Hepatitis B Virus HBx Protein Interactions with the Ubiquitin Proteasome System

Abstract
:1. Introduction
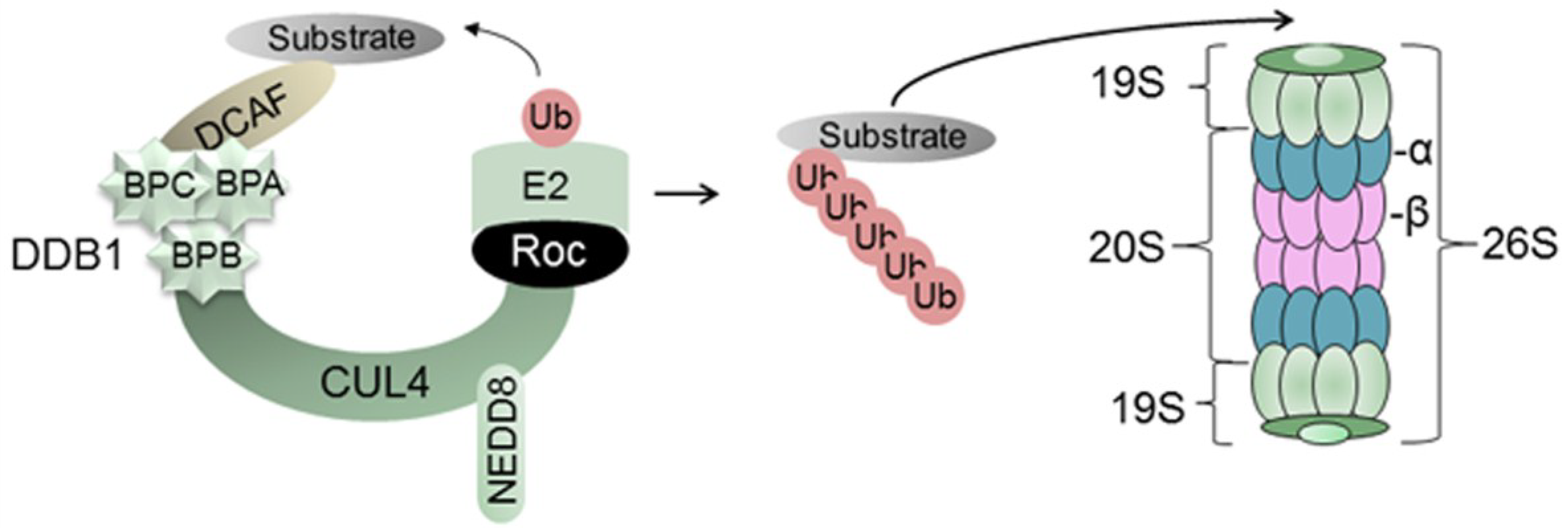
2. The UPS
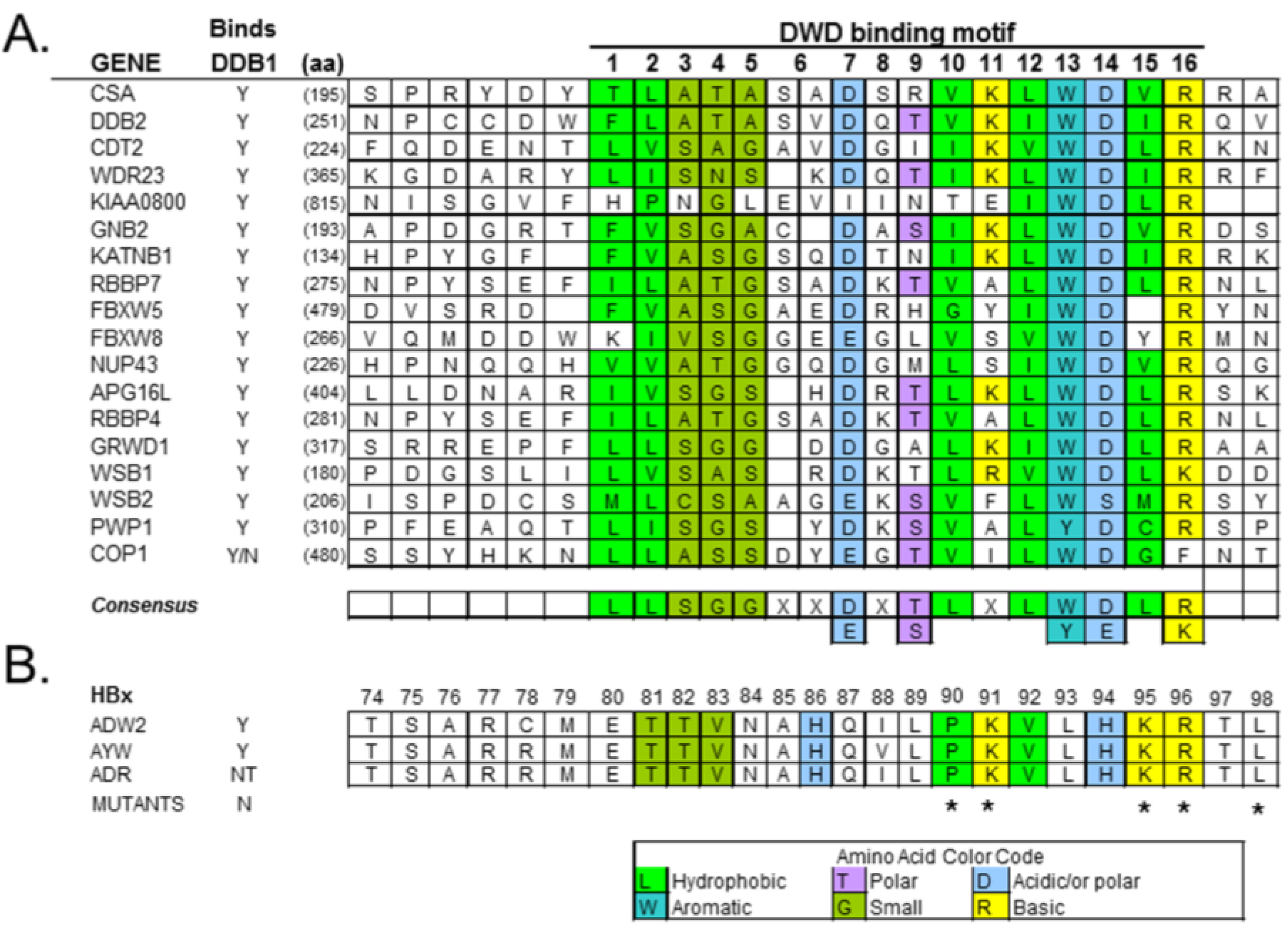
2.1. Damaged DNA Binding Protein 1 (DDB1) and DDB1-Cullin-Associated Factors (DCAFs)
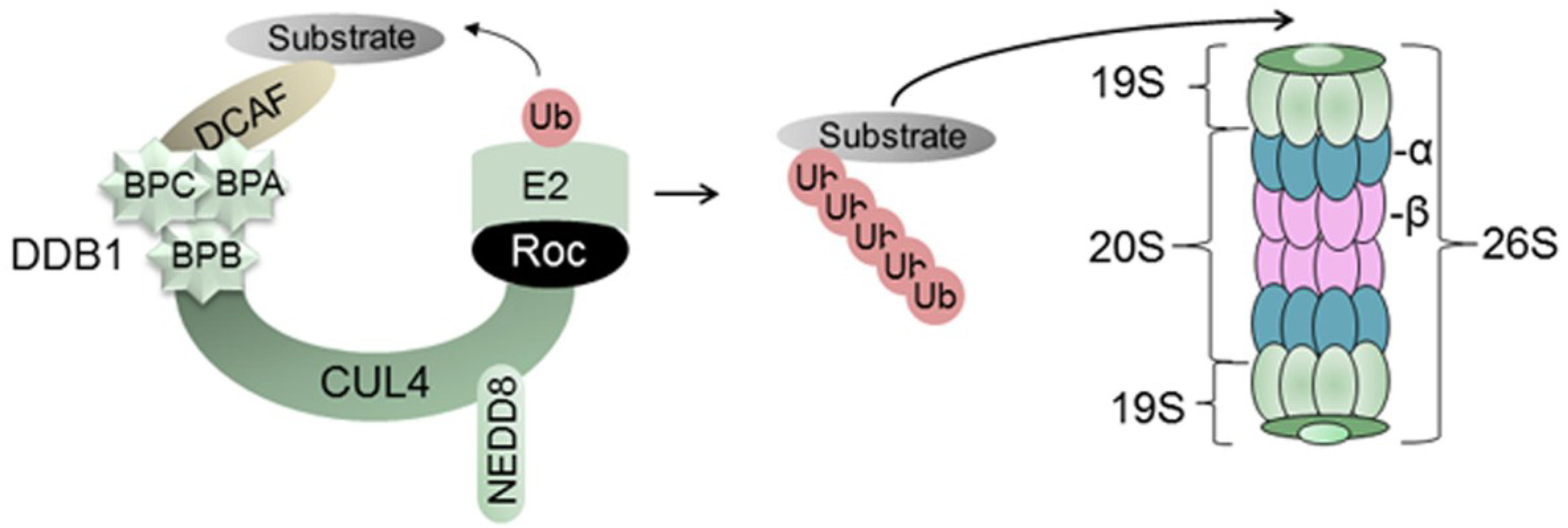
2.2. Regulation of CRLs
2.3. The 26S Proteasome
2.4. The UPS and Cancer
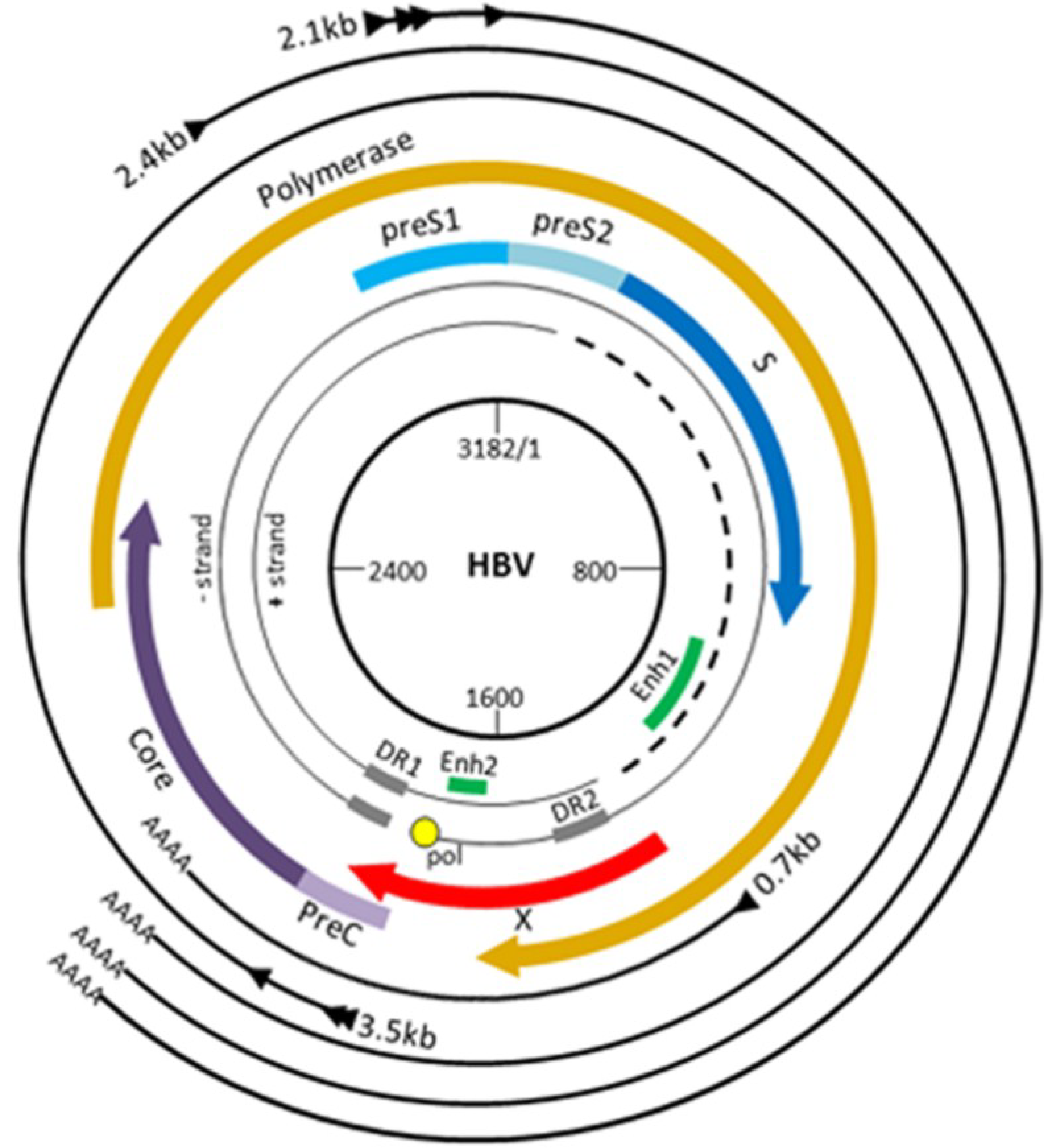
3. The HBV Life Cycle and HBx
3.1. HBV Replication
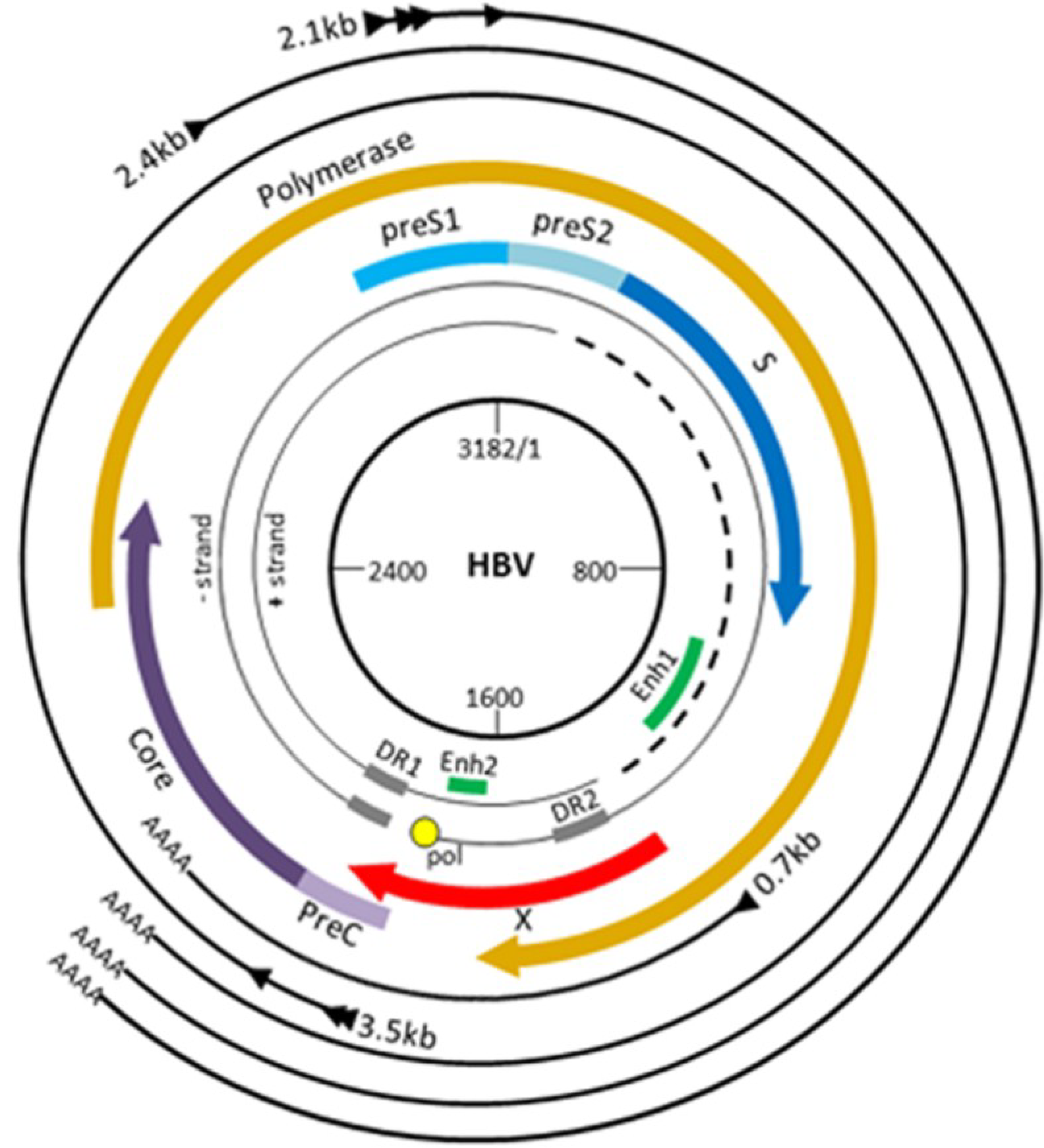
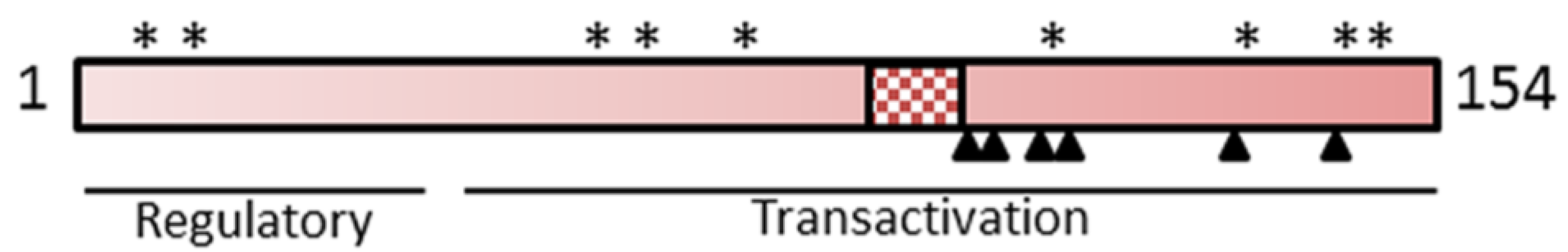
3.2. The Regulatory HBx Protein
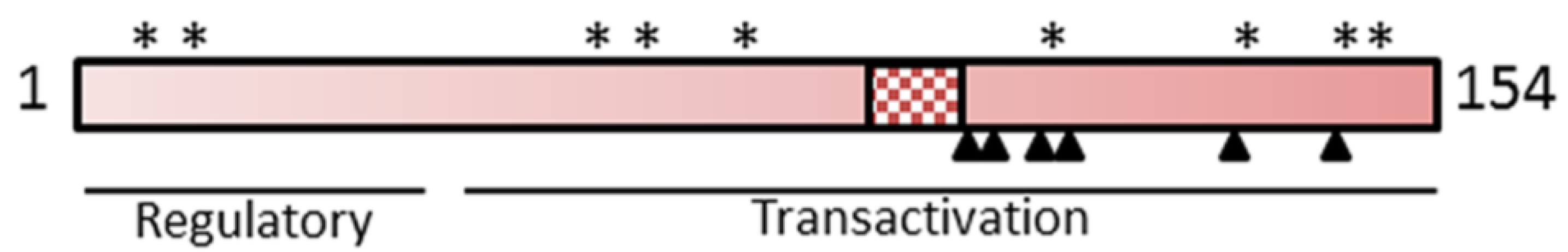
3.3. HBV Replication Requires HBx
4. HBx Interactions with CRL4
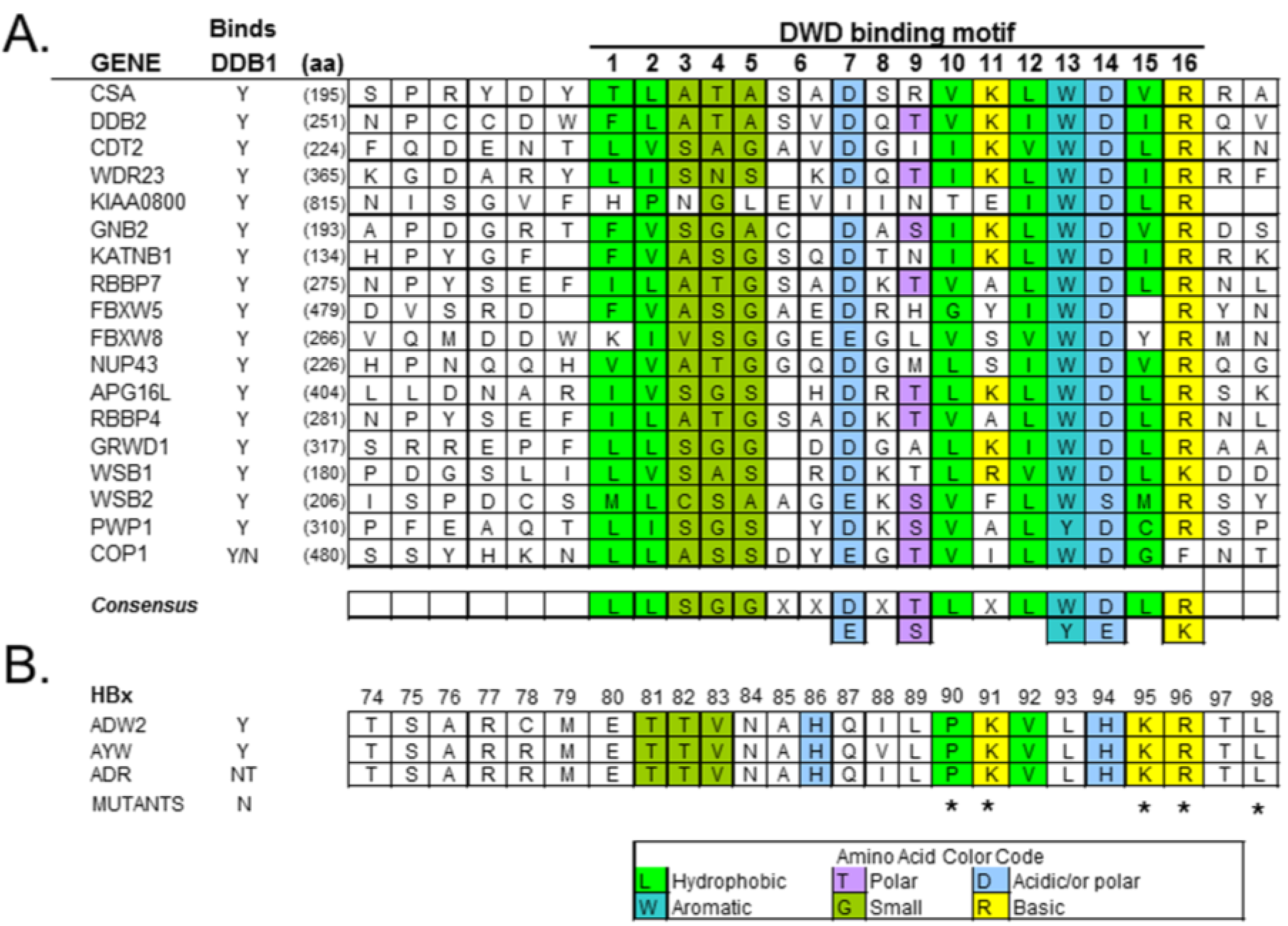
4.1. HBx Binds Cellular DDB1
4.2. HBx Is a Viral DCAF
4.3. HBx Binds the CSN Signalosome
5. HBx Interactions with the Proteasome
6. Lessons from Other Viruses
6.1. E3 Ligases and Innate Immunity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus family | Virus (protein) 1 | Cellular pathway altered | Reference |
|---|---|---|---|
| Paramyxoviridae | Simian Virus 5 (V) | Innate immunity | [97] |
| HPIV2 (V) | Innate immunity | [96] | |
| Mumps (V) | Innate immunity | [104] | |
| Hepadnaviridae | HBV (HBx) | Unknown | - |
| Retroviridae | HIV-1 (Vpr) | Cell cycle | [105,106] |
| HIV-2 (Vpx) | Cell cycle | [107] | |
| Flaviviridae | HCV (NS3/4A) | Unknown | - |
| Herpesviridae | MHV-68 (M2) | Apoptosis | [108] |
| EBV (BPLF1) | Cell cycle | [88] | |
| BHV-1 (VP8) | Unknown | - |
6.2. E3 Ligases and the Cell Cycle
6.3. E3 Ligases and the Damaged DNA Response
7. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Pickart, C.M. Back to the future with ubiquitin. Cell 2004, 116, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. Ubiquitin, ubiquitination and the ubiquitin-proteasome system in cancer. Atlas Genet. Cytogenet. Oncol. Haematol. 2010, 14, 1088–1099. [Google Scholar]
- Brooks, P.; Fuertes, G.; Murray, R.Z.; Bose, S.; Knecht, E.; Rechsteiner, M.C.; Hendil, K.B.; Tanaka, K.; Dyson, J.; Rivett, J. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem. J. 2000, 346, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of Cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gustin, J.K.; Moses, A.V.; Fruh, K.; Douglas, J.L. Viral takeover of the host ubiquitin system. Front. Microbiol. 2011, 2, 161. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.; Fruh, K. Viral modulators of cullin RING ubiquitin ligases: Culling the host defense. Sci. Stke 2006, 335, 1–5. [Google Scholar]
- Gao, G.; Luo, H. The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 2006, 84, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Randow, F.; Lehner, P.J. Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 2009, 11, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.G.; Wong, J.; Marchant, D.; Luo, H. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev. Med. Virol. 2013, 23, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Zoulim, F.; Mason, W.S. Hepadnaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 2185–2221. [Google Scholar]
- Hodgson, A.J.; Slagle, B.L. Molecular biology of HBV-related hepatocellular carcinoma. In Chronic Hepatitis B and C; Shih, C., Ed.; World Scientific Publishing Co. Pte. Ltd.: Singapore, Singapore, 2012; pp. 99–131. [Google Scholar]
- Bosu, D.R.; Kipreos, E.T. Cullin-RING ubiquitin ligases: Global regulation and activation cycles. Cell Div. 2008, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Nag, A. CUL4A ubiquitin ligase: A promising drug target for cancer and other human diseases. Open Biol. 2014, 4, 130217. [Google Scholar] [CrossRef] [PubMed]
- Iovine, B.; Iannella, M.L.; Bevilacqua, M.A. Damage-specific DNA binding protein 1 (DDB1): A protein with a wide range of functions. Int. J. Biochem. Cell Biol. 2011, 43, 1664–1667. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Xiong, Y. CRL4s: The CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 2009, 34, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [PubMed]
- He, Y.J.; McCall, C.M.; Hu, J.; Zeng, Y.; Xiong, Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006, 20, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Higa, L.A.; Wu, M.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 2006, 8, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, P. DCAFs, the missing link of the CUL4-DDB1 Ubiquitin Ligase. Mol. Cell 2007, 26, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Kalra, N.; Kumar, V. The X protein of hepatitis B virus binds to the F box protein Skp2 and inhibits the ubiquitination and proteasomal degradation of c-Myc. FEBS Lett. 2006, 580, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Osaka, F.; Chiba, T.; Miyamoto, C.; Okabayashi, K.; Shimbara, N.; Kato, S.; Tanaka, K. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 1999, 18, 6829–6834. [Google Scholar] [CrossRef] [PubMed]
- Rabut, G.; Peter, M. Function and regulation of protein neddylation. “Protein modifications: Beyond the usual suspects” review series. EMBO Rep. 2008, 9, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Higa, L.A.; Banks, D.; Min, W.; Kobayashi, R.; Hong, S.; Hui, Z. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 2006, 5, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Colantoni, A.; Bianchi, V.; Gherardini, P.F.; Tomba, G.S.; Ausiello, G.; Helmer-Citterich, M.; Ferre, F. Alternative splicing tends to avoid partial removals of protein-protein interaction sites. BMC Genomics 2013, 14, 379. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; McCall, C.M.; Ohta, T.; Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 2004, 6, 1003. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Saeki, Y.; Tanaka, K. Assembly and function of the proteasome. Methods Mol. Biol 2012, 832, 315–337. [Google Scholar] [PubMed]
- Kniepert, A.; Groettrup, M. The unique functions of tissue-specific proteasomes. Trends Biochem. Sci. 2014, 39, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Arii, S.; Zhao, C.; Imoto, I.; Ueda, M.; Nagai, H.; Emi, M.; Inazawa, J. TFDP1, Cul4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology 2002, 35, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Schindl, M.; Gnant, M.; Schoppmann, S.F.; Horvat, R.; Birner, P. Overexpression of the human homologue for Caenorhabditis elegans cul-4 gene is associated with poor outcome in node-negative breast cancer. Anticancer Res. 2007, 27, 949–952. [Google Scholar] [PubMed]
- Birner, P.; Schoppmann, A.; Schindl, M.; Dinhof, C.; Jesch, B.; Berghoff, A.S.; Schoppmann, S.F. Human homologue for Caenorhabditis elegans CUL-4 protein overexpression is associated with malignant potential of epithelial ovarian tumours and poor outcome in carcinoma. J. Clin. Pathol. 2012, 65, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Dagher, R.; Farrell, A.; Ko, C.W.; Sridhara, R.; Justice, R.; Pazdur, R. Bortezomib for the treatment of mantle cell lymphoma. Clin. Cancer Res. 2007, 13, 5291–5294. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Locarnini, S. New insight in the pathobiology of hepatitis B virus infection. Gut 2012, 61 (Suppl. 1), i6–i17. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Yan, H.; Wang, H.; He, W.; Jing, Z.; Qi, Y.; Fu, L.; Gao, Z.; Huang, Y.; Xu, G.; et al. Sodium taurocholate cotransporting polypeptide mediates woolly monkey hepatitis B virus infection of Tupaia hepatocytes. J. Virol. 2013, 87, 7176–7184. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2013, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Funk, A.; Mhamdi, M.; Will, H.; Sirma, H. Avian hepatitis B viruses: Molecular and cellular biology, phylogenesis, and host tropism. World J. Gastroenterol. 2007, 13, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Tennant, B.C.; Toshkov, I.A.; Peek, S.F.; Jacob, J.R.; Menne, S.; Hornbuckle, W.E.; Schinazi, R.D.; Korba, B.E.; Cote, P.J.; Gerin, J.L. Hepatocellular carcinoma in the woodchuck model of hepatitis B virus infection. Gastroenterology 2004, 127, S283–S293. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-S.; Kaneko, S.; Girones, R.; Anderson, R.W.; Hornbuckle, W.E.; Tennant, B.C.; Cote, P.J.; Gerin, J.L.; Purcell, R.H.; Miller, R.H. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J. Virol. 1993, 67, 1218–1226. [Google Scholar] [PubMed]
- Zoulim, F.; Saputelli, J.; Seeger, C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 1994, 68, 2026–2030. [Google Scholar] [PubMed]
- Scaglioni, P.P.; Melegari, M.; Wands, J.R. Posttranscriptional regulation of hepatitis B virus replication by the precore protein. J. Virol. 1997, 71, 345–353. [Google Scholar] [PubMed]
- Tsuge, M.; Hiraga, N.; Akiyama, R.; Tanaka, S.; Matsushita, M.; Mitsui, F.; Abe, H.; Kitamura, S.; Hatakeyama, T.; Kimura, T.; et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 2010, 91, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Melegari, M.; Scaglioni, P.P.; Wands, J.R. Cloning and characterization of a novel hepatitis B virus X binding protein that inhibits viral replication. J. Virol. 1998, 72, 1737–1743. [Google Scholar] [PubMed]
- Bouchard, M.J.; Wang, L.-H.; Schneider, R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2002, 294, 2376–2378. [Google Scholar] [CrossRef]
- Hoare, J.; Henkler, F.; Dowling, J.J.; Errington, W.; Goldin, R.D.; Fish, D.; McGarvey, M.J. Subcellular localization of the X protein in HBV infected hepatocytes. J. Med. Virol. 2001, 64, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Ryu, D.K.; Jung, H.S.; Chang, H.E.; Ryu, W.S. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 2006, 90, 978–986. [Google Scholar] [CrossRef]
- Henkler, F.; Hoare, J.; Waseem, N.; Goldin, R.D.; McGarvey, M.J.; Koshy, R.; King, I.A. Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol 2001, 82, 871–882. [Google Scholar] [PubMed]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular functions and biological roles of hepatitis B virus X protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Duan, L.-X. Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am. J. Pathol. 1997, 150, 1141–1157. [Google Scholar] [PubMed]
- Cougot, D.; Neuveut, C.; Buendia, M.A. HBV induced carcinogenesis. J. Clin. Virol. 2005, 34 (Suppl. 1), S75–S78. [Google Scholar] [CrossRef]
- Fallot, G.; Neuveut, C.; Buendia, M.A. Diverse roles of hepatitis B virus in liver cancer. Curr. Opin. Virol. 2012, 2, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Benhenda, S.; Cougot, D.; Buendia, M.A.; Neuveut, C. Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv. Cancer Res. 2009, 103, 75–109. [Google Scholar] [PubMed]
- Wei, Y.; Neuveut, C.; Tiollais, P.; Buendia, M.A. Molecular biology of the hepatitis B virus and role of the X gene. Pathol. Biol. Paris 2010, 58, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Protzer, U. Hepatitis B virus X protein: A key regulator of the virus life cycle. In Viral Genomes—Molecular Structure, Diversity, Gene Expression Mechanisms and Host-Virus Interactions; Garcia, M.L., Romanowski, V., Eds.; InTech: Rijeka, Croatia, 2012; pp. 141–154. [Google Scholar]
- Seeger, C.; Zoulim, F.; Mason, W.S. Hepadnaviruses. In Fields Virology, 5th ed.; Knipe, D.M., Griffin, D.E., Lamb, R.A., Strauss, S.E., Howley, P.M., Martin, M.A., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2977–3029. [Google Scholar]
- Du, J.; Zhou, Y.; Fu, Q.X.; Gong, W.L.; Zhao, F.; Peng, J.C.; Zhan, L.S. Bioluminescence imaging of hepatitis B virus enhancer and promoter activities in mice. FEBS Lett. 2008, 582, 3552–3556. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sarkar, D.P. Hepatitis B virus X protein: Structure-Function relationships and role in viral pathogenesis. In Transcription Factors; Gossen, M., Kaufmann, J., Triezenberg, S.J., Eds.; Springer-Verlag: Berlin Heidelberg, Germany, 2004; pp. 377–407. [Google Scholar]
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mal, T.K.; Jayasuryan, N.; Chauhan, V.S. Assignment of disulphide bonds in the X protein (HBx) of hepatitis B virus. Biochem. Biophys. Res. Commun. 1995, 212, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, K.; Kumar, S.; Reddy, V.S.; Kumar, V. Mass spectrometric determination of disulfide bonds in the biologically active recombinant HBx protein of hepatitis B virus. Biochemistry 2014, 53, 4685–4695. [Google Scholar] [CrossRef] [PubMed]
- Leon, D.A.; Herberg, F.W.; Banky, P.; Taylor, S.S. A stable alpha-helical domain at the N terminus of the RIalpha subunits of cAMP-dependent protein kinase is a novel dimerization/docking motif. J. Biol. Chem. 1997, 272, 28431–28437. [Google Scholar] [CrossRef] [PubMed]
- Locker, J.K.; Griffiths, G. An unconventional role for cytoplasmic disulfide bonds in vaccinia virus proteins. J. Cell Biol. 1999, 144, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Saaranen, M.J.; Ruddock, L.W. Disulfide bond formation in the cytoplasm. Antioxid. Redox. Signal. 2013, 19, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Torii, N.; Furusaka, A.; Malayaman, N.; Hu, Z.Y.; Liang, T.J. Structural and functional characterization of interaction between hepatitis B virus X protein and the proteasome complex. J. Biol. Chem. 2000, 275, 15157–15165. [Google Scholar] [CrossRef] [PubMed]
- Kidd-Ljunggren, K.; Oberg, M.; Kidd, A.H. The hepatitis B virus X gene: Analysis of functional domain variation and gene phylogeny using multiple sequences. J. Gen. Virol. 1995, 76, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Sohn, S.Y.; Yen, T.S.B.; Ahn, B.Y. Ubiquitin-dependent and -independent proteasomal degradation of hepatitis B virus X protein. Biochem. Biophys. Res. Commun. 2008, 366, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Andrisani, O.M.; Bouchard, M.J.; Lee, C.G.; Ou, J.H.; Siddiqui, A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology 2014. [Google Scholar] [CrossRef]
- Zhang, Z.-S.; Torii, N.; Hu, Z.; Jacob, J.; Liang, T.J. X-deficient woodchuck hepatitis virus mutants behave like attenuated viruses and induce protective immunity in vivo. J. Clin. Investig. 2001, 108, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Hildt, E.; Eckerskorn, C.; Sirma, H.; Kekule, A.; Hofschneider, P.H. Isolation and molecular characterization of hepatitis B virus X-protein from a baculovirus expression system. Hepatology 1997, 26, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Song, C.Z.; Bai, Z.L.; Song, C.C.; Wang, Q.W. Aggregate formation of hepatitis B virus X protein affects cell cycle and apoptosis. World J. Gastroenterol. 2003, 9, 1521–1524. [Google Scholar] [PubMed]
- Lee, T.-H.; Elledge, S.J.; Butel, J.S. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J. Virol. 1995, 69, 1107–1114. [Google Scholar] [PubMed]
- Keasler, V.V.; Slagle, B.L. The interaction of HBx with cellular DDB1. In The Pleiotropic Functions of the Viral Protein HBx in Hepatitis B Virus Infection and the Development of Liver Cancer; Kobarg, J., Ed.; Research Signpost: Kerala, India, 2008; pp. 91–103. [Google Scholar]
- Li, T.; Robert, E.I.; van Breugel, P.C.; Strubin, M.; Zheng, N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Rho, H.M.; Choi, C.Y.; Choi, B.H.; Paik, N.W. The interference of HBx in transcriptional regulation. In The Pleiotropic Functions of the Viral Protein HBx in Hepatitis B Virus Infection and the Development of Liver Cancer; Kobarg, J., Ed.; Research Signpost: Kerala, India, 2008; pp. 11–41. [Google Scholar]
- Hodgson, A.J.; Hyser, J.M.; Keasler, V.V.; Cang, Y.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology 2012, 426, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Sitterlin, D.; Lee, T.H.; Prigent, S.; Tiollais, P.; Butel, J.S.; Transy, C. Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X protein is conserved among mammalian hepadnaviruses and restricted to transactivation-proficient X-insertion mutants. J. Virol. 1997, 71, 6194–6199. [Google Scholar] [PubMed]
- Sitterlin, D.; Bergametti, F.; Tiollais, P.; Tennant, B.C.; Transy, C. Correct binding of viral X protein to UVDDB-p127 cellular protein is critical for efficient infection by hepatitis B viruses. Oncogene 2000, 19, 4427–4431. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Bontron, S.; Lin-Marq, N.; Strubin, M. Hepatitis B virus X protein associated with UV-DDB1 induces cell death in the nucleus and is functionally antagonized by UV-DDB2. J. Biol. Chem. 2002, 277, 38847–38854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Sun, D.C.; Lou, S.; Bo, X.C.; Lu, Z.; Qian, X.H.; Wang, S.Q. HBx protein of hepatitis B virus (HBV) can form complex with mitochondrial HSP60 and HSP70. Arch. Virol. 2005, 150, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kanai, F.; Ichimura, T.; Tateishi, K.; Asaoka, Y.; Guleng, B.; Jazag, A.; Ohta, M.; Imamura, J.; Ikenoue, T.; et al. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene 2006, 25, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Gastaldello, S.; Hildebrand, S.; Faridani, O.; Callegari, S.; Palmkvist, M.; Di, G.C.; Masucci, M.G. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat. Cell Biol. 2010, 12, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kwong, J.; Sun, E.C.Y.; Liang, T.J. Proteasome complex as a potential cellular target of hepatitis B virus X protein. J. Virol. 1996, 70, 5582–5591. [Google Scholar] [PubMed]
- Hu, Z.Y.; Zhang, Z.S.; Doo, E.; Coux, O.; Goldberg, A.L.; Liang, T.J. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J. Virol. 1999, 73, 7231–7240. [Google Scholar] [PubMed]
- Zhang, Z.; Sun, E.; Ou, J.H.; Liang, T.J. Inhibition of cellular proteasome activities mediates HBX-independent hepatitis B virus replication in vivo. J. Virol. 2010, 84, 9326–9331. [Google Scholar] [CrossRef] [PubMed]
- Bandi, P.; Garcia, M.L.; Booth, C.J.; Chisari, F.V.; Robek, M.D. Bortezomib inhibits hepatitis B virus replication in transgenic mice. Antimicrob. Agents Chemother. 2010, 54, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, P. Pathogenic role of the CRL4 ubiquitin ligase in human disease. Front. Oncol. 2012, 2, 21. [Google Scholar] [PubMed]
- Stohwasser, R.; Holzhutter, H.G.; Lehmann, U.; Henklein, P.; Kloetzel, P.M. Hepatitis B virus HBx peptide 116–138 and proteasome activator PA28 compete for binding to the proteasome alpha4/MC6 subunit. Biol. Chem. 2003, 384, 39–49. [Google Scholar] [PubMed]
- Lin, G.Y.; Paterson, R.G.; Richardson, C.D.; Lamb, R.A. The V protein of Paramyxovirus SV5 interacts with damage-specific DNA binding protein. Virology 1998, 249, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Ulane, C.M.; Horvath, C.M. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 2002, 304, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Precious, B.; Childs, K.; Fitzpatrick-Swallow, V.; Goodbourn, S.; Randall, R.E. Simian virus 5 V protein acts as an adaptor, linking DDB1 to STAT2, to facilitate the ubiquitination of STAT1. J. Virol. 2005, 79, 13434–13441. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M. Weapons of STAT destruction. Interferon evasion by paramyxovirus V protein. Eur. J. Biochem. 2004, 271, 4621–4628. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Hao, C.; Yan, J.; DeLucia, M.; Mehrens, J.; Wang, C.; Gronenborn, A.M.; Skowronski, J. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 2012, 287, 12550–12558. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Chisari, F.V. Stealth and cunning: Hepatitis B and hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y.; et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Jung, S.Y.; Hodgson, A.J.; Madden, C.R.; Qin, J.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J. Virol. 2011, 85, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 2011, 1, 1106–1117. [Google Scholar] [CrossRef]
- Ulane, C.M.; Rodriguez, J.J.; Parisien, J.P.; Horvath, C.M. STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J. Virol. 2003, 77, 6385–6393. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, E.; Belaidouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C.; Margottin-Goguet, F. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the CUL4-DDB1 ubiquitin ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Schrofelbauer, B.; Hakata, Y.; Landau, N.R. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. USA 2007, 104, 4130–4135. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Swanson, S.K.; Manel, N.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008, 4, e1000059. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.Z.; Pickering, M.T.; Cho, N.H.; Chang, H.; Volkert, M.R.; Kowalik, T.F.; Jung, J.U. Deregulation of DNA damage signal transduction by herpesvirus latency-associated M2. J. Virol. 2006, 80, 5862–5874. [Google Scholar] [CrossRef] [PubMed]
- Gearhart, T.L.; Bouchard, M.J. Replication of the hepatitis B virus requires a calcium-dependent HBx-induced G1 phase arrest of hepatocytes. Virology 2010, 407, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Casciano, J.C.; Bagga, S.; Yang, B.; Bouchard, M.J. Modulation of cell proliferation pathways by the hepatitis B virus X protein: A potential contributor to the development of hepatocellular carcinoma. In Hepatocellular Carcinoma—Basic Research; Lau, W.Y., Ed.; InTech: Rijeka, Croatia, 2012; pp. 103–152. [Google Scholar]
- Wang, W.H.; Hullinger, R.L.; Andrisani, O.M. Hepatitis B virus X protein via the p38MAPK pathway induces E2F1 release and ATR kinase activation mediating p53 apoptosis. J. Biol. Chem. 2008, 283, 25455–25467. [Google Scholar] [CrossRef] [PubMed]
- Studach, L.; Wang, W.H.; Weber, G.; Tang, J.; Hullinger, R.L.; Malbrue, R.; Liu, X.; Andrisani, O. Polo-like kinase 1 activated by the hepatitis B virus X protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. J. Biol. Chem. 2010, 285, 30282–30293. [Google Scholar] [CrossRef] [PubMed]
- Rakotomalala, L.; Studach, L.; Wang, W.H.; Gregori, G.; Hullinger, R.L.; Andrisani, O. Hepatitis B virus X protein increases the Cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J. Biol. Chem. 2008, 283, 28729–28740. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.A.; Lee, T.H.; Butel, J.S.; Slagle, B.L. Hepatitis B virus X protein interferes with cellular DNA repair. J. Virol. 1998, 72, 266–272. [Google Scholar] [PubMed]
- Qadri, I.; Fatima, K.; AbdeL-Hafiz, H. Hepatitis B virus X protein impedes the DNA repair via its association with transcription factor, TFIIH. BMC Microbiol. 2011, 11, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Warmerdam, D.O.; Kanaar, R. Dealing with DNA damage: Relationships between checkpoint and repair pathways. Mutat. Res. 2010, 704, 2–11. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minor, M.M.; Slagle, B.L. Hepatitis B Virus HBx Protein Interactions with the Ubiquitin Proteasome System. Viruses 2014, 6, 4683-4702. https://doi.org/10.3390/v6114683
Minor MM, Slagle BL. Hepatitis B Virus HBx Protein Interactions with the Ubiquitin Proteasome System. Viruses. 2014; 6(11):4683-4702. https://doi.org/10.3390/v6114683
Chicago/Turabian StyleMinor, Marissa M., and Betty L. Slagle. 2014. "Hepatitis B Virus HBx Protein Interactions with the Ubiquitin Proteasome System" Viruses 6, no. 11: 4683-4702. https://doi.org/10.3390/v6114683
APA StyleMinor, M. M., & Slagle, B. L. (2014). Hepatitis B Virus HBx Protein Interactions with the Ubiquitin Proteasome System. Viruses, 6(11), 4683-4702. https://doi.org/10.3390/v6114683