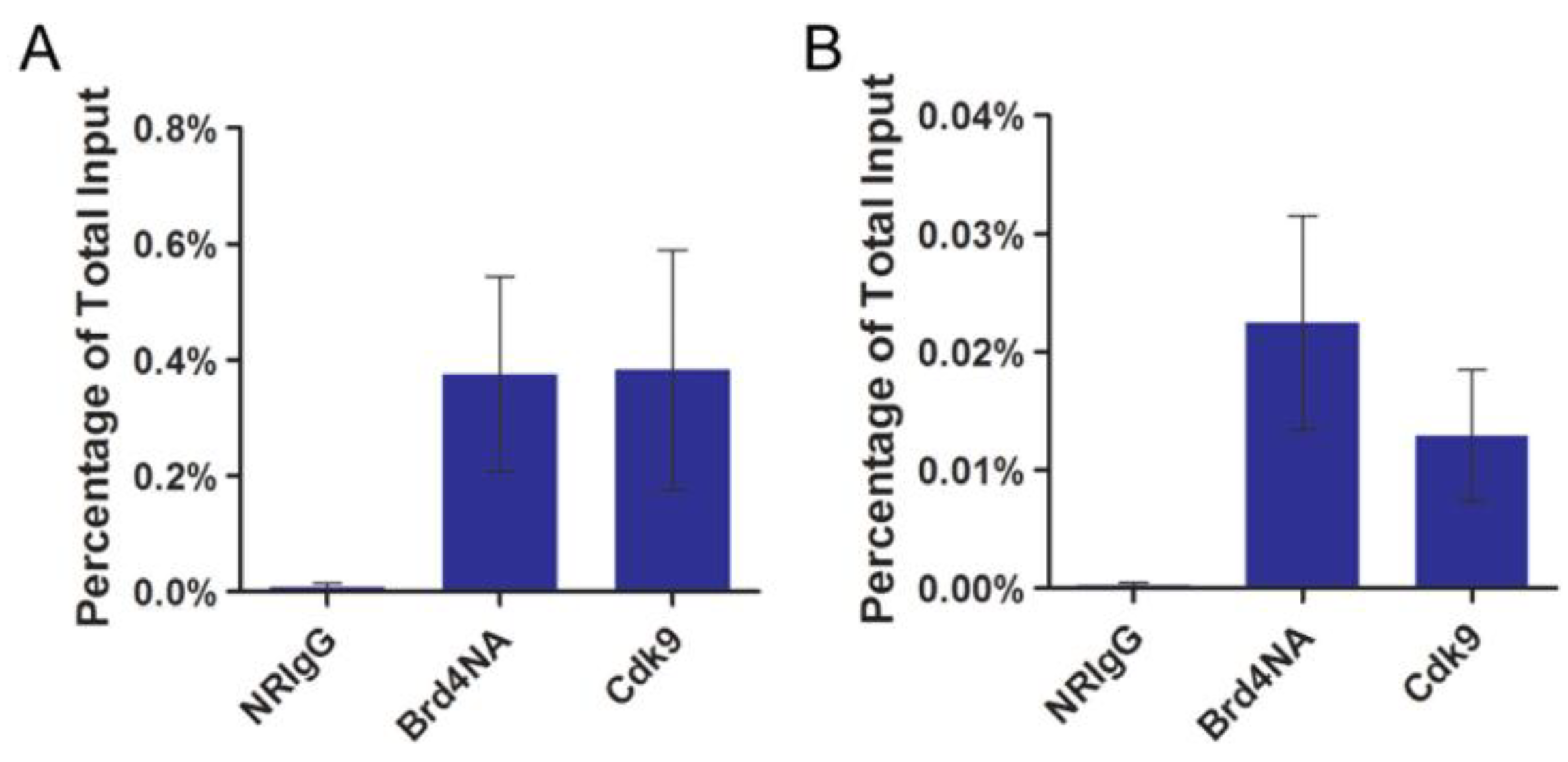
2.2. P-TEFb Is Recruited to the Papillomavirus Genome
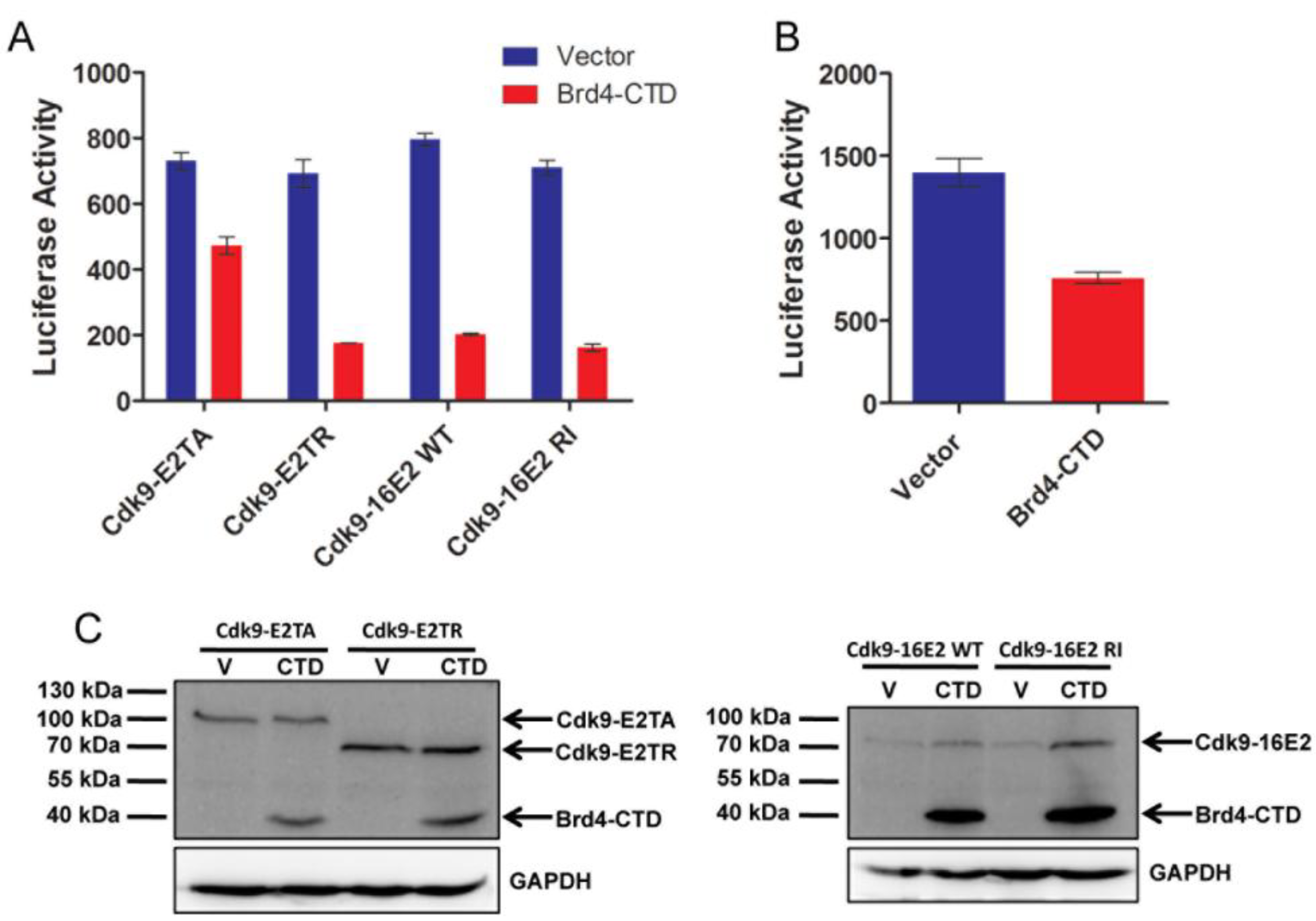
2.3. Brd4-CTD Disrupts Papillomavirus Transcription Activation by the Cdk9-E2 Fusion Proteins
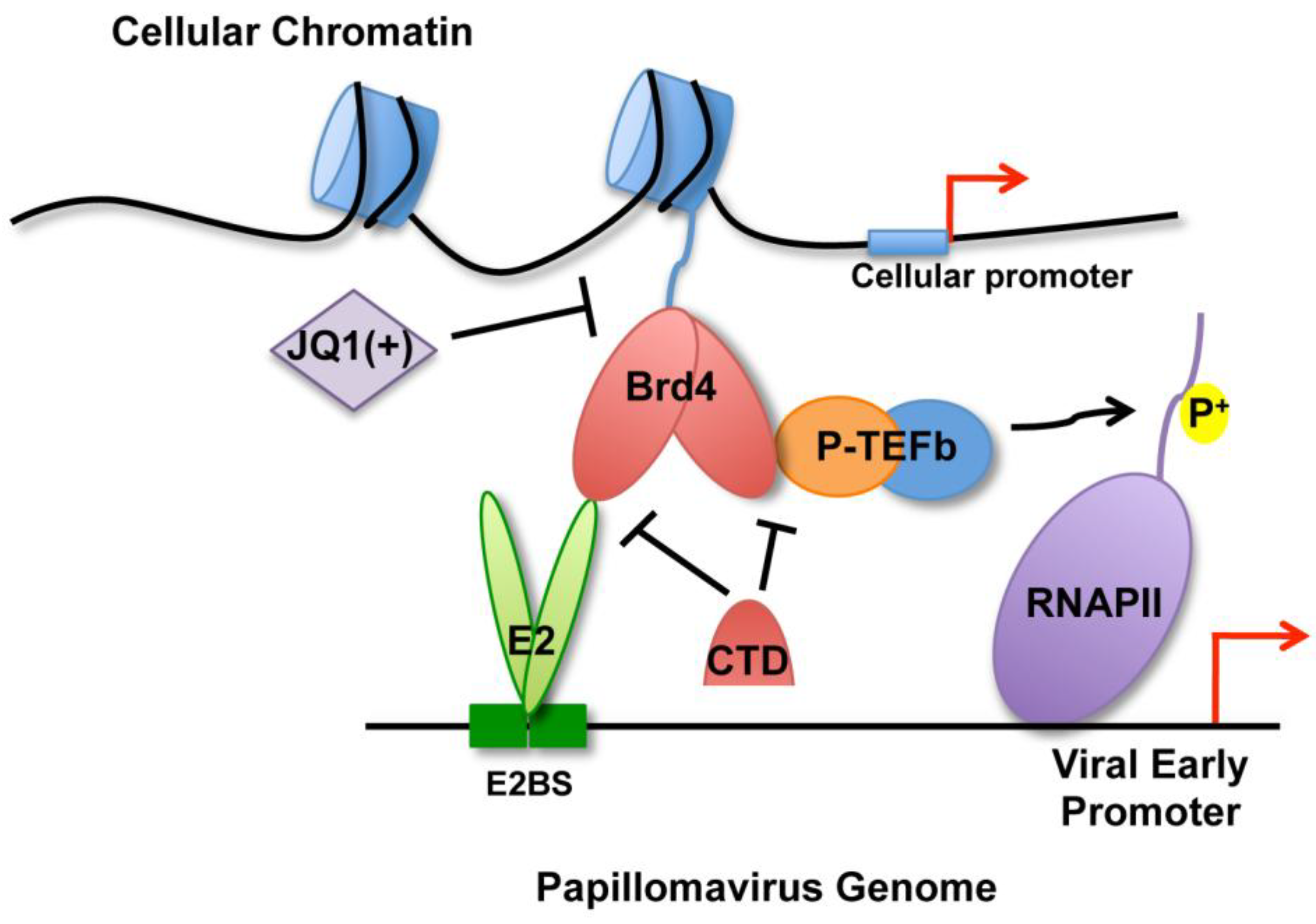
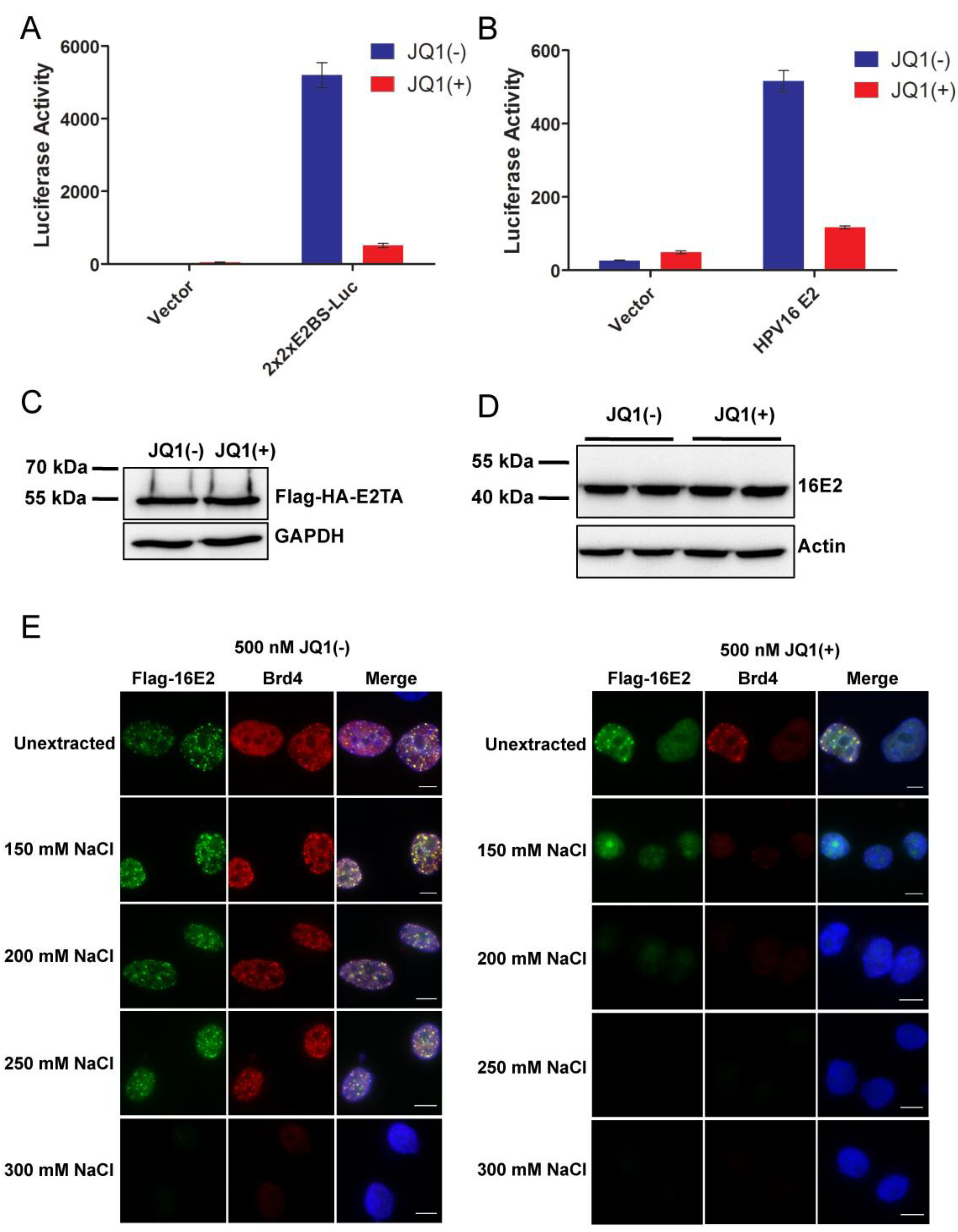
2.4. Releasing Brd4 from Chromatin by JQ1(+) Correlates with Diminished E2-Mediated Transcription Activation
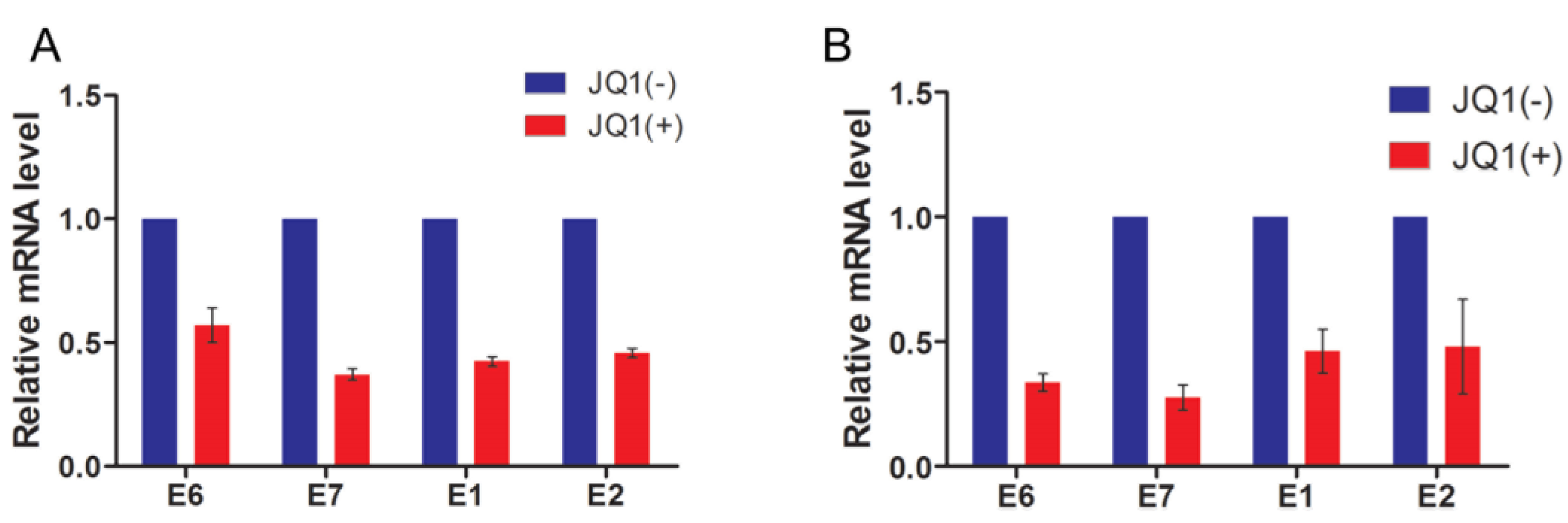
2.5. JQ1(+) Treatment Reduces Papillomavirus Gene Expression
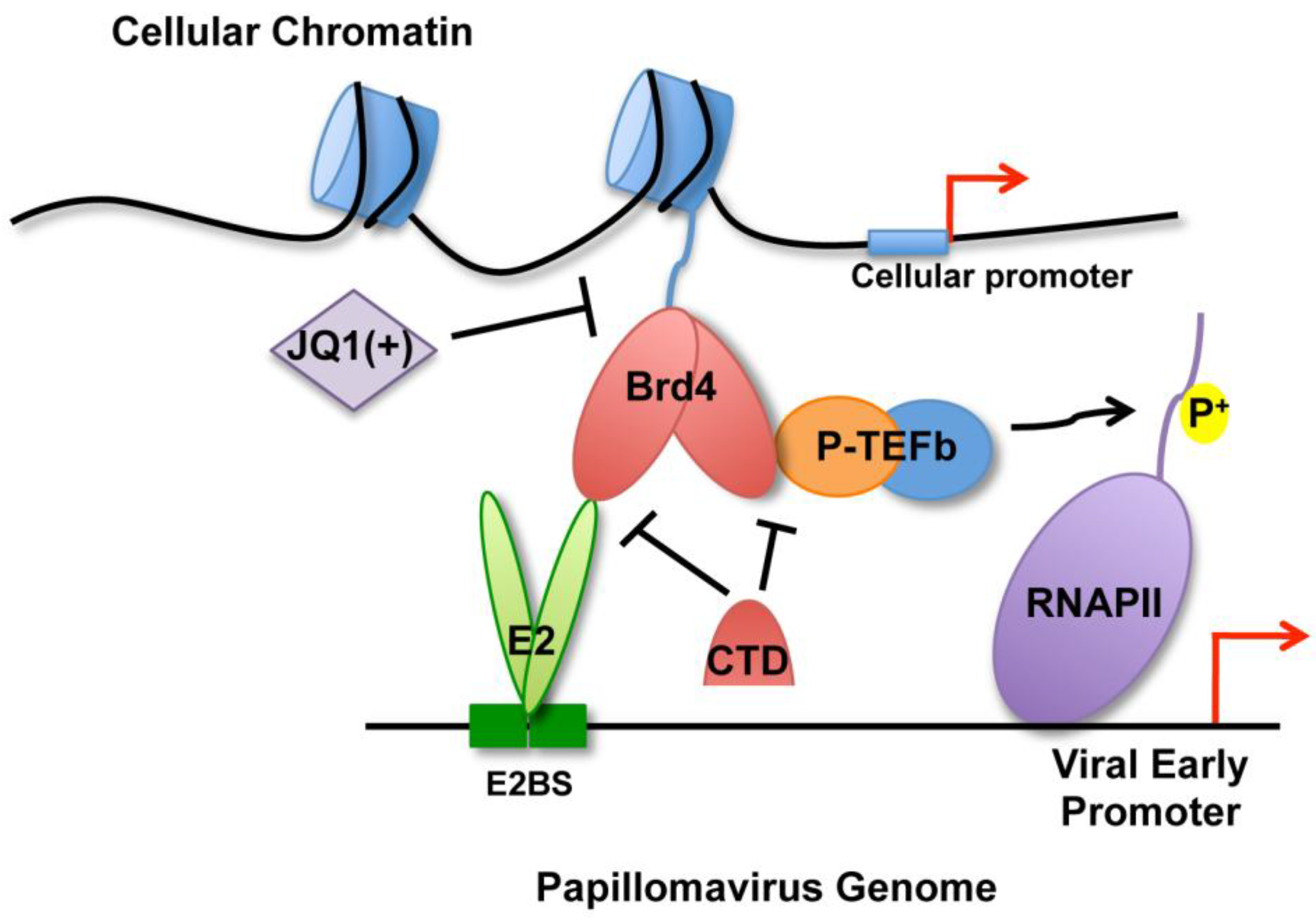
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Lines
4.2. Transient Transfections
4.3. Reagents
4.4. Recombinant Plasmid Construction
4.5. Luciferase Transactivation Assay
4.6. Quantitative Reverse Transcription PCR (RT-qPCR)
4.7. Immunoprecipitation
4.8. Chromatin Immunoprecipitation (ChIP)
4.9. Whole Genomic Extraction
4.10. Immunoblotting
4.11. Immunofluorescence Staining
4.12. Microscopy and Image Analysis
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- zur Hausen, H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology 2009, 384, 260–265. [Google Scholar]
- Ault, K.A. Long-term efficacy of human papillomavirus vaccination. Gynecol Oncol 2007, 107, S27–S30. [Google Scholar] [CrossRef] [PubMed]
- Trimble, C.L.; Peng, S.; Kos, F.; Gravitt, P.; Viscidi, R.; Sugar, E.; Pardoll, D.; Wu, T.C. A phase i trial of a human papillomavirus DNA vaccine for hpv16+ cervical intraepithelial neoplasia 2/3. Clin. Cancer Res. 2009, 15, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U. Established and potential strategies against papillomavirus infections. J. Antimicrob. Chemother. 2004, 53, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Lowy, D.R. Papillomaviruses and their replication. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; p. 2197. [Google Scholar]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. (Lond) 2006, 110, 525–541. [Google Scholar] [CrossRef]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Zheng, Z.M. Regulation of bovine papillomavirus type 1 gene expression by rna processing. Front. Biosci. (Landmark Ed.) 2009, 14, 1270–1282. [Google Scholar]
- Johansson, C.; Schwartz, S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat. Rev. Microbiol. 2013, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [PubMed]
- Spink, K.M.; Laimins, L.A. Induction of the human papillomavirus type 31 late promoter requires differentiation but not DNA amplification. J. Virol. 2005, 79, 4918–4926. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Spalholz, B.A.; Yang, Y.C.; Howley, P.M. Transactivation of a bovine papilloma virus transcriptional regulatory element by the e2 gene product. Cell 1985, 42, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Brandsma, J.L.; Peng, X.; Srimatkandada, S.; Li, L.; Canaan, A.; Deisseroth, A.B. High and low levels of cottontail rabbit papillomavirus e2 protein generate opposite effects on gene expression. J. Biol. Chem. 2001, 276, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Steger, G.; Corbach, S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral e2 protein. J. Virol.. 1997, 71, 50–58. [Google Scholar] [PubMed]
- Thierry, F.; Yaniv, M. The bpv1-e2 trans-acting protein can be either an activator or a repressor of the hpv18 regulatory region. EMBO J. 1987, 6, 3391–3397. [Google Scholar] [PubMed]
- Kim, J.; Lee, D.; Gwan Hwang, S.; Hwang, E.S.; Choe, J. Brca1 associates with human papillomavirus type 18 e2 and stimulates e2-dependent transcription. Biochem Biophys Res. Commun 2003, 305, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.A.; Naidu, S.R.; Wang, X.; Imbalzano, A.N.; Androphy, E.J. Interaction of papillomavirus e2 protein with the brm chromatin remodeling complex leads to enhanced transcriptional activation. J. Virol. 2007, 81, 2213–2220. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Hwang, S.G.; Kim, J.; Choe, J. Functional interaction between p/caf and human papillomavirus e2 protein. J. Biol. Chem. 2002, 277, 6483–6489. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lee, B.; Kim, J.; Kim, D.W.; Choe, J. Camp response element-binding protein-binding protein binds to human papillomavirus e2 protein and activates e2-dependent transcription. J. Biol. Chem. 2000, 275, 7045–7051. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.R.; You, J.; Howley, P.M. Bromodomain protein 4 mediates the papillomavirus e2 transcriptional activation function. J. Virol. 2006, 80, 4276–4285. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide sirna screen identifies smcx, ep400, and brd4 as e2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved p-tefb-interacting domain of brd4 inhibits hiv transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein brd4 is a positive regulatory component of p-tefb and stimulates rna polymerase ii-dependent transcription. Mol. Cell. 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wang, S.; Nguyen, T.; Shire, K.; Frappier, L. The ebna1 protein of epstein-barr virus functionally interacts with brd4. J. Virol. 2008, 82, 12009–12019. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of p-tefb for stimulation of transcriptional elongation by the bromodomain protein brd4. Mol. Cell. 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to hpv transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef] [PubMed]
- Senechal, H.; Poirier, G.G.; Coulombe, B.; Laimins, L.A.; Archambault, J. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus e2 affect binding to the long isoform of brd4. Virology 2007, 358, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, Q.; Lievens, S.; Tavernier, J.; You, J. Abrogation of the brd4-positive transcription elongation factor b complex by papillomavirus e2 protein contributes to viral oncogene repression. J. Virol. 2010, 84, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Kovelman, R.; Bilter, G.K.; Glezer, E.; Tsou, A.Y.; Barbosa, M.S. Enhanced transcriptional activation by e2 proteins from the oncogenic human papillomaviruses. J. Virol. 1996, 70, 7549–7560. [Google Scholar] [PubMed]
- Helfer, C.M.; Yan, J.; You, J. Brd4 and papillomavirus transcription activation. Unpublished work. 2014. [Google Scholar]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus e2 protein with brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Majello, B.; Napolitano, G.; Giordano, A.; Lania, L. Transcriptional regulation by targeted recruitment of cyclin-dependent cdk9 kinase in vivo. Oncogene 1999, 18, 4598–4605. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Q.; Helfer, C.M.; Jiao, J.; You, J. Bromodomain protein brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012, 287, 10738–10752. [Google Scholar] [CrossRef] [PubMed]
- Law, M.F.; Lowy, D.R.; Dvoretzky, I.; Howley, P.M. Mouse cells transformed by bovine papillomavirus contain only extrachromosomal viral DNA sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 2727–2731. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A.; Browne, H.M.; Appleby, M.; Minson, A.C. Properties of a non-tumorigenic human cervical keratinocyte cell line. Int. J. Cancer 1989, 43, 672–676. [Google Scholar] [PubMed]
- Agrawal, R.; Pelkonen, J.; Mantyjarvi, R.A. Bovine papillomavirus type 1-transformed primary mouse fibroblasts show no correlation between tumorigenicity and viral gene expression, but c-myc gene expression is elevated in tumorigenic cell lines. J. Gen. Virol. 1992, 73, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Alazawi, W.; Pett, M.; Arch, B.; Scott, L.; Freeman, T.; Stanley, M.A.; Coleman, N. Changes in cervical keratinocyte gene expression associated with integration of human papillomavirus 16. Cancer Res. 2002, 62, 6959–6965. [Google Scholar] [PubMed]
- Scarpini, C.G.; Groves, I.J.; Pett, M.R.; Ward, D.M.; Coleman, N. Virus transcript levels and cell growth rates after naturally-occurring hpv16 integration events in basal cervical keratinocytes. J. Pathol 2014. [Google Scholar]
- You, J.; Schweiger, M.R.; Howley, P.M. Inhibition of e2 binding to brd4 enhances viral genome loss and phenotypic reversion of bovine papillomavirus-transformed cells. J. Virol. 2005, 79, 14956–14961. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chiang, C.M. Chromatin adaptor brd4 modulates e2 transcription activity and protein stability. J. Biol. Chem. 2009, 284, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Kwon, D.; McBride, A.A. Papillomavirus e2 proteins and the host brd4 protein associate with transcriptionally active cellular chromatin. J. Virol. 2009, 83, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with brd4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Helfer, C.M.; Wang, R.; You, J. Analysis of the papillomavirus e2 and bromodomain protein brd4 interaction using bimolecular fluorescence complementation. PLoS One 2013, 8, e77994. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 2013, 87, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of bet bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Ozato, K.; McBride, A.A. Interaction of bovine papillomavirus e2 protein with brd4 stabilizes its association with chromatin. J. Virol. 2005, 79, 8920–8932. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Breitburd, F.; Croissant, O.; Orth, G. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J. Virol. 1977, 21, 1205–1209. [Google Scholar] [PubMed]
- Rosl, F.; Waldeck, W.; Sauer, G. Isolation of episomal bovine papillomavirus chromatin and identification of a dnase i-hypersensitive region. J. Virol. 1983, 46, 567–574. [Google Scholar] [PubMed]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (bet) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (p-tefb) from 7sk small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. Bet bromodomain-targeting compounds reactivate hiv from latency via a tat-independent mechanism. Cell. Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. Bet bromodomain inhibition as a therapeutic strategy to target c-myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. Bet bromodomain inhibition targets both c-myc and il7r in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, W.; Helfer, C.M.; Bradner, J.E.; Hornick, J.L.; Janicki, S.M.; French, C.A.; You, J. Activation of sox2 expression by brd4-nut oncogenic fusion drives neoplastic transformation in nut midline carcinoma. Cancer Res. 2014, 74, 3332–3343. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. Rnai screen identifies brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; You, J. Brd4 phosphorylates papillomavirus e2. Unpublished work. 2014. [Google Scholar]
- Flores, E.R.; Lambert, P.F. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997, 71, 7167–7179. [Google Scholar] [PubMed]
- Slidebook 5.0., 5.0.0.1 ed.; Olympus America Inc.: Denver, USA, 2009.
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Helfer, C.M.; Yan, J.; You, J. The Cellular Bromodomain Protein Brd4 has Multiple Functions in E2-Mediated Papillomavirus Transcription Activation. Viruses 2014, 6, 3228-3249. https://doi.org/10.3390/v6083228
Helfer CM, Yan J, You J. The Cellular Bromodomain Protein Brd4 has Multiple Functions in E2-Mediated Papillomavirus Transcription Activation. Viruses. 2014; 6(8):3228-3249. https://doi.org/10.3390/v6083228
Chicago/Turabian StyleHelfer, Christine M., Junpeng Yan, and Jianxin You. 2014. "The Cellular Bromodomain Protein Brd4 has Multiple Functions in E2-Mediated Papillomavirus Transcription Activation" Viruses 6, no. 8: 3228-3249. https://doi.org/10.3390/v6083228
APA StyleHelfer, C. M., Yan, J., & You, J. (2014). The Cellular Bromodomain Protein Brd4 has Multiple Functions in E2-Mediated Papillomavirus Transcription Activation. Viruses, 6(8), 3228-3249. https://doi.org/10.3390/v6083228