1. Introduction
Cell cultures have become indispensable tools for biological and medical research. Advances in cell culture and development of various in vitro models have found a number of applications in studying tissue development and function in health and disease. However, to provide reliable and reproducible results, cell cultures must be healthy and above all uncontaminated. Handling with cell cultures always poses the risk of contamination, either with eukaryotic cells from other cell cultures or, more frequently, with microbiological organisms including fungi and bacteria, and sometimes with persistent viral infections. Therefore, to maintain experiment integrity, any risks of contamination should be managed effectively.
Contamination with bacteria or fungi usually causes visible effects on cell cultures, viruses are on the contrary, due to their small size and lack of visual cues of their presence, difficult to detect by routine light microscopy (LM) and thus might easily be overlooked [
1]. Frequently contamination with viruses remains unrecognized, unless viral infection leads to cytopathological changes of the cultured cells, such as atypical cell morphology or increased cell death. The cell culture laboratory environment, the personnel or rarely already contaminated cell lines could be the source of viruses. However, most commonly, the viral infection originates from infected donor animals, either by serum or when using an animal tissue as a source of cells for primary and subsequent cell cultures [
1].
Adenoviruses (AdVs) are non-enveloped, icosahedral viruses, with a linear double stranded DNA genome that can infect all five major vertebrate classes [
2]. Porcine adenoviruses (PAdVs) are classified within the genus
Mastadenovirus in the
Adenoviridae family [
2], and are regarded as low grade pathogens, infecting the porcine populations worldwide. They often do not cause any disease [
3], or the infection is only manifested in a milder diarrhea [
4] or respiratory signs [
5], with no other associated clinical symptoms. There are at least five types of PAdV circulating in domestic pig populations internationally [
6], among which PAdV types 1 to 3 are closely related, whereas types 4 and 5 are less similar, both to this group and to each other [
2].
AdVs enter the host cell by receptor-mediated endocytosis. They can bind to one of the adenovirus receptors, e.g., coxsackievirus and adenovirus receptor (CAR) at the cell surface and locally activate the αv superficial cell integrins, which triggers the clathrin-mediated endocytosis [
7,
8]. Once inside the endosome, they rapidly lyse the endosomal membrane and escape to the cytosol. By trafficking along the microtubules they reach the nucleus, where they bind to the nuclear envelope and release the viral genome into the nucleus through the nuclear pore [
9]. After the selective transcription and translation of viral genes, the AdVs assembly in the nucleus and leave the host cell via the induced cell lysis [
10].
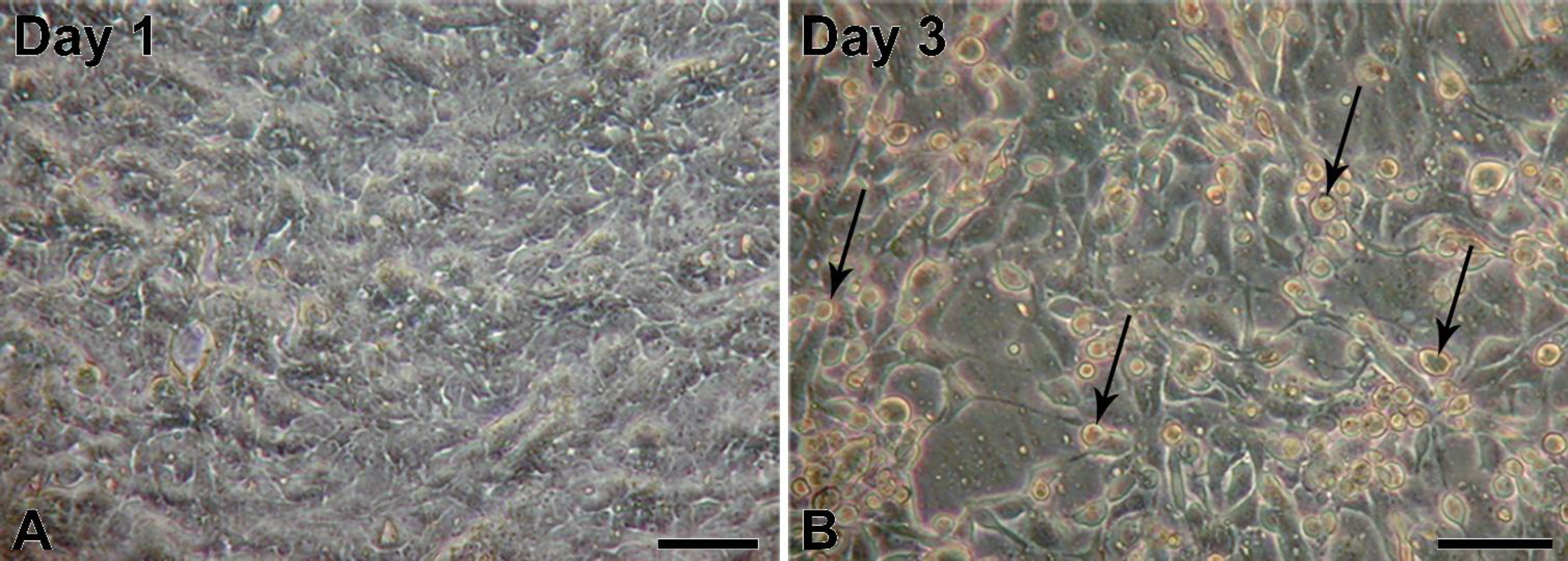
The current study describes the detection of AdV in subsequent cultures of normal porcine urothelial (NPU) cells isolated form urinary bladders of domestic pigs (Sus scrofa domestica). To detect the AdV, first the negative staining of NPU cell cultures was performed. The PAdV was further confirmed by polymerase chain reaction (PCR) amplification of AdV hexon gene and additionally characterized by transmission electron microscopy (TEM). According to the cell culture origin and the unique molecular characteristics of the analyzed AdV isolate, it was designated as PAdV-SVN1 (Porcine Adenovirus strain SVN1). Since most PAdV infections are asymptomatic, the PAdV detection methods described herein are advisable always when establishing the primary and subsequent cell cultures from porcine tissue. Overall, only early recognition of viral contamination can reduce the risk of transferring the infection also to the other cell cultures in the cell laboratory.
2. Materials and Methods
2.1. Porcine Urinary Bladders and Establishment of Primary and Secondary NPU Cell Culture
The experiments were approved by the Veterinary Administration of the Slovenian Ministry of Agriculture and Forestry in compliance with the Animal Health Protection Act and the Instructions for Granting Permits for Animal Experimentation for Scientific Purposes.
Porcine urinary bladders (
n = 7) were obtained from a local slaughterhouse. The urine, urothelial, connective, and muscle tissue were tested for presence of adenoviruses with PCR. For harvesting of primary and subsequent NPU cell cultures, porcine urinary bladder was cut in large segments and NPU cells were gently scraped from urothelium, filtered through the 40 µL Cell Strainer (BD Falcon, Heidelberg, Germany), collected and seeded onto polystyrene Tissue Culture Flasks (TPP, Trasadingen, Switzerland) at a density of 2 × 10
5 viable cells/cm
2. At 80%–100% confluence, the NPU cells were harvested with TripLE™ Select (Gibco, Life technologies, Wien, Austria) and reseeded onto new Tissue Culture Flasks. Cells were sub-cultured until the XIII passage. The NPU cell cultures were cultured in UroM medium, which does not allow the growth of fibroblasts [
11] and is adapted for the growth of porcine urothelial cells, at 37 °C in a 95% humidified atmosphere of 5% CO
2 in air as in [
12,
13]. The medium was changed on alternate days. The UroM medium consisted of equal parts of MCDB153 medium (Sigma-Aldrich, Taufkirchen, Germany) and Advanced-Dulbecco’s modified essential medium (Invitrogen, Life technologies, Wien, Austria), supplemented with 2.5% fetal bovine serum (Gibco, Life technologies, Wien, Austria), 0.1 mM phosphoethanolamine (Sigma-Aldrich, Taufkirchen, Germany), 15 µg/mL adenine (Sigma-Aldrich, Taufkirchen, Germany), 0.5 µg/mL hydrocortisone (Sigma-Aldrich, Taufkirchen, Germany), 5 µg/mL insulin (Sigma-Aldrich, Taufkirchen, Germany), 4 mM glutamax (Gibco, Life technologies, Wien, Austria), 100 µg/mL streptomycin and 100 U/mL penicillin.
To confirm the presence of adenovirus, the NPU cells of I, X, XI and XIII passages were analyzed by PCR. NPU cells of the XIII passage were also prepared for negative staining and TEM of ultrathin sections.
2.2. Negative Staining Electron Microscopy
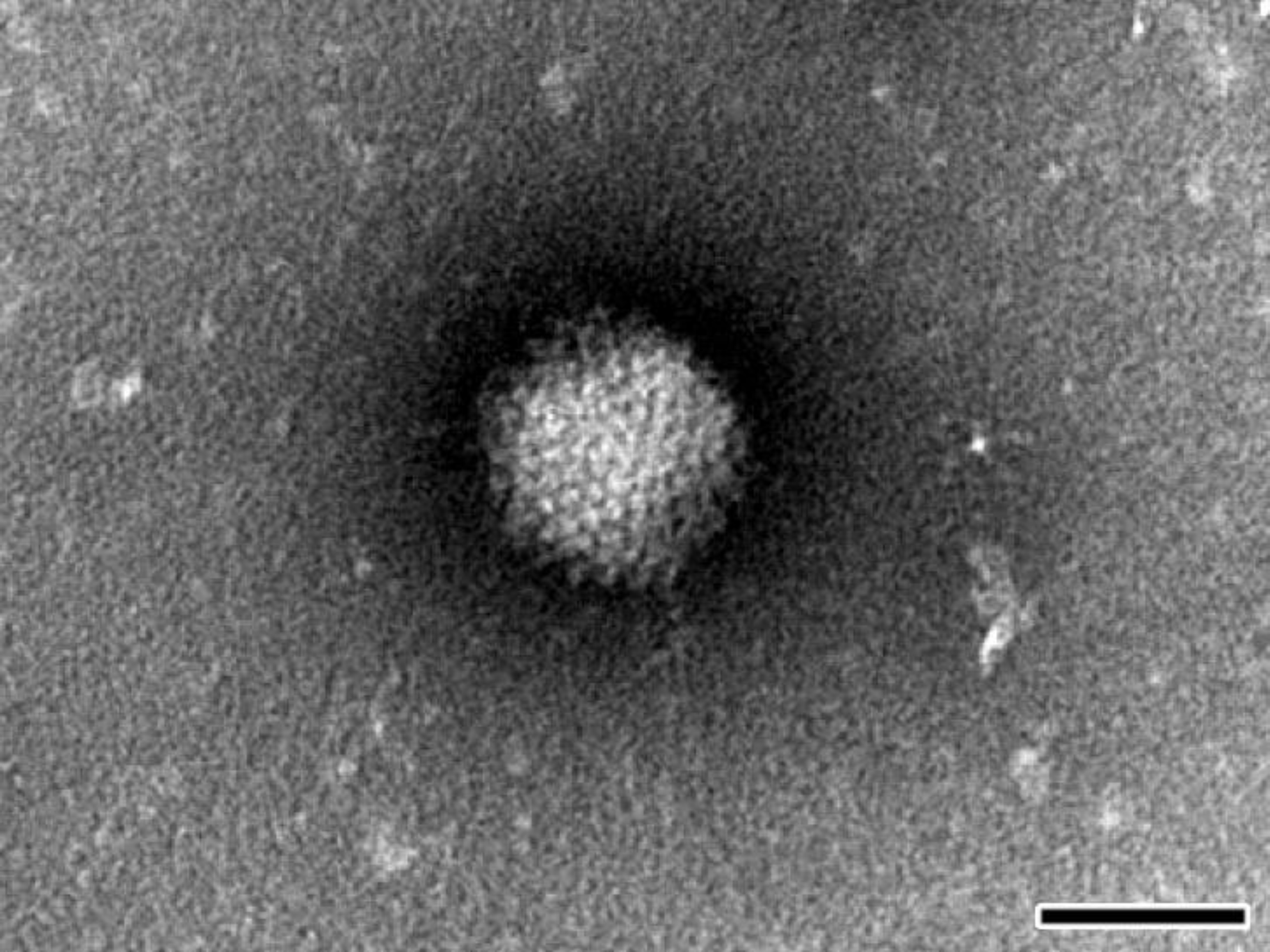
For negative staining NPU cell cultures were frozen, thawed three times and clarified by centrifugation at 1000× g. Clarified supernatant was ultracentrifuged at 100,000× g for 1 h. The resulting pellet was resuspended in saline solution, placed onto formvar (SPI Supplies, West Chester, PA, USA) coated with carbon stabilized grids and negatively stained using phosphotungstic acid. The grids were examined with TEM (JEM-1200 EX II, JEOL, Tokyo, Japan) at 80 kV.
2.3. DNA Extraction and PCR
For nucleic acid extraction, NPU cell cultures were frozen and thawed three times. Volume of 400 µL suspension was used for total viral nucleic acid extraction with iPrep™ PureLink® Virus Kit (Life technologies, Invitrogen Division, Carlsbad, CA, USA). Extracted nucleic acid was stored at −80 °C.
Adenoviral nucleic acid was detected by semi-nested touchdown polymerase chain reaction (TD-PCR) via broad-spectrum primers as already described [
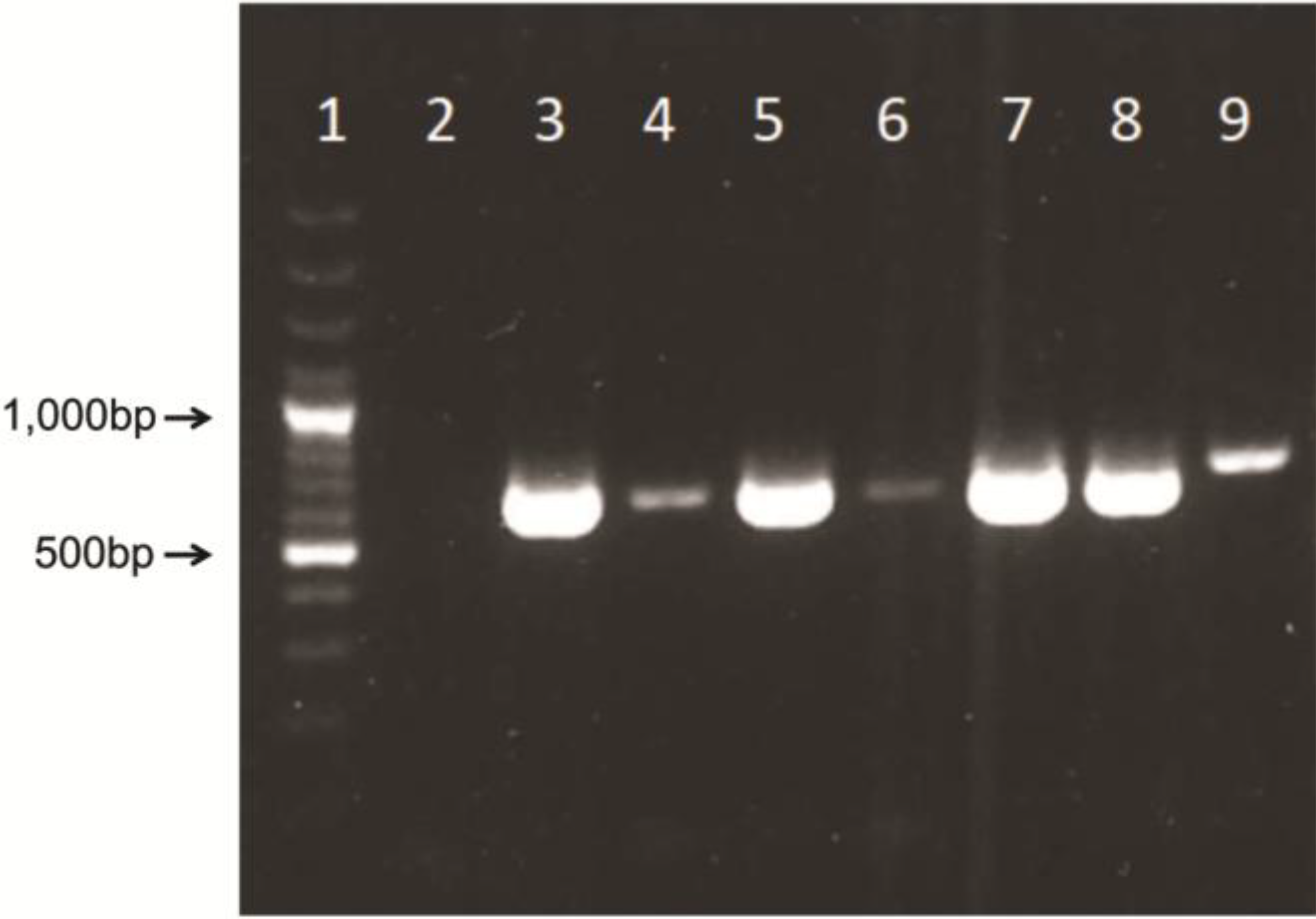
14]. Briefly, 5’ region of AdV hexon gene was amplified using MaAdF1/R, AtAdF1/R primer pairs and MaAdF2/R, AtAdF2/R primer pairs for semi-nested TD-PCR, using Tfi polymerase enzyme system (Life technologies, Invitrogen Division). Identical TD-PCR programs were used for both rounds of semi-nested amplification as follows: 94 °C for 4 min, followed by 10 cycles of 94 °C for 30 s, 65 °C for 30 s (with a decrement of 1 °C per cycle), and 72 °C for 1 min. An additional 30 cycles were completed as follows: 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min, finishing with a 72 °C, 7-min extension. PCR products were confirmed based on PCR amplified fragments that can vary between 599 and 725 bp in size because of amplification of hypervariable region V1 [
15]. Fragments were visualized in the agarose gel electrophoresis (1.5%) and SYBR Safe staining (Life Science, Invitrogen Division, Carlsbad, CA, USA).
For accurate characterization and classification of our isolate, a DNA-dependent DNA polymerase gene (polymerase gene) region was included in molecular analysis. The partial polymerase gene was amplified with consensus primer pair polFouter/polRouter [
16]. A fragment of 553 nucleotide sequence was amplified following the protocol published by Wellehan
et al. [
16]. As positive control we used human adenovirus (HAdV) DNA isolated from a clinical sample. As negative control, the VII passage of our previously prepared NPU cells originating from another non-infected urinary bladder was analyzed. For the inhibition control, an eukaryotic 18S assay was carried out (Life Technologies, Applied Biosystems Division, Foster City, CA, USA), using OneStep real-time RT-PCR system (Life Technologies, Invitrogen Division, Carlsbad, CA, USA).
2.4. Sequencing
To confirm the specificity of the PCR product, amplified MaAdV hexon gene and polymerase gene fragments were purified with Wizard SV Gel and PCR Clean-Up system (Promega, Madison, WI, USA). All genome fragments were directly sequenced with initial PCR primers, used in the first round TD-PCR of the MaAdV hexon gene and PCR of polymerase gene amplification reaction. For the sequencing reaction ABI PRISM BigDyeTM 3.1 terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems) was used in a 20 µL reaction and purified with BigDye Xterminator Purification Kit (Applied Biosystems). Sequence data were generated in ABI 3500 Genetic Analyser (Life Technologies, Applied Biosystems Division, Foster City, CA, USA).
Nucleotide sequences were aligned and assembled in CLC software (CLC Bio, Aarhus, Denmark) [
17] and deposited in GenBank under accession number KJ499459 (hexon gene), KJ933482 (polymerase gene).
2.5. Phylogenetic Analysis
The hexon and polymerase gene fragments were further included in the phylogenetic analysis. Deduced amino acid sequences of both genes were prepared and additional amino acid sequences of representative strains from GenBank were selected to ensure accurate phylogenetic analysis. Multiple alignments were performed and Neighbor-Joining phylogenetic trees were constructed with bootstrap test of 1000 replicates, using Mega software package version 5.2.2. [
18].
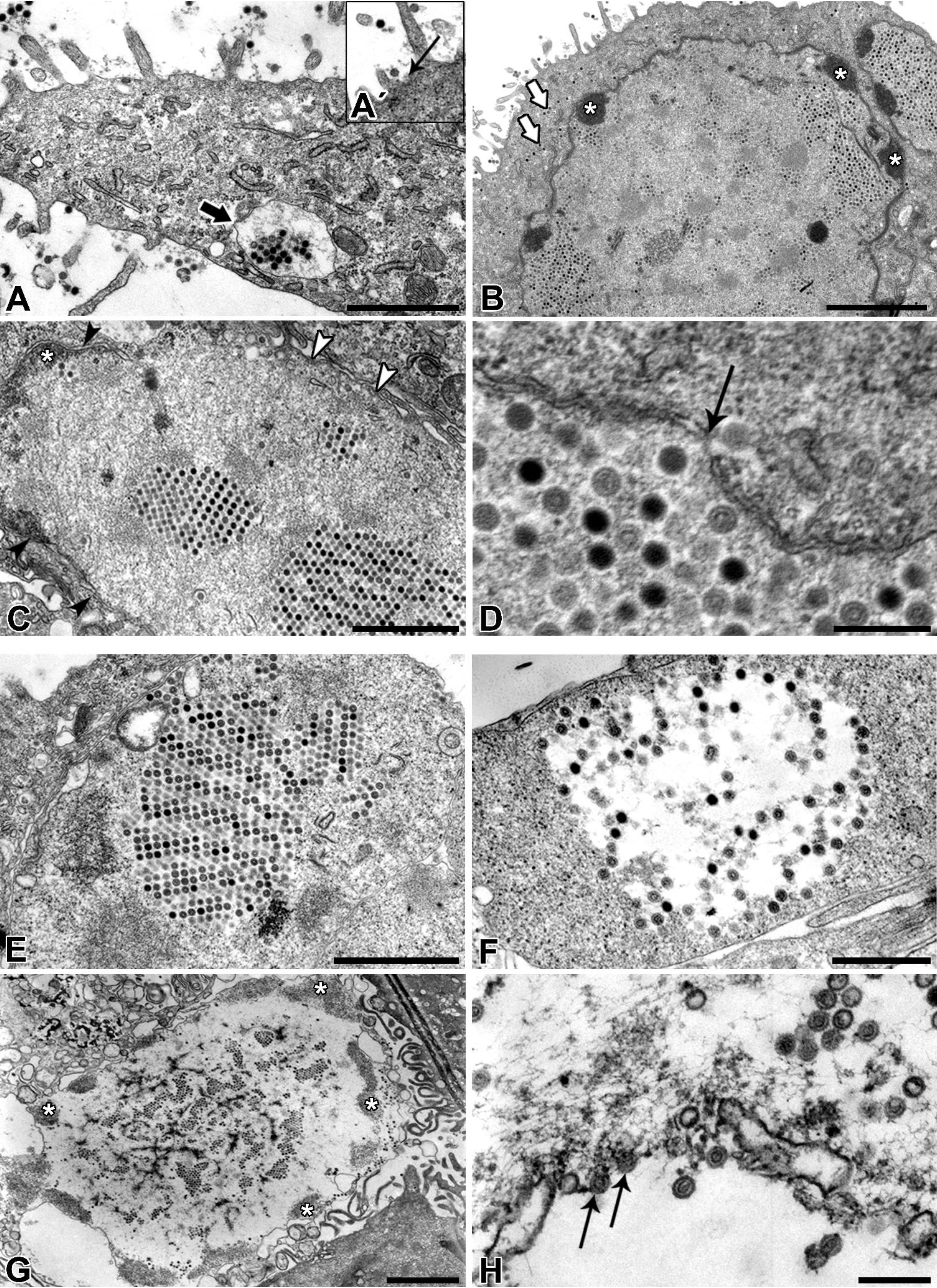
2.6. Ultrathin Section Electron Microscopy
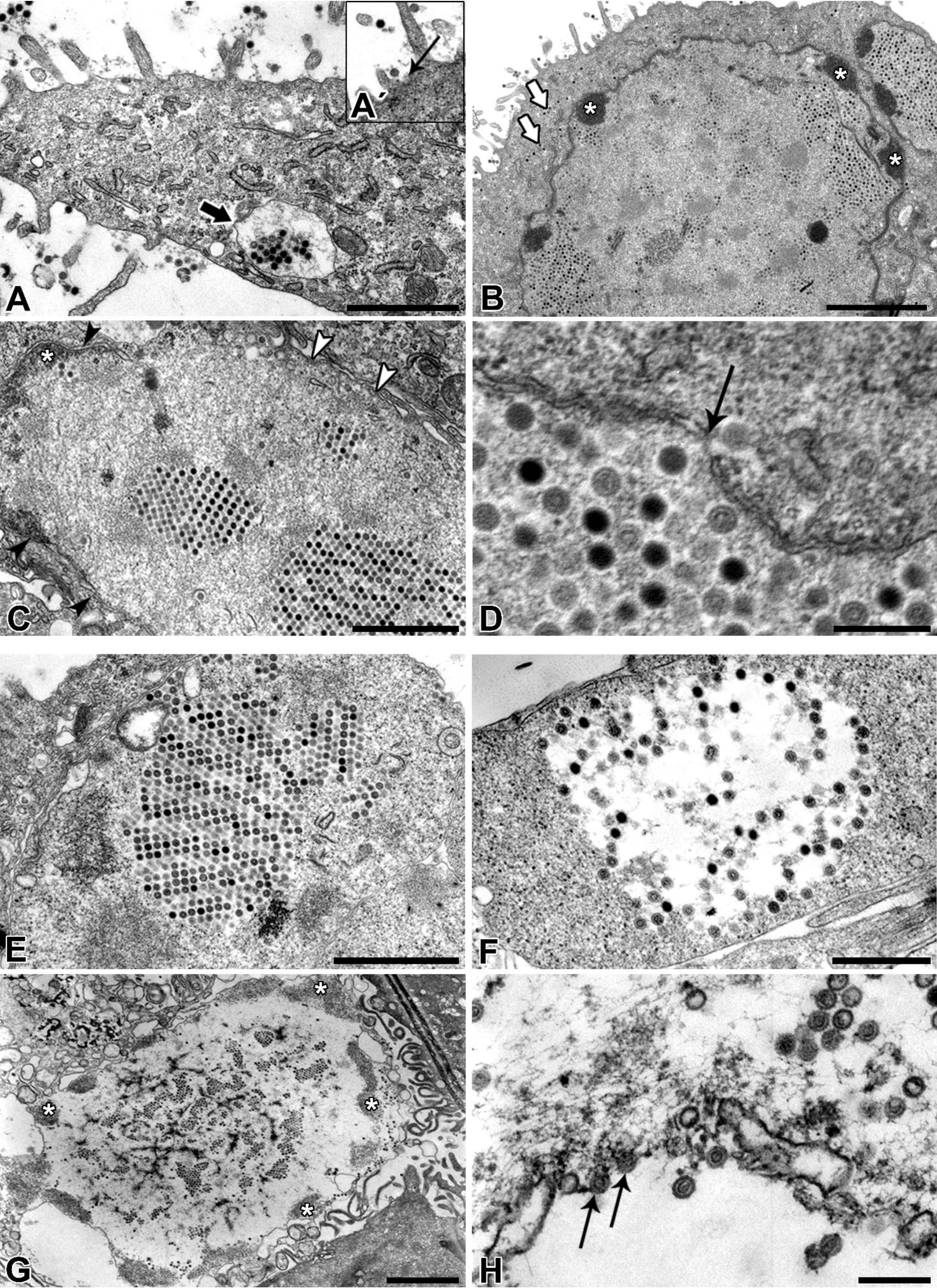
NPU cells of the XIII passage were seeded onto porous membranes (BD Falcon) at a density of 2 × 105 viable cells/cm2. After three weeks in culture, NPU cells were fixed with 4% (w/v) paraformaldehyde and 2.5% (v/v) glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4 for 2 h 45 min. The fixation was followed by overnight rising in 0.33% sucrose in cacodylate buffer and a post-fixation in 1% (w/v) osmium tetroxide for 1 h at 4 °C. The samples were afterwards dehydrated in a graded series of ethanol and embedded in Epon (Serva Electrophoresis, Heidelberg, Germany). Ultrathin sections were contrasted with uranyl acetate and lead citrate and examined with TEM (Philips CM100, Eindhoven, Netherlands) at 80 kV.
4. Discussion
HAdV is most frequently associated with respiratory infections, hemorrhagic cystitis and gastroenteritis, particularly in infants and young children [
19]. Similarly, PAdV in piglets often causes respiratory and gastrointestinal infections [
4,
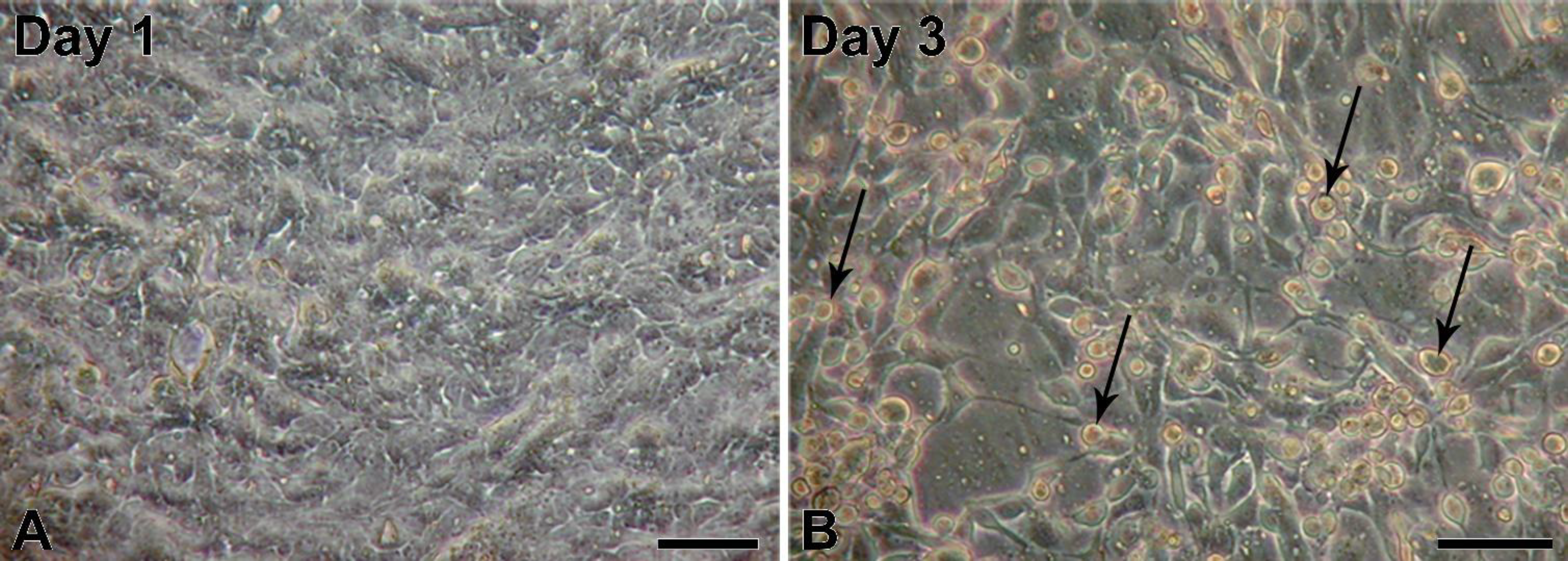
5]; however, to our knowledge, infections of porcine urinary bladder have not been reported yet. The obtained porcine urinary bladders did not show any visible signs of infection; therefore, we assume that the adenoviral infection was milder and without clinical signs. First passages of NPU cells harvested from the infected urinary bladder showed normal epithelial morphology. Interestingly, also with the PCR, the adenoviral contamination of the NPU cells of the first passage was not detected, suggesting on a very low viral load just after the cell harvest. However, with cell subculturing the amount of infectious viruses seemed to increase, which resulted in NPU cell desquamation and their alteration into fibroblast-like morphology. Additionally, adenoviral DNA in the X and the following passages was successfully detected. Many factors, viral and hosts can contribute to the regulation of virus replication cycle. For the dynamic of viral load through the cell culture passages, a more sensitive method, like real-time PCR should be used, including the normalization of the cell number.
For adenoviral detection and identification different approaches can be used: the conventional methods, which include TEM, viral antigen detection and virus isolation in cell culture, and methods based on molecular techniques that focus on the amplification and detection of the viral genome [
20]. In the last decades TEM has made a major contribution to virology, including the discovery of many viruses and enlightenment of the fundamental virus-host cell interactions [
21]. Even though it is nowadays gradually being overruled by the more sensitive molecular detection approaches and live cell imaging fluorescence based imaging techniques [
22], it still remains one of the inevitable identification tools in virology as it enables fast morphology-based identification of viruses [
23] and provides the insight into the viral life cycle and its influence on a host cell fate [
21]. The PCR is frequently referred to as a gold standard for molecular detection of nucleic acids in virology [
24]. The method is highly sensitive and accurate, and is especially applicable when dealing with noninfectious viruses [
20]. However, one of the major challenges for the development of a sensitive and universal PCR capable of detecting all viral strains still remains the high degree of heterogeneity among the various serotypes of the viral family [
25]. Additionally, the molecular approach lacks the information of viral infectivity.
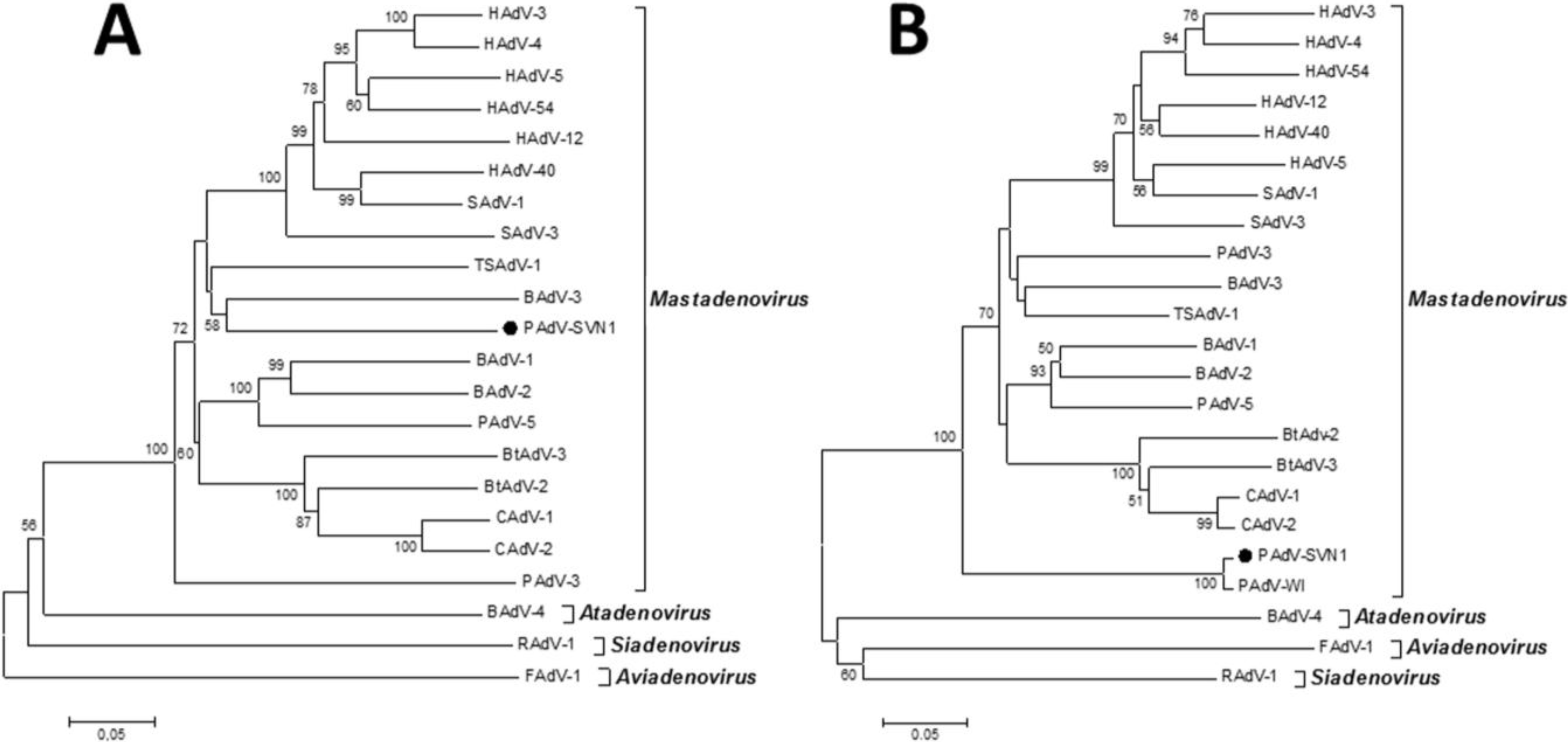
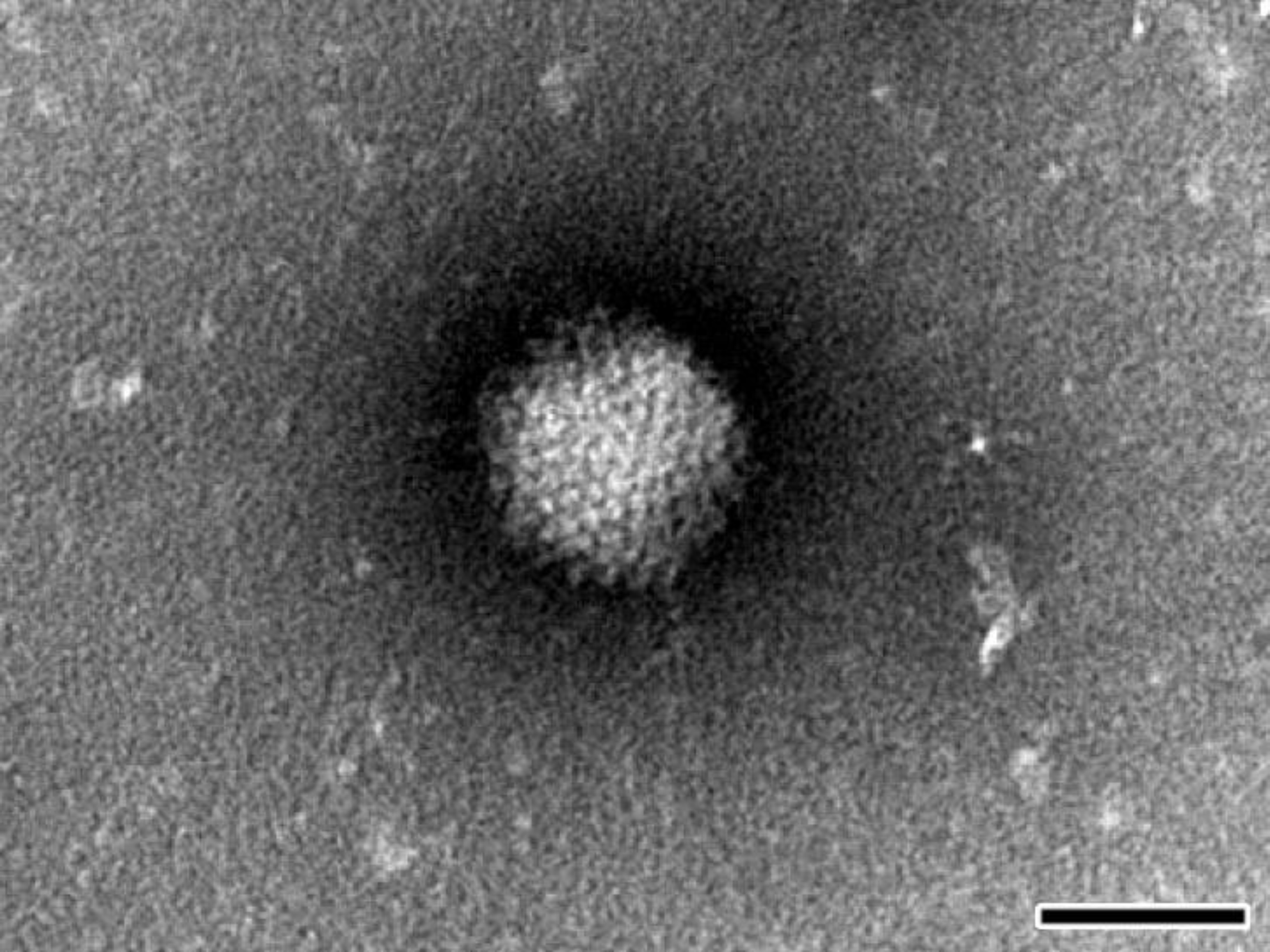
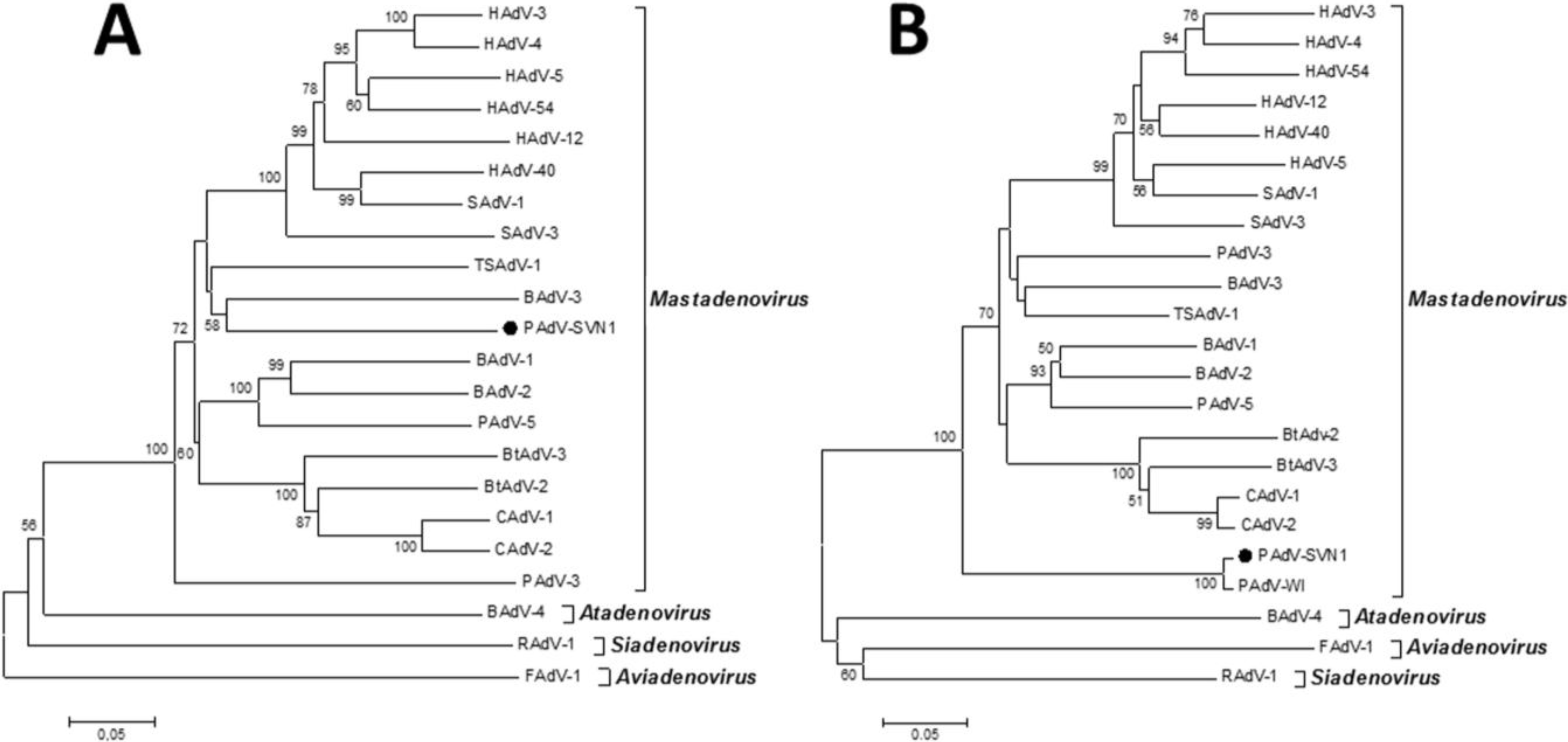
Considering all of the above, the most effective way to detect and fully characterize a virus is the combination of different approaches. In the present study the suspicion of viral contamination of NPU cells isolated from porcine urinary bladder, was confirmed with conventional (TEM) and molecular based (PCR) methods. Initially, TEM (negative staining) was used to detect and morphologically identify the virus belonging to the
Adenoviriade family. Later on, the detailed classification of the adenoviral isolate was made by nucleotide sequence analysis. For this purpose a broad range of adenovirus primers was used, enabling successful amplification of the hexon and polymerase genes of various types of mammal adenoviruses. The sequencing and sequence analysis of the amplified hexon gene fragment of adenovirus isolate showed the highest similarity to recently described novel porcine adenovirus strain PAdV-WI, which was so far detected only in pen wash water of newborn to finisher pigs and has been proposed as a prototype of a new species in the
Mastadenovirus genus [
14]. According to the short nucleotide sequence of the hexon gene analyzed in this study and the comparison to the initially recognized PAdV-WI, both strains showed a clear separation from other, even the most similar representatives in the
Mastadenovirus genus obtained from the GenBank (
Figure 4B,
Table 1). For the polymerase gene fragment similar pattern was observed. Strain PAdV-SVN1 was distantly related to other
Mastadenovirus strains (
Figure 4A). Unfortunately, not all species representatives within the
Mastadenovirus genus were available for both of the analyzed genome regions. Thus, clear classification of PAdV-SVN1 as well as PAdV-WI is not possible until the whole genome sequence is available and comparable analysis with all the representative strains of
Mastadenovirus genus is performed.
Afterwards, the TEM of ultrathin sections additionally confirmed the ultrastructure and life-cycle characteristic for AdVs. Nevertheless, according to our TEM results it remains unclear how the virions left the nucleus and re-entered the cytoplasm. We assume that the virions caused degradation of the nuclear envelope leading to the enhanced transfer of virions between nucleus and cytoplasm or they even escaped from nucleus through the nuclear pores.
Overall, an interdisciplinary approach gave us a holistic view of the viral ultrastructure, its genotype and interactions with the host cell. The present study is the first describing the isolation of the novel strain of porcine adenovirus lineage most probably originating from the urothelial tissue intended for use in the cell culture experiments. Thus when establishing cell cultures from animal or human tissue one should consider animal or human tissues as a potential source of silent viral infection, which can become apparent only after few passages in culture. The establishment and cultivation of cell cultures can be a demanding, time consuming, and costly expenditure. To avoid such cultivation of seemingly uninfected cell cultures, we propose that the ultrastructural and molecular examination of tissues and harvested cells should be an essential part in providing uncontaminated healthy cell cultures that ensure reliable and reproducible results.
In conclusion, since cell culture contamination is a frequently encountered problem, the only way to keep cell cultures as a trustworthy research tool is the effective management and prevention of its contamination. The animal tissue is a potential source of contamination, thus when choosing donor animals special care should be taken to choose only healthy animals from controlled breeding that have been regularly inspected by the veterinarian.
The PAdV-WI adenoviral strain has been just recently described, and hence many questions regarding its infectivity are still to be answered. To elucidate the risk of PAdV-WI and PAdV-SVN1 infections, screening of the pig stool and urine samples for the presence of these viruses, and also seroprevalance study on different age groups of pigs in order to determine the age group of pigs with high infection rate should be performed. Nevertheless, although the donor animal or human has no clinical signs of infection, in order to avoid potential viral contamination a multi-step approach, combining molecular and electron microscopy detection techniques is highly recommended always when considering establishing the cell culture de novo.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}