Current Perspectives on HIV-1 Antiretroviral Drug Resistance

Abstract
:
1. Introduction
2. Viral Fitness and Its Influence on Drug Resistance
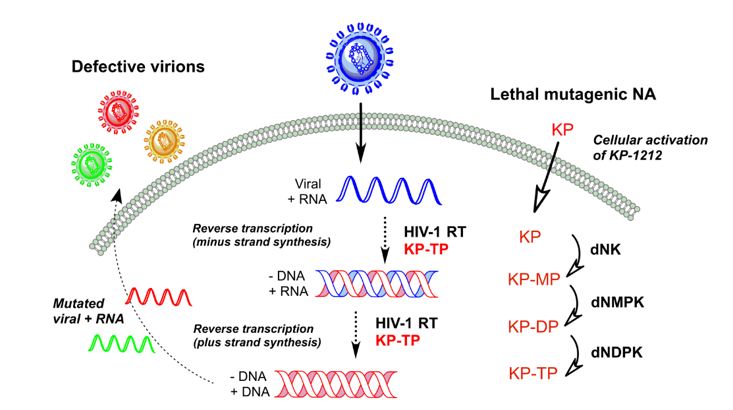
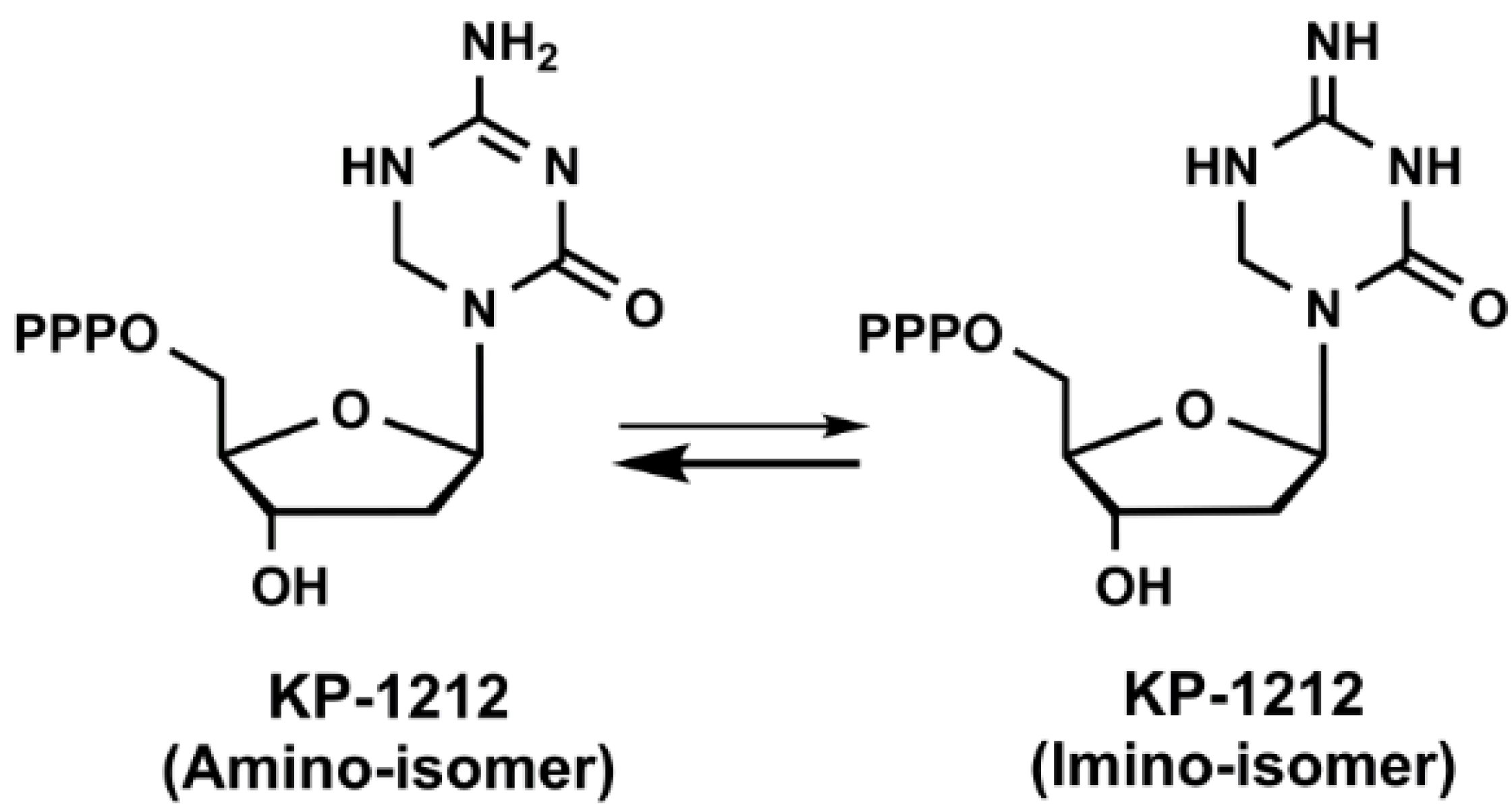
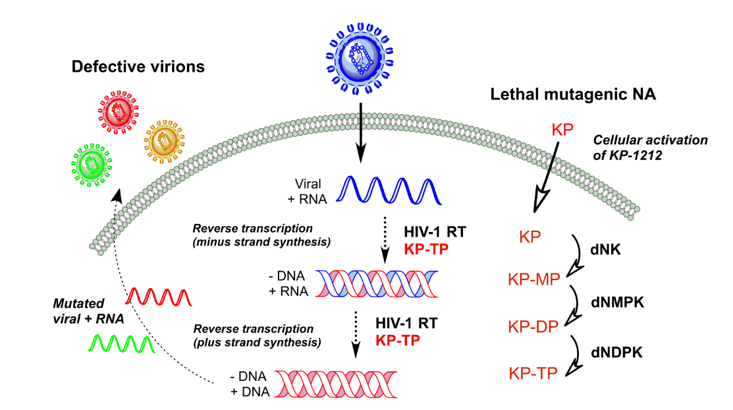
3. Lethal Mutagenesis

4. Antiretroviral Drug Classes and Acquired HIV-1 Drug Resistance
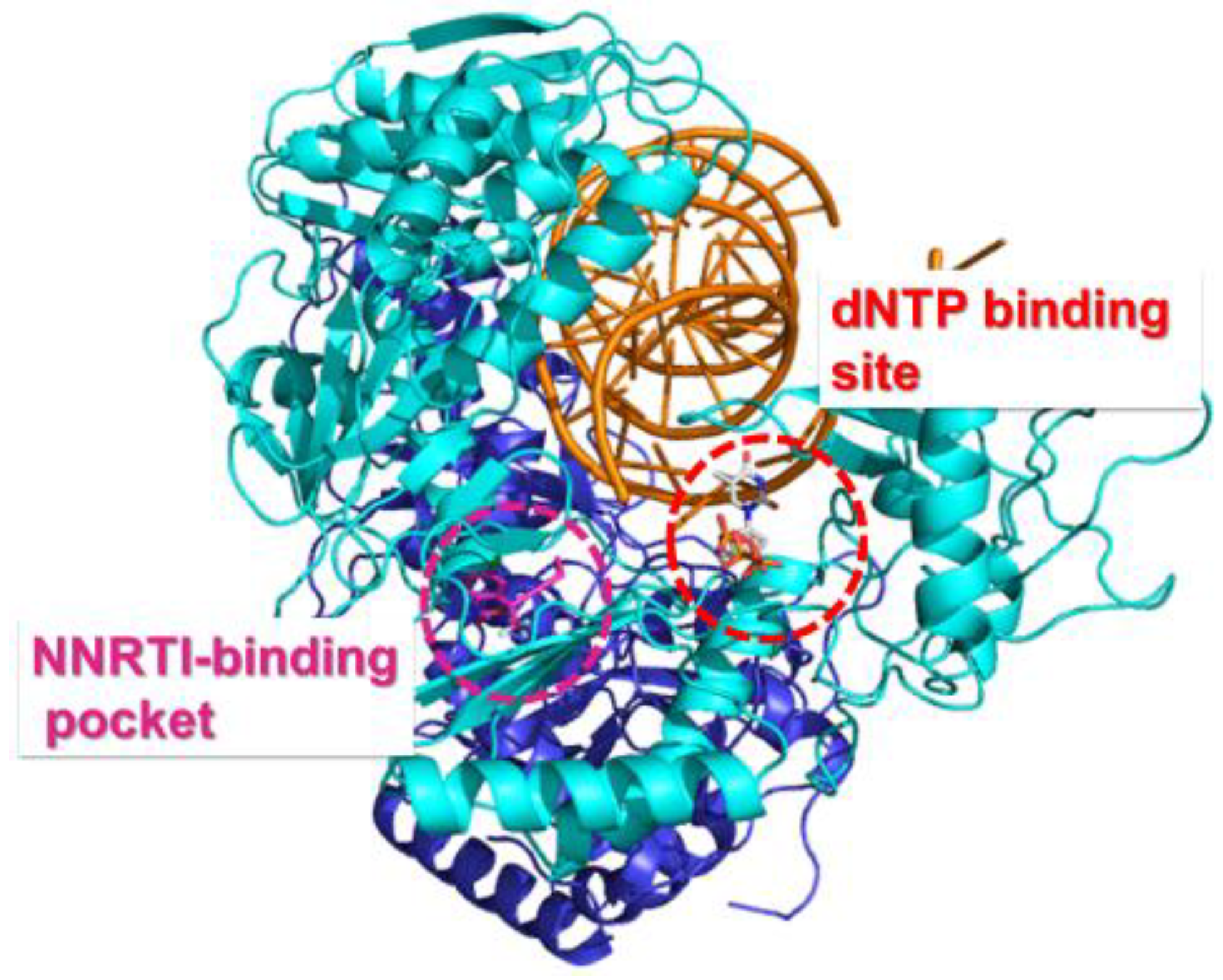
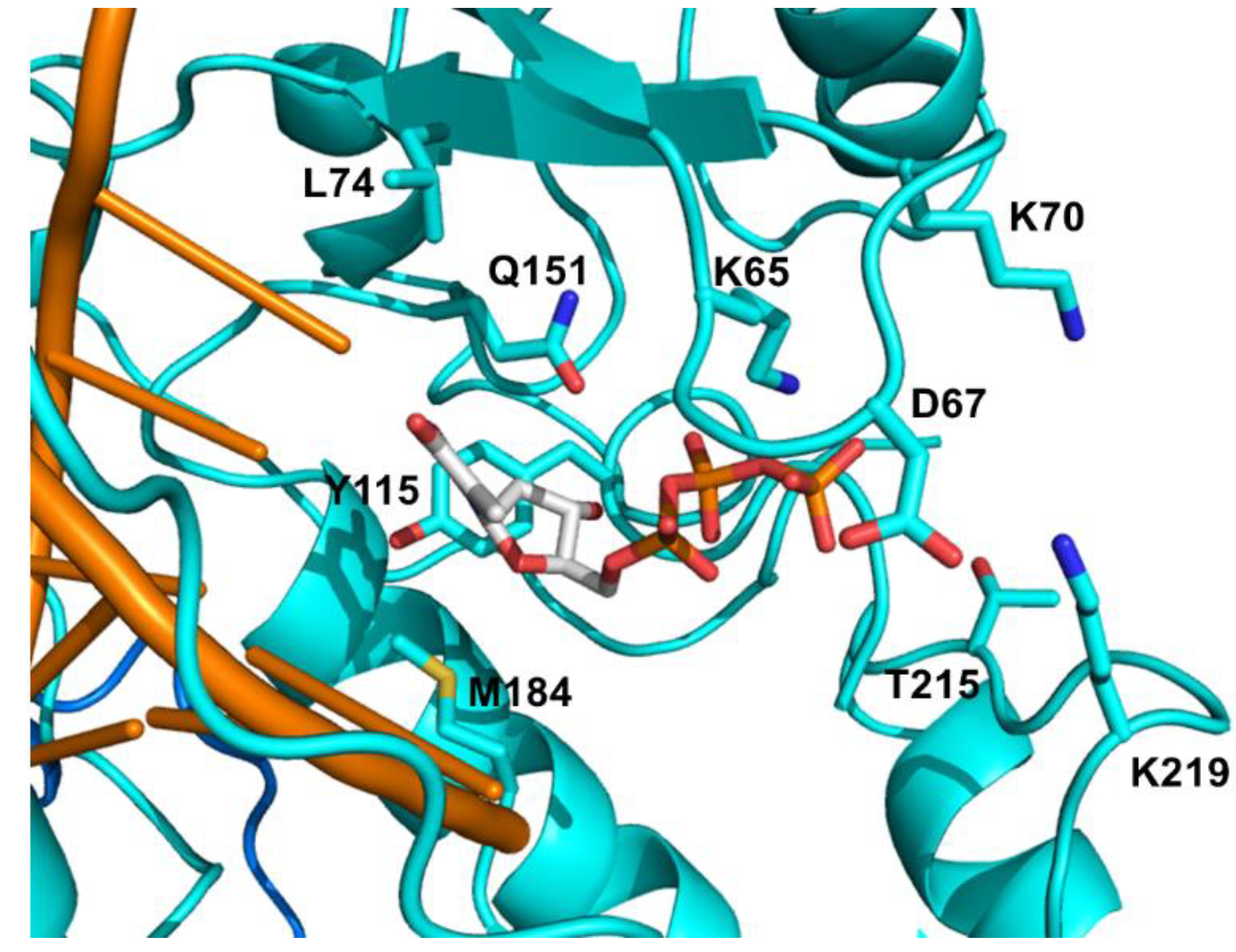
4.1. FDA Approved NRTIs and Mechanisms of Resistance
4.1.1. NRTI Specific Discriminatory Mutations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Consensus | Amino acid mutations associated with | ||||||
|---|---|---|---|---|---|---|---|
| FTC | 3TC | TDF | ABC | ddI | d4T | AZT | |
| M41 | L | L | |||||
| K65 | R | R | R | R | R | R | L |
| D67 | N | N | |||||
| K70 | E | E | E | E | E | R | R |
| L74 | V,I | V,I | V,I | V,I | |||
| Y115 | F | F | |||||
| M184 | V,I | V,I | V,I | V,I | |||
| L210 | W | W | |||||
| T215 | Y, F | Y, F | |||||
| K219 | Q,E | Q,E | |||||
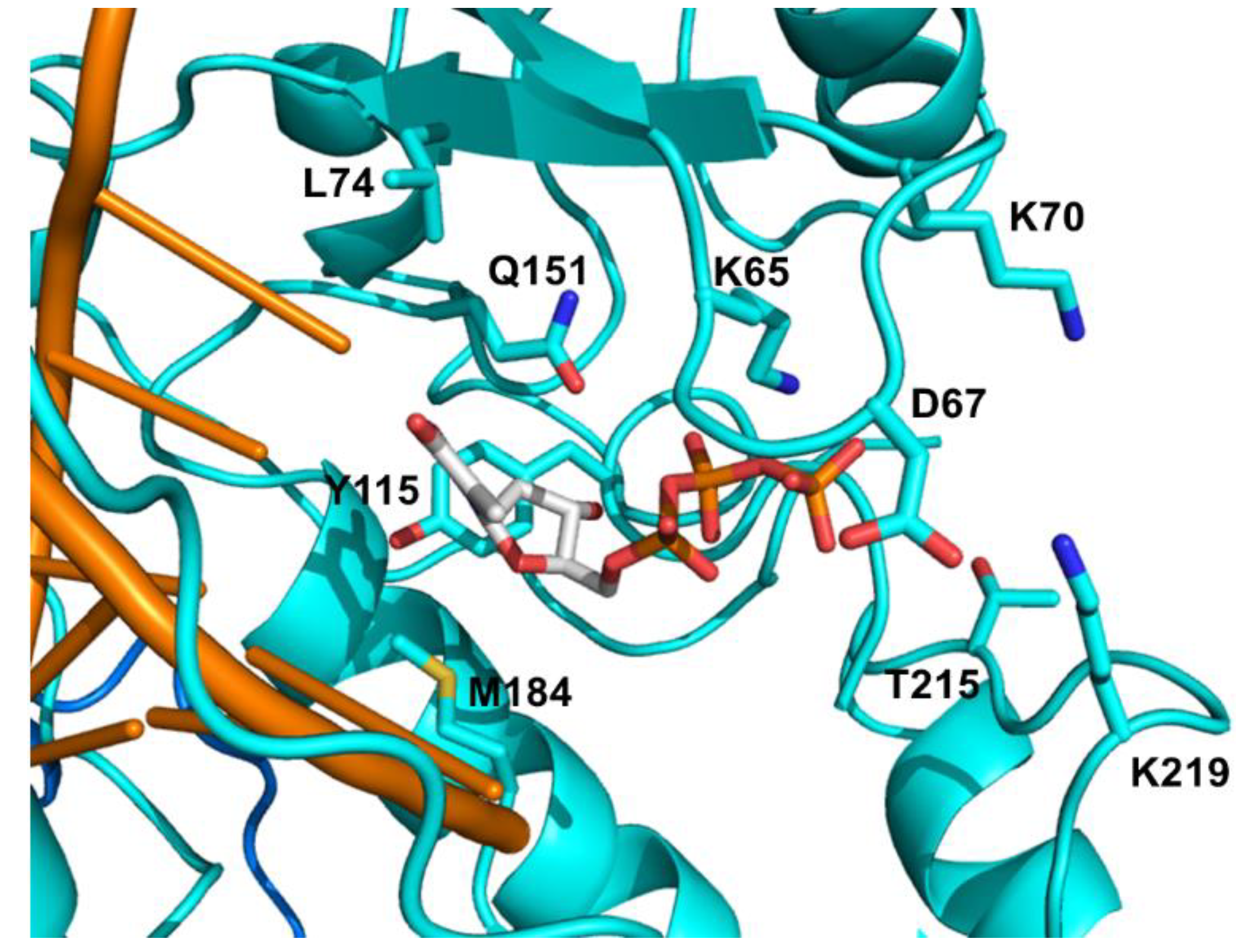
4.1.2. Excision of NRTIs
4.1.3. Multi-NRTI Resistance Mutations
| Consensus | Amino acid mutations associated with | ||
|---|---|---|---|
| 69 insertion complex | Q151 complex | TAMs | |
| M41 | L | L | |
| A62 | V | V | |
| D67 | N | ||
| T69 | Insert | ||
| K70 | R | R | |
| V75 | I | ||
| F77 | L | ||
| F116 | Y | ||
| Q151 | M | ||
| L210 | W | W | |
| T215 | Y,F | Y,F | |
| K219 | Q,E | Q,E | |
4.1.4. Investigational NRTIs and Emergence of Potential Resistance Mutations
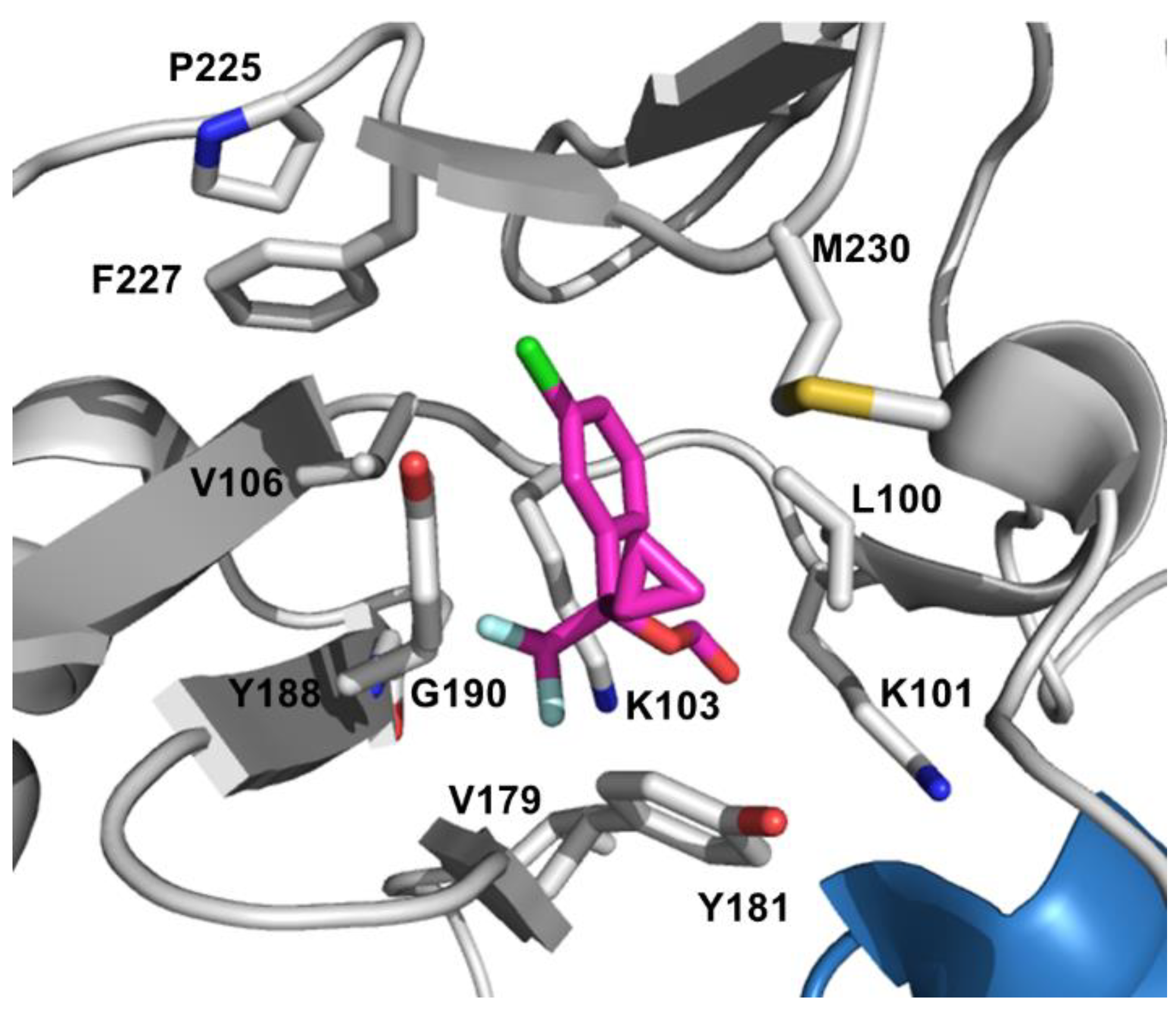
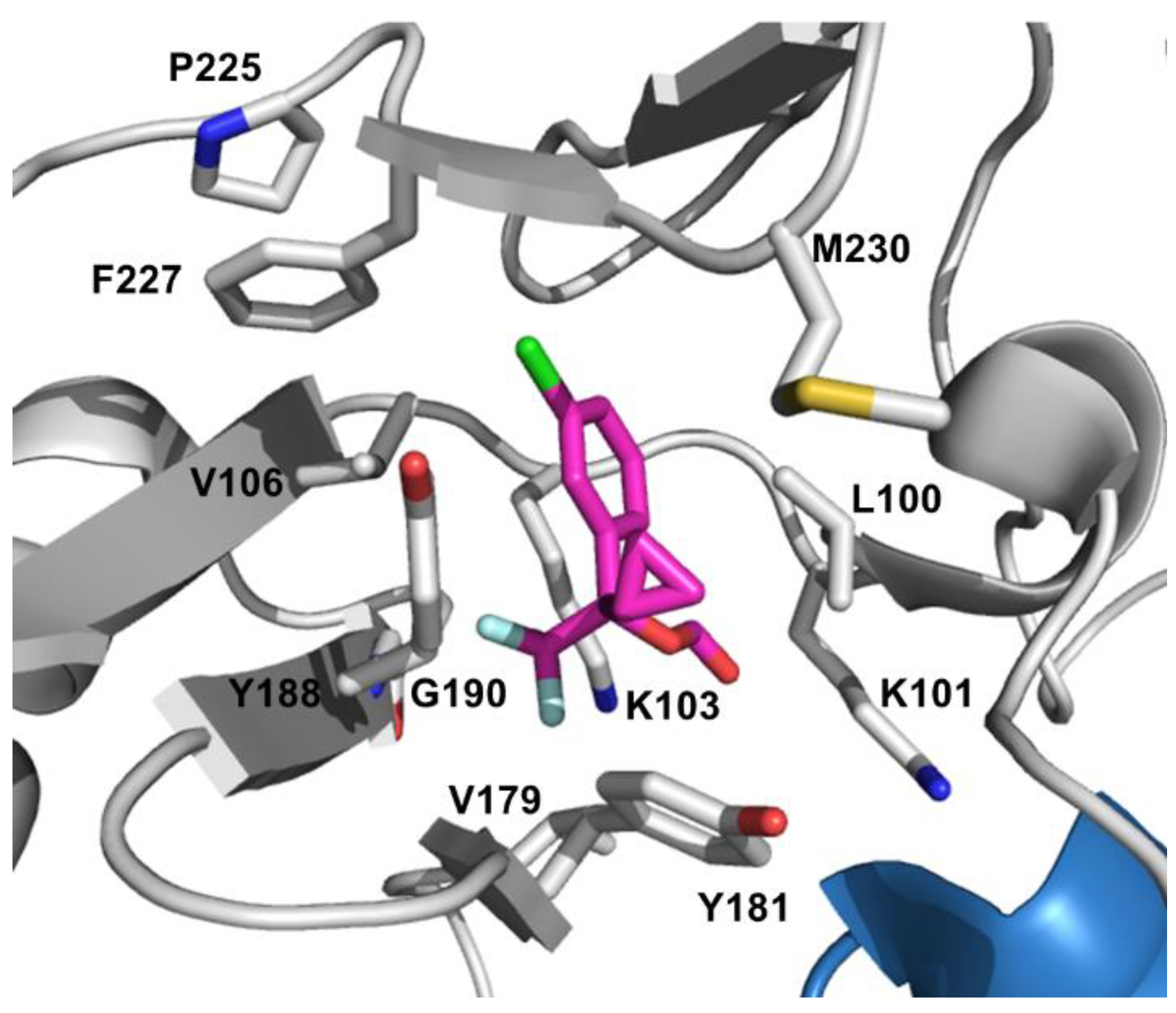
4.2. NNRTI Resistance
| Consensus | Amino acid mutations | |||
|---|---|---|---|---|
| NVP | EFV | ETR | RPV | |
| V90 | I | |||
| A98 | G | |||
| L100 | I | I | I | I |
| K101 | P,E,H | P,E,H | P,E,H | P,E,H |
| K103 | N,S | N,S | ||
| V106 | A,M | M, A | I | |
| V108 | I | |||
| E138 | A,G,K,Q | K,A,G,Q,R | ||
| V179 | D,E,F | D,E,F | D,F,T,E | L,D,E,F |
| Y181 | C,I,V | C | C,I,V | C,I,V |
| Y188 | C,L,H | L,C,H | L | |
| G190 | A,S,E,Q | A,S,E,Q | S,A,E,Q | A,S,E,Q |
| H221 | Y | |||
| P225 | H | |||
| F227 | L,C | L,C | C | C |
| M230 | L | L | L | I,L |
4.2.1. Resistance to First-generation NNRTIs
4.2.2. Resistance to Later-generation NNRTIs
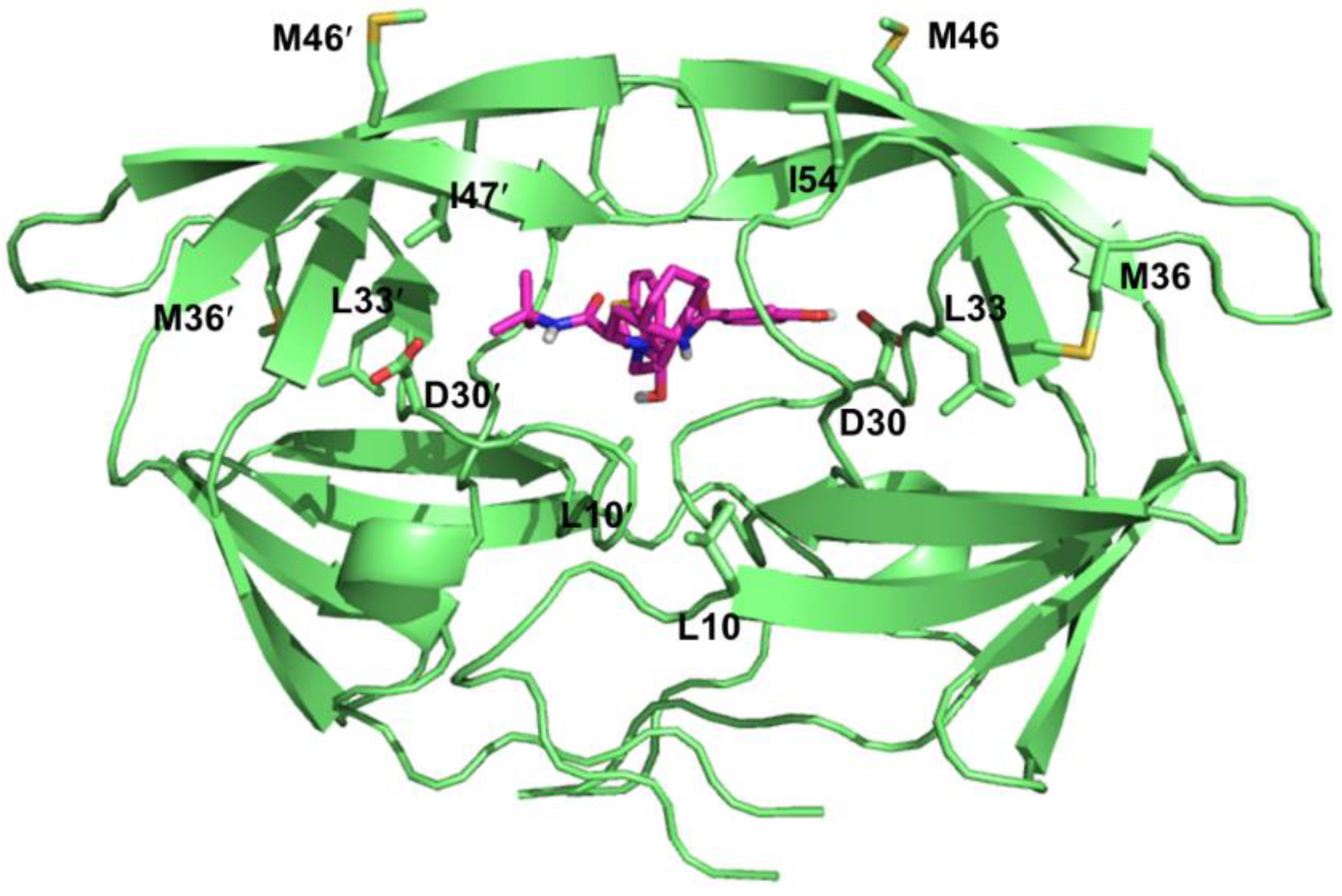
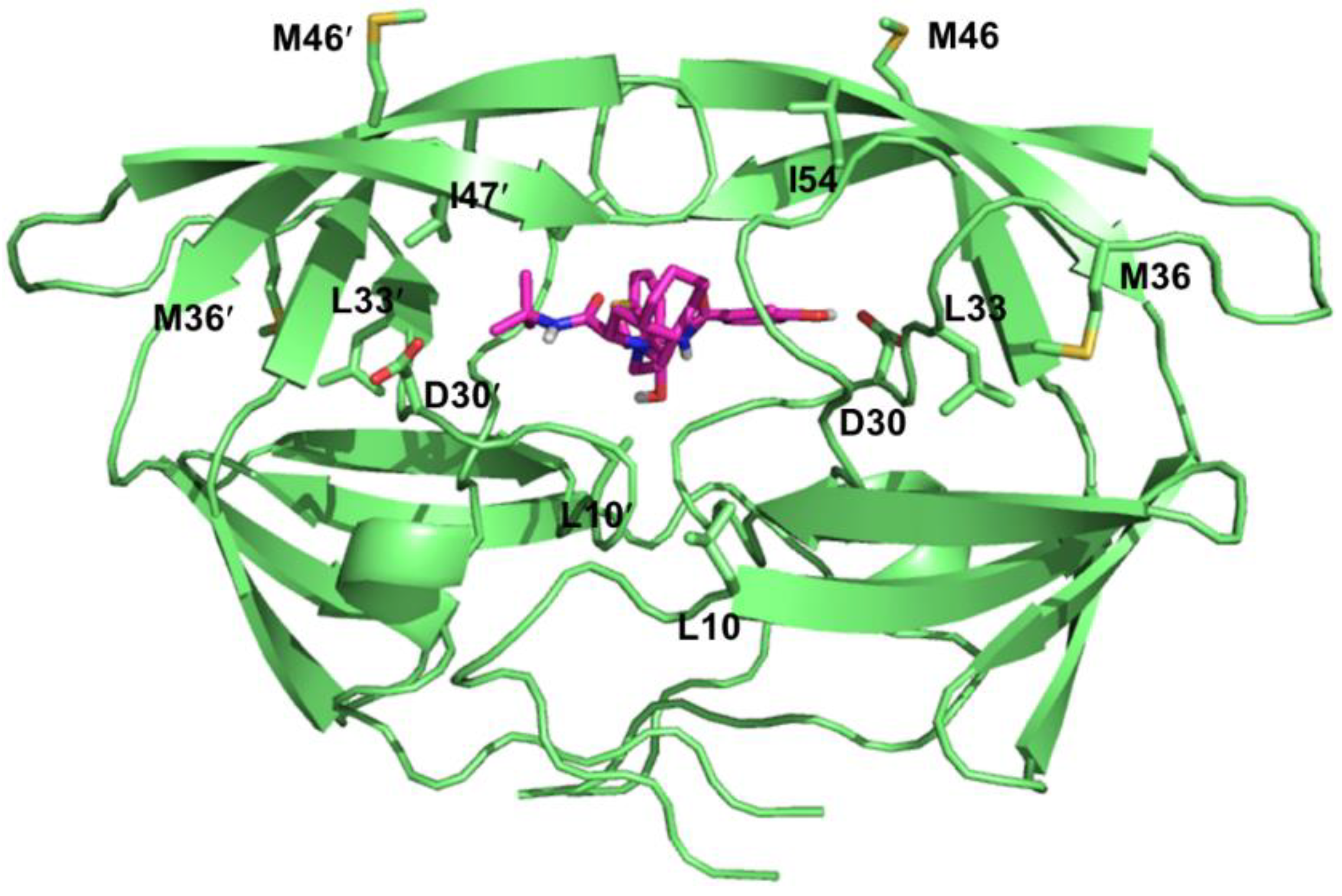
4.3. Resistance to HIV-1 Protease Inhibitors
| Consensus | Amino acid mutations | |||||||
|---|---|---|---|---|---|---|---|---|
| ATV/r | DRV/r | FPV/r | IDV/r | LPV/r | NFV | SQV/r | TPV/r | |
| L10 | I, F, V,C | F,I,R,V | I,R,V | I,R,V | F,I | I,R,V | I | |
| V11 | I | |||||||
| G16 | E | |||||||
| K20 | R,M,I,T,V | M,R | M,R | |||||
| L24 | I | I | I | I | ||||
| D30 | N | |||||||
| V32 | I | I | I | I | I | I | ||
| L33 | I,F,V | F | F | F | F | F | ||
| E34 | Q | |||||||
| M36 | I,L,V | I | I | I,L,V | ||||
| K43 | T | |||||||
| M46 | I,L | I,L | I,L | I,L | I,L | I,L | ||
| I47 | V | V,A | V,A | V,A | V,A | V,A | V,A | |
| G48 | V,M | V,M | V,M | V,M | ||||
| I50 | L | V | V | V | ||||
| F53 | L,Y | L | ||||||
| I54 | L,V,M,T,A | M,L | L,V,M,T,A | V,T,A,L,M | V,L,A,M,T,S | V,T,A,L,M | V,L,T,A,M | A,M,V,T |
| Q58 | E | |||||||
| D60 | E | |||||||
| I62 | V | V | ||||||
| L63 | P | |||||||
| I64 | L,M,V | |||||||
| H69 | K,R | |||||||
| A71 | V,I,T,L | V,T | V,T | V,T | V,T | |||
| G73 | C,S,T,A | S | S,A | S | S | |||
| L74 | P | P | ||||||
| L76 | V | V | V | V | ||||
| V77 | I | I | I | |||||
| V82 | A,T,F,I,S | F | A,F,S,T | A,F,T,S | A,F,T,S | A,F,T,S | A,F,T,S | L,T,S |
| N83 | D | |||||||
| I84 | V | V | V | V | V | V | V | V |
| I85 | V | |||||||
| N88 | S | S | D,S | S | ||||
| L89 | V | I,M,V | ||||||
| L90 | M | M | M | M | M | M | ||
| I93 | L,M | |||||||
4.4. Resistance to HIV-1 Integrase Inhibitors
| Consensus | Amino acid mutations | ||
|---|---|---|---|
| RAL | EVG | DTG | |
| T66 | A | I,A,K | |
| L74 | M | ||
| E92 | Q | Q,G | Q |
| T97 | A | A | |
| E138 | A,K | K,A | A,K |
| G140 | A,S,C | A,S,C | A,S,C |
| Y143 | R,H,C | ||
| S147 | G | ||
| Q148 | H,K,R | R,H,K | H,R,K |
| N155 | H | H | |
4.5. Resistance to Viral Entry Inhibitors
| Consensus | Amino acid mutations |
|---|---|
| Enfuvirtide | |
| G36 | D,S |
| I37 | V |
| V38 | A,M,E |
| Q39 | R |
| Q40 | H |
| N42 | T |
| N43 | D |
6. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Barre-Sinoussi, F. Hiv as the cause of aids. Lancet 1996, 348, 31–35. [Google Scholar] [CrossRef] [PubMed]
- WHO. Unaids Report on the Global Aids Epidemic 2013. Available online: http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf (accessed on 16 June 2014).
- De Clercq, E. Human viral diseases: What is next for antiviral drug discovery? Curr. Opin. Virol. 2012, 2, 572–579. [Google Scholar] [CrossRef]
- De Clercq, E. Anti-hiv drugs: 25 compounds approved within 25 years after the discovery of hiv. Int. J. Antimicrob. Agents 2009, 33, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Quinones-Mateu, M.E.; Mansky, L.M. Hiv-1 mutagenesis during antiretroviral therapy: Implications for successful drug treatment. Front. Biosci. 2005, 10, 743–750. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burke, D.S. Recombination in hiv: An important viral evolutionary strategy. Emerg. Infect. Dis. 1997, 3, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [PubMed]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and cd4 lymphocytes in hiv-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Ghosh, S.K.; Taylor, M.E.; Johnson, V.A.; Emini, E.A.; Deutsch, P.; Lifson, J.D.; Bonhoeffer, S.; Nowak, M.A.; Hahn, B.H.; et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 1995, 373, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Perelson, A.S.; Neumann, A.U.; Markowitz, M.; Leonard, J.M.; Ho, D.D. Hiv-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science 1996, 271, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in hiv-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.; Maier, R.; Vartanian, J.P.; Bocharov, G.; Jung, V.; Fischer, U.; Meese, E.; Wain-Hobson, S.; Meyerhans, A. Recombination: Multiply infected spleen cells in hiv patients. Nature 2002, 418, 144. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary hiv-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M. Is hiv unique or merely different? J. Acquir. Immune Defic. Syndr. 1989, 2, 1–9. [Google Scholar]
- Coffin, J.M. Hiv population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. Rna virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. On the nature of virus quasispecies. Trends Microbiol. 1996, 4, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.J.; de la Torre, J.C.; Steinhauer, D.A. Rna virus populations as quasispecies. Curr. Top. Microbiol. Immunol. 1992, 176, 1–20. [Google Scholar] [PubMed]
- Eigen, M.; Schuster, P. The Hypercycle: A Principle of Natural Self-Organization; Springer: Berlin, Germany, 1979. [Google Scholar]
- Domingo, E.; Wu, H.; Lien, E.J.; Lien, L.L.; Schultz, R.M.; Ram, V.J.; Spence, P.; Gupta, S.P.; Bhat, S.P.; Villarreal, E.C. Quasispecies and the development of new antiviral strategies. In Progress Drug Research; Jucker, E., Ed.; Birkhäuser Verlag: Basel, Switzerland, 2003; Volume 60, pp. 133–158. [Google Scholar]
- Biebricher, C.K.; Eigen, M. What is a quasispecies? Curr. Top. Microbiol. Immunol. 2006, 299, 1–31. [Google Scholar]
- Rouzine, I.M.; Brunet, E.; Wilke, C.O. The traveling-wave approach to asexual evolution: Muller’s ratchet and speed of adaptation. Theor. Popul. Biol. 2008, 73, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.J.; Sanjuan, R.; Wilke, C.O. Theory of lethal mutagenesis for viruses. J. Virol. 2007, 81, 2930–2939. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. Retrovirus mutation rates and their role in genetic variation. J. Gen. Virol. 1998, 79, (Pt 6). 1337–1345. [Google Scholar] [PubMed]
- Nowak, M.A.; Anderson, R.M.; McLean, A.R.; Wolfs, T.F.; Goudsmit, J.; May, R.M. Antigenic diversity thresholds and the development of aids. Science 1991, 254, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. Hiv mutagenesis and the evolution of antiretroviral drug resistance. Drug Resist. Updates 2002, 5, 219–223. [Google Scholar] [CrossRef]
- Preston, B.D.; Dougherty, J.P. Mechanisms of retroviral mutation. Trends Microbiol. 1996, 4, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L. Molecular basis of fidelity of DNA synthesis and nucleotide specificity of retroviral reverse transcriptases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 71, 91–147. [Google Scholar] [PubMed]
- Jonckheere, H.; Anne, J.; de Clercq, E. The hiv-1 reverse transcription (rt) process as target for rt inhibitors. Med. Res. Rev. 2000, 20, 129–154. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The accuracy of reverse transcriptase from hiv-1. Science 1988, 242, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Preston, B.D.; Poiesz, B.J.; Loeb, L.A. Fidelity of hiv-1 reverse transcriptase. Science 1988, 242, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Preston, B.D.; Johnston, L.A.; Soni, A.; Loeb, L.A.; Kunkel, T.A. Fidelity of two retroviral reverse transcriptases during DNA-dependent DNA synthesis in vitro. Mol. Cell. Biol. 1989, 9, 469–476. [Google Scholar] [PubMed]
- Steinhauer, D.A.; Domingo, E.; Holland, J.J. Lack of evidence for proofreading mechanisms associated with an rna virus polymerase. Gene 1992, 122, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Host rna polymerase ii makes minimal contributions to retroviral frame-shift mutations. J. Gen. Virol. 2004, 85, 2389–2395. [Google Scholar] [CrossRef]
- O’Neil, P.K.; Sun, G.; Yu, H.; Ron, Y.; Dougherty, J.P.; Preston, B.D. Mutational analysis of hiv-1 long terminal repeats to explore the relative contribution of reverse transcriptase and rna polymerase ii to viral mutagenesis. J. Biol. Chem. 2002, 277, 38053–38061. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Liddament, M.T. Retroviral restriction by apobec proteins. Nat. Rev. Immunol. 2004, 4, 868–877. [Google Scholar] [CrossRef]
- Malim, M.H.; Emerman, M. Hiv-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Greene, W.C. The apobec3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits hiv-1 infection and is suppressed by the viral vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human apobec3g through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Mariani, R.; Chen, D.; Schrofelbauer, B.; Navarro, F.; Konig, R.; Bollman, B.; Munk, C.; Nymark-McMahon, H.; Landau, N.R. Species-specific exclusion of apobec3g from hiv-1 virions by vif. Cell 2003, 114, 21–31. [Google Scholar] [CrossRef]
- KewalRamani, V.N.; Coffin, J.M. Virology. Weapons of mutational destruction. Science 2003, 301, 923–925. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, A.S. DNA-cytosine deaminases: From antibody maturation to antiviral defense. DNA Repair 2004, 3, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Aires da Silva, F.; Santa-Marta, M.; Freitas-Vieira, A.; Mascarenhas, P.; Barahona, I.; Moniz-Pereira, J.; Gabuzda, D.; Goncalves, J. Camelized rabbit-derived vh single-domain intrabodies against vif strongly neutralize hiv-1 infectivity. J. Mol. Biol. 2004, 340, 525–542. [Google Scholar]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Lecossier, D.; Bouchonnet, F.; Clavel, F.; Hance, A.J. Hypermutation of hiv-1 DNA in the absence of the vif protein. Science 2003, 300, 1112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase cem15 induces hypermutation in newly synthesized hiv-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.P. Death by deamination: A novel host restriction system for hiv-1. Cell 2003, 114, 281–283. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lavens, D.; Peelman, F.; van der Heyden, J.; Uyttendaele, I.; Catteeuw, D.; Verhee, A.; van Schoubroeck, B.; Kurth, J.; Hallenberger, S.; Clayton, R.; et al. Definition of the interacting interfaces of apobec3g and hiv-1 vif using mappit mutagenesis analysis. Nucleic Acids Res. 2010, 38, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, J.; Santa-Marta, M. Hiv-1 vif and apobec3g: Multiple roads to one goal. Retrovirology 2004, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Cadima-Couto, I.; Goncalves, J. Towards inhibition of vif-apobec3g interaction: Which protein to target? Adv. Virol. 2010, 2010, 649315. [Google Scholar]
- Munk, C.; Jensen, B.E.; Zielonka, J.; Haussinger, D.; Kamp, C. Running loose or getting lost: How hiv-1 counters and capitalizes on apobec3-induced mutagenesis through its vif protein. Viruses 2012, 4, 3132–3161. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.A. The rate of adaptation in asexuals. Genetics 2000, 155, 961–968. [Google Scholar] [PubMed]
- Zhuang, J.; Jetzt, A.E.; Sun, G.; Yu, H.; Klarmann, G.; Ron, Y.; Preston, B.D.; Dougherty, J.P. Human immunodeficiency virus type 1 recombination: Rate, fidelity, and putative hot spots. J. Virol. 2002, 76, 11273–11282. [Google Scholar] [CrossRef] [PubMed]
- Mulder, L.C.; Harari, A.; Simon, V. Cytidine deamination induced hiv-1 drug resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 5501–5506. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Temin, H.M. Retroviral recombination and reverse transcription. Science 1990, 250, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tang, L.Y.; Li, T.; Ma, Y.; Sapp, C.M. Most retroviral recombinations occur during minus-strand DNA synthesis. J. Virol. 2000, 74, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jetzt, A.E.; Ron, Y.; Preston, B.D.; Dougherty, J.P. The nature of human immunodeficiency virus type 1 strand transfers. J. Biol. Chem. 1998, 273, 28384–28391. [Google Scholar] [CrossRef] [PubMed]
- Jetzt, A.E.; Yu, H.; Klarmann, G.J.; Ron, Y.; Preston, B.D.; Dougherty, J.P. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 2000, 74, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.N.; Aldrovandi, G.M.; Kutsch, O.; Shaw, G.M. Dynamics of hiv-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4204–4209. [Google Scholar] [CrossRef] [PubMed]
- Althaus, C.L.; Bonhoeffer, S. Stochastic interplay between mutation and recombination during the acquisition of drug resistance mutations in human immunodeficiency virus type 1. J. Virol. 2005, 79, 13572–13578. [Google Scholar] [CrossRef] [PubMed]
- Nora, T.; Charpentier, C.; Tenaillon, O.; Hoede, C.; Clavel, F.; Hance, A.J. Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment. J. Virol. 2007, 81, 7620–7628. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, M.T.; Althaus, C.L.; Muller, V.; Bonhoeffer, S. Recombination in hiv and the evolution of drug resistance: For better or for worse? BioEssays 2004, 26, 180–188. [Google Scholar] [CrossRef]
- Perales, C.; Iranzo, J.; Manrubia, S.C.; Domingo, E. The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol. 2012, 20, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Mullins, J.I.; Jensen, M.A. Evolutionary dynamics of hiv-1 and the control of aids. Curr. Top. Microbiol. Immunol. 2006, 299, 171–192. [Google Scholar] [PubMed]
- Gao, F.; Chen, Y.; Levy, D.N.; Conway, J.A.; Kepler, T.B.; Hui, H. Unselected mutations in the human immunodeficiency virus type 1 genome are mostly nonsynonymous and often deleterious. J. Virol. 2004, 78, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Essigmann, J.M.; Kazazi, F.; Zhang, J.; Rose, K.D.; Mullins, J.I. Lethal mutagenesis of hiv with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA 1999, 96, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Grande-Perez, A.; Lazaro, E.; Lowenstein, P.; Domingo, E.; Manrubia, S.C. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. USA 2005, 102, 4448–4452. [Google Scholar] [CrossRef] [PubMed]
- Huber-Ruano, I.; Pastor-Anglada, M. Transport of nucleoside analogs across the plasma membrane: A clue to understanding drug-induced cytotoxicity. Curr. Drug Metab. 2009, 10, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.; Wang, L. The role of the cellular deoxynucleoside kinases in activation of nucleoside analogs used in chemotherapy. In Recent Advances in Nucleosides: Chemistry and Chemotherapy; Chu, C.K., Ed.; Elsevier: New York, NY, USA, 2002; pp. 465–468. [Google Scholar]
- Loeb, L.A.; Mullins, J.I. Lethal mutagenesis of hiv by mutagenic ribonucleoside analogs. AIDS Res. Hum. Retrovir. 2000, 16, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kamath-Loeb, A.S.; Hizi, A.; Tabone, J.; Solomon, M.S.; Loeb, L.A. Inefficient repair of rna x DNA hybrids. Eur. J. Biochem./FEBS 1997, 250, 492–501. [Google Scholar] [CrossRef]
- Suen, W.; Spiro, T.G.; Sowers, L.C.; Fresco, J.R. Identification by uv resonance raman spectroscopy of an imino tautomer of 5-hydroxy-2'-deoxycytidine, a powerful base analog transition mutagen with a much higher unfavored tautomer frequency than that of the natural residue 2'-deoxycytidine. Proc. Natl. Acad. Sci. USA 1999, 96, 4500–4505. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Loeb, L.A.; Preston, B.D. Lethal mutagenesis of hiv. Virus Res. 2005, 107, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Daifuku, R.; Loeb, L.A. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 2004, 58, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Kantarjian, H. Azacitidine. Nat. Rev. 2005, Suppl, S6–S7. [Google Scholar]
- Bouchard, J.; Walker, M.C.; Leclerc, J.M.; Lapointe, N.; Beaulieu, R.; Thibodeau, L. 5-azacytidine and 5-azadeoxycytidine inhibit human immunodeficiency virus type 1 replication in vitro. Antimicrob. Agents Chemother. 1990, 34, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.J.; Clouser, C.L.; Patterson, S.; Mansky, L.M. 5-azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 2009, 83, 11950–11958. [Google Scholar] [CrossRef] [PubMed]
- Clouser, C.L.; Patterson, S.E.; Mansky, L.M. Exploiting drug repositioning for discovery of a novel hiv combination therapy. J. Virol. 2010, 84, 9301–9309. [Google Scholar] [CrossRef] [PubMed]
- Rogstad, D.K.; Herring, J.L.; Theruvathu, J.A.; Burdzy, A.; Perry, C.C.; Neidigh, J.W.; Sowers, L.C. Chemical decomposition of 5-aza-2'-deoxycytidine (decitabine): Kinetic analyses and identification of products by nmr, hplc, and mass spectrometry. Chem. Res. Toxicol. 2009, 22, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Grusby, L.; Laird, P.W.; Magge, S.N.; Moeller, B.J.; Jaenisch, R. Mutagenicity of 5-aza-2'-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci. USA 1997, 94, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Pathak, V.K.; Temin, H.M. 5-azacytidine and rna secondary structure increase the retrovirus mutation rate. J. Virol. 1992, 66, 3093–3100. [Google Scholar]
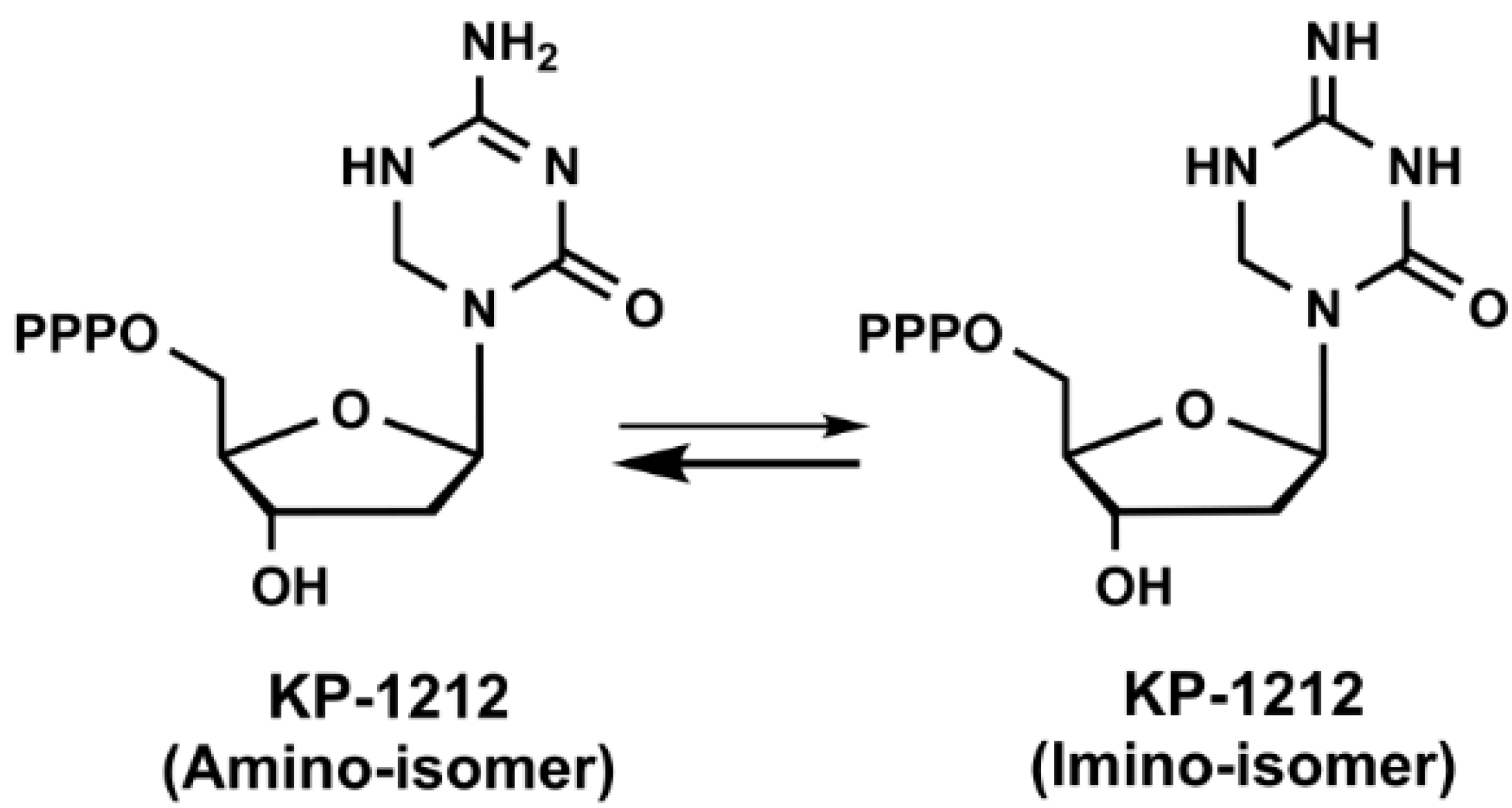
- Harris, K.S.; Brabant, W.; Styrchak, S.; Gall, A.; Daifuku, R. Kp-1212/1461, a nucleoside designed for the treatment of hiv by viral mutagenesis. Antivir. Res. 2005, 67, 1–9. [Google Scholar] [CrossRef]
- Murakami, E.; Basavapathruni, A.; Bradley, W.D.; Anderson, K.S. Mechanism of action of a novel viral mutagenic covert nucleotide: Molecular interactions with hiv-1 reverse transcriptase and host cell DNA polymerases. Antivir. Res. 2005, 67, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Ray, A.S.; Hanes, J.; Suo, Z.; Colacino, J.M.; Anderson, K.S.; Johnson, K.A. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 2001, 276, 40847–40857. [Google Scholar] [CrossRef]
- Clay, P.G.; McRae, M.; Laurent, J.P. Safety, tolerability, and pharmacokinetics of kp-1461 in phase i clinical studies: A single oral dose study in non-hiv-infected adults, and a 14-day dose-escalating study in highly antiretroviral-experienced hiv-infected adults. J. Int. Assoc. Physicians AIDS Care 2011, 10, 232–238. [Google Scholar] [CrossRef]
- Mullins, J.I.; Heath, L.; Hughes, J.P.; Kicha, J.; Styrchak, S.; Wong, K.G.; Rao, U.; Hansen, A.; Harris, K.S.; Laurent, J.P.; et al. Mutation of hiv-1 genomes in a clinical population treated with the mutagenic nucleoside kp1461. PLoS One 2011, 6, e15135. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.; Clay, P.; Redfield, R.; Lalezari, J.; Liporace, R.; Schneider, S.; Sension, M.; McRae, M.; Laurent, J.P. Safety, tolerability, and efficacy of kp-1461 as monotherapy for 124 days in antiretroviral-experienced, hiv type 1-infected subjects. AIDS Res. Hum. Retrovir. 2013, 29, 250–255. [Google Scholar]
- Mehellou, Y.; de Clercq, E. Twenty-six years of anti-hiv drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of hiv-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Kaushik, N.; McCormick, S.; Borkow, G.; Parniak, M.A. Phenotypic mechanism of hiv-1 resistance to 3'-azido-3'-deoxythymidine (azt): Increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 1998, 37, 15908–15917. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Matsuura, S.E.; So, A.G.; Scott, W.A. Unblocking of chain-terminated primer by hiv-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. USA 1998, 95, 13471–13476. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of hiv-1 reverse transcriptase: Implicaiton fro drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of hiv-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Selmi, B.; Boretto, J.; Sarfati, S.R.; Guerreiro, C.; Canard, B. Mechanism-based suppression of dideoxynucleotide resistance by k65r human immunodeficiency virus reverse transcriptase using an alpha-boranophosphate nucleoside analogue. J. Biol Chem. 2001, 276, 48466–48472. [Google Scholar] [PubMed]
- Deval, J.; Alvarez, K.; Selmi, B.; Bermond, M.; Boretto, J.; Guerreiro, C.; Mulard, L.; Canard, B. Mechanistic insights into the suppression of drug resistance by human immunodeficiency virus type 1 reverse transcriptase using alpha-boranophosphate nucleoside analogs. J. Biol. Chem. 2005, 280, 3838–3846. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N.; Sheen, C.W.; Zelina, S.; Torres, P.S.; Parikh, U.M.; Mellors, J.W. Molecular mechanism by which the k70e mutation in human immunodeficiency virus type 1 reverse transcriptase confers resistance to nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2007, 51, 48–53. [Google Scholar] [CrossRef]
- Deval, J.; White, K.L.; Miller, M.D.; Parkin, N.T.; Courcambeck, J.; Halfon, P.; Selmi, B.; Boretto, J.; Canard, B. Mechanistic basis for reduced viral and enzymatic fitness of hiv-1 reverse transcriptase containing both k65r and m184v mutations. J. Biol. Chem. 2004, 279, 509–516. [Google Scholar] [CrossRef]
- Feng, J.Y.; Myrick, F.T.; Margot, N.A.; Mulamba, G.B.; Rimsky, L.; Borroto-Esoda, K.; Selmi, B.; Canard, B. Virologic and enzymatic studies revealing the mechanism of k65r- and q151m-associated hiv-1 drug resistance towards emtricitabine and lamivudine. Nucleosides Nucleotides Nucleic Acids 2006, 25, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Das, K.; Clark, A.D., Jr.; Ding, J.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Lamivudine (3tc) resistance in hiv-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc. Natl. Acad. Sci. USA 1999, 96, 10027–10032. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.S.; Murakami, E.; Peterson, C.N.; Shi, J.; Schinazi, R.F.; Anderson, K.S. Interactions of enantiomers of 2',3'-didehydro-2',3'-dideoxy-fluorocytidine with wild type and m184v mutant hiv-1 reverse transcriptase. Antivir. Res. 2002, 56, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.Q.; Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. The role of steric hindrance in 3tc resistance of human immunodeficiency virus type-1 reverse transcriptase. J. Mol. Biol. 2000, 300, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liang, C.; Gotte, M.; Wainberg, M.A. Negative effect of the m184v mutation in hiv-1 reverse transcriptase on initiation of viral DNA synthesis. Virology 2003, 311, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Van Cor-Hosmer, S.K.; Daddacha, W.; Kim, B. Mechanistic interplay among the m184i hiv-1 reverse transcriptase mutant, the central polypurine tract, cellular dntp concentrations and drug sensitivity. Virology 2010, 406, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, A.G.; Charpentier, C.; Wirden, M.; Landman, R.; Valantin, M.A.; Simon, A.; Katlama, C.; Yeni, P.; Descamps, D.; Aubron-Olivier, C.; et al. Resistance profiles of emtricitabine and lamivudine in tenofovir-containing regimens. J. Antimicrob. Chemother. 2012, 67, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.S.; Basavapathruni, A.; Anderson, K.S. Mechanistic studies to understand the progressive development of resistance in human immunodeficiency virus type 1 reverse transcriptase to abacavir. J. Biol. Chem. 2002, 277, 40479–40490. [Google Scholar] [CrossRef] [PubMed]
- Frangeul, A.; Bussetta, C.; Deval, J.; Barral, K.; Alvarez, K.; Canard, B. Gln151 of hiv-1 reverse transcriptase acts as a steric gate towards clinically relevant acyclic phosphonate nucleotide analogues. Antivir. Therapy 2008, 13, 115–124. [Google Scholar]
- Mbisa, J.L.; Gupta, R.K.; Kabamba, D.; Mulenga, V.; Kalumbi, M.; Chintu, C.; Parry, C.M.; Gibb, D.M.; Walker, S.A.; Cane, P.A.; et al. The evolution of hiv-1 reverse transcriptase in route to acquisition of q151m multi-drug resistance is complex and involves mutations in multiple domains. Retrovirology 2011, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, A.; Kodama, E.N.; Schuckmann, M.M.; Kirby, K.A.; Michailidis, E.; Sakagami, Y.; Oka, S.; Singh, K.; Sarafianos, S.G. K70q adds high-level tenofovir resistance to “q151m complex” hiv reverse transcriptase through the enhanced discrimination mechanism. PLoS One 2011, 6, e16242. [Google Scholar] [CrossRef] [PubMed]
- Kisic, M.; Mendieta, J.; Puertas, M.C.; Parera, M.; Martinez, M.A.; Martinez-Picado, J.; Menendez-Arias, L. Mechanistic basis of zidovudine hypersusceptibility and lamivudine resistance conferred by the deletion of codon 69 in the hiv-1 reverse transcriptase coding region. J. Mol. Biol. 2008, 382, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Das, K.; Hsiou, Y.; Sarafianos, S.G.; Clark, A.D., Jr.; Jacobo-Molina, A.; Tantillo, C.; Hughes, S.H.; Arnold, E. Structure and functional implications of the polymerase active site region in a complex of hiv-1 rt with a double-stranded DNA template-primer and an antibody fab fragment at 2.8 a resolution. J. Mol. Biol. 1998, 284, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Lanier, E.R.; Givens, N.; Stone, C.; Griffin, P.; Gibb, D.; Walker, S.; Tisdale, M.; Irlbeck, D.; Underwood, M.; St Clair, M.; et al. Effect of concurrent zidovudine use on the resistance pathway selected by abacavir-containing regimens. HIV Med. 2004, 5, 394–399. [Google Scholar] [CrossRef]
- Waters, L.; Nelson, M.; Mandalia, S.; Bower, M.; Powles, T.; Gazzard, B.; Stebbing, J. The risks and incidence of k65r and l74v mutations and subsequent virologic responses. Clin. Infect. Dis. 2008, 46, 96–100. [Google Scholar] [CrossRef]
- Miller, M.D.; Margot, N.; Lu, B.; Zhong, L.; Chen, S.S.; Cheng, A.; Wulfsohn, M. Genotypic and phenotypic predictors of the magnitude of response to tenofovir disoproxil fumarate treatment in antiretroviral-experienced patients. J. Infect. Dis. 2004, 189, 837–846. [Google Scholar] [CrossRef]
- Menendez-Arias, L. Molecular basis of human immunodeficiency virus drug resistance: An update. Antivir. Res. 2010, 85, 210–231. [Google Scholar] [CrossRef]
- Meyer, P.R.; Matsuura, S.E.; Tolun, A.A.; Pfeifer, I.; So, A.G.; Mellors, J.W.; Scott, W.A. Effects of specific zidovudine resistance mutations and substrate structure on nucleotide-dependent primer unblocking by human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 2002, 46, 1540–1545. [Google Scholar] [CrossRef]
- Ray, A.S.; Murakami, E.; Basavapathruni, A.; Vaccaro, J.A.; Ulrich, D.; Chu, C.K.; Schinazi, R.F.; Anderson, K.S. Probing the molecular mechanisms of azt drug resistance mediated by hiv-1 reverse transcriptase using a transient kinetic analysis. Biochemistry 2003, 42, 8831–8841. [Google Scholar] [CrossRef]
- Menendez-Arias, L. Mechanisms of resistance to nucleoside analogue inhibitors of hiv-1 reverse transcriptase. Virus Res. 2008, 134, 124–146. [Google Scholar] [CrossRef] [PubMed]
- Larder, B.A.; Coates, K.E.; Kemp, S.D. Zidovudine-resistant human immunodeficiency virus selected by passage in cell culture. J. Virol. 1991, 65, 5232–5236. [Google Scholar]
- Hanna, G.J.; Johnson, V.A.; Kuritzkes, D.R.; Richman, D.D.; Brown, A.J.; Savara, A.V.; Hazelwood, J.D.; D’Aquila, R.T. Patterns of resistance mutations selected by treatment of human immunodeficiency virus type 1 infection with zidovudine, didanosine, and nevirapine. J. Infect. Dis. 2000, 181, 904–911. [Google Scholar] [CrossRef]
- Marcelin, A.G.; Delaugerre, C.; Wirden, M.; Viegas, P.; Simon, A.; Katlama, C.; Calvez, V. Thymidine analogue reverse transcriptase inhibitors resistance mutations profiles and association to other nucleoside reverse transcriptase inhibitors resistance mutations observed in the context of virological failure. J. Med. Virol. 2004, 72, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Selective excision of aztmp by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 2001, 75, 4832–4842. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Matsuura, S.E.; Schinazi, R.F.; So, A.G.; Scott, W.A. Differential removal of thymidine nucleotide analogues from blocked DNA chains by human immunodeficiency virus reverse transcriptase in the presence of physiological concentrations of 2'-deoxynucleoside triphosphates. Antimicrob. Agents Chemother. 2000, 44, 3465–3472. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Matsuura, S.E.; Mian, A.M.; So, A.G.; Scott, W.A. A mechanism of azt resistance: An increase in nucleotide-dependent primer unblocking by mutant hiv-1 reverse transcriptase. Mol. Cell 1999, 4, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Lu, C.D.; Sharma, S.K.; Matsuura, S.; So, A.G.; Scott, W.A. Nucleotide-induced stable complex formation by hiv-1 reverse transcriptase. Biochemistry 1997, 36, 5749–5757. [Google Scholar] [CrossRef]
- Marchand, B.; Gotte, M. Site-specific footprinting reveals differences in the translocation status of hiv-1 reverse transcriptase. Implications for polymerase translocation and drug resistance. J. Biol. Chem. 2003, 278, 35362–35372. [Google Scholar] [CrossRef]
- Marchand, B.; White, K.L.; Ly, J.K.; Margot, N.A.; Wang, R.; McDermott, M.; Miller, M.D.; Gotte, M. Effects of the translocation status of human immunodeficiency virus type 1 reverse transcriptase on the efficiency of excision of tenofovir. Antimicrob. Agents Chemother. 2007, 51, 2911–2919. [Google Scholar] [CrossRef]
- Iyidogan, P.; Anderson, K.S. Understanding the molecular mechanism of sequence dependent tenofovir removal by hiv-1 reverse transcriptase: Differences in primer binding site versus polypurine tract. Antivir. Res. 2012, 95, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Das, K.; Han, Q.; Bauman, J.D.; Clark, A.D., Jr.; Hou, X.; Frenkel, Y.V.; Gaffney, B.L.; Jones, R.A.; Boyer, P.L.; et al. Structural basis of hiv-1 resistance to azt by excision. Nat. Struct. Mol. Biol. 2010, 17, 1202–1209. [Google Scholar] [CrossRef]
- Dharmasena, S.; Pongracz, Z.; Arnold, E.; Sarafianos, S.G.; Parniak, M.A. 3'-azido-3'-deoxythymidine-(5')-tetraphospho-(5')-adenosine, the product of atp-mediated excision of chain-terminating aztmp, is a potent chain-terminating substrate for hiv-1 reverse transcriptase. Biochemistry 2007, 46, 828–836. [Google Scholar] [CrossRef]
- Meyer, P.R.; Smith, A.J.; Matsuura, S.E.; Scott, W.A. Chain-terminating dinucleoside tetraphosphates are substrates for DNA polymerization by human immunodeficiency virus type 1 reverse transcriptase with increased activity against thymidine analogue-resistant mutants. Antimicrob. Agents Chemother. 2006, 50, 3607–3614. [Google Scholar] [CrossRef]
- Eggink, D.; Huigen, M.C.; Boucher, C.A.; Gotte, M.; Nijhuis, M. Insertions in the beta3-beta4 loop of reverse transcriptase of human immunodeficiency virus type 1 and their mechanism of action, influence on drug susceptibility and viral replication capacity. Antivir. Res. 2007, 75, 93–103. [Google Scholar] [CrossRef]
- Cases-Gonzalez, C.E.; Franco, S.; Martinez, M.A.; Menendez-Arias, L. Mutational patterns associated with the 69 insertion complex in multi-drug-resistant hiv-1 reverse transcriptase that confer increased excision activity and high-level resistance to zidovudine. J. Mol. Biol. 2007, 365, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Lennerstrand, J.; Matsuura, S.E.; Larder, B.A.; Scott, W.A. Effects of dipeptide insertions between codons 69 and 70 of human immunodeficiency virus type 1 reverse transcriptase on primer unblocking, deoxynucleoside triphosphate inhibition, and DNA chain elongation. J. Virol. 2003, 77, 3871–3877. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Nucleoside analog resistance caused by insertions in the fingers of human immunodeficiency virus type 1 reverse transcriptase involves atp-mediated excision. J. Virol. 2002, 76, 9143–9151. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, A.; Kodama, E.; Sarafianos, S.G.; Sakagami, Y.; Kohgo, S.; Kitano, K.; Ashida, N.; Iwai, Y.; Hayakawa, H.; Nakata, H.; et al. 2'-deoxy-4'-c-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell biol. 2008, 40, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Kodama, E.I.; Kohgo, S.; Kitano, K.; Machida, H.; Gatanaga, H.; Shigeta, S.; Matsuoka, M.; Ohrui, H.; Mitsuya, H. 4'-ethynyl nucleoside analogs: Potent inhibitors of multidrug-resistant human immunodeficiency virus variants in vitro. Antimicrob. Agents Chemother. 2001, 45, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, E.; Marchand, B.; Kodama, E.N.; Singh, K.; Matsuoka, M.; Kirby, K.A.; Ryan, E.M.; Sawani, A.M.; Nagy, E.; Ashida, N.; et al. Mechanism of inhibition of hiv-1 reverse transcriptase by 4'-ethynyl-2-fluoro-2'-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 2009, 284, 35681–35691. [Google Scholar] [CrossRef] [PubMed]
- Nakata, H.; Amano, M.; Koh, Y.; Kodama, E.; Yang, G.; Bailey, C.M.; Kohgo, S.; Hayakawa, H.; Matsuoka, M.; Anderson, K.S.; et al. Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4'-ethynyl-2-fluoro-2'-deoxyadenosine. Antimicrob. Agents Chemother. 2007, 51, 2701–2708. [Google Scholar] [CrossRef]
- Ohrui, H.; Kohgo, S.; Hayakawa, H.; Kodama, E.; Matsuoka, M.; Nakata, T.; Mitsuya, H. 2'-deoxy-4'-c-ethynyl-2-fluoroadenosine: A nucleoside reverse transcriptase inhibitor with highly potent activity against wide spectrum of hiv-1 strains, favorable toxic profiles, and stability in plasma. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Sohl, C.D.; Singh, K.; Kasiviswanathan, R.; Copeland, W.C.; Mitsuya, H.; Sarafianos, S.G.; Anderson, K.S. Mechanism of interaction of human mitochondrial DNA polymerase gamma with the novel nucleoside reverse transcriptase inhibitor 4'-ethynyl-2-fluoro-2'-deoxyadenosine indicates a low potential for host toxicity. Antimicrob. Agents Chemother. 2012, 56, 1630–1634. [Google Scholar] [CrossRef]
- Muftuoglu, Y.; Sohl, C.D.; Mislak, A.C.; Mitsuya, H.; Sarafianos, S.G.; Anderson, K.S. Probing the molecular mechanism of action of the hiv-1 reverse transcriptase inhibitor 4'-ethynyl-2-fluoro-2'-deoxyadenosine (efda) using pre-steady-state kinetics. Antivir. Res. 2014, 106, 1–4. [Google Scholar] [CrossRef]
- Yang, G.; Dutschman, G.E.; Wang, C.J.; Tanaka, H.; Baba, M.; Anderson, K.S.; Cheng, Y.C. Highly selective action of triphosphate metabolite of 4'-ethynyl d4t: A novel anti-hiv compound against hiv-1 rt. Antivir. Res. 2007, 73, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Dutschman, G.E.; Grill, S.P.; Gullen, E.A.; Haraguchi, K.; Takeda, S.; Tanaka, H.; Baba, M.; Cheng, Y.C. Novel 4'-substituted stavudine analog with improved anti-human immunodeficiency virus activity and decreased cytotoxicity. Antimicrob. Agents Chemother. 2004, 48, 1640–1646. [Google Scholar] [CrossRef]
- Li, Z.; Terry, B.; Olds, W.; Protack, T.; Deminie, C.; Minassian, B.; Nowicka-Sans, B.; Sun, Y.; Dicker, I.; Hwang, C.; et al. In vitro cross-resistance profile of nucleoside reverse transcriptase inhibitor (nrti) bms-986001 against known nrti resistance mutations. Antimicrob. Agents Chemother. 2013, 57, 5500–5508. [Google Scholar]
- Yang, G.; Paintsil, E.; Dutschman, G.E.; Grill, S.P.; Wang, C.J.; Wang, J.; Tanaka, H.; Hamasaki, T.; Baba, M.; Cheng, Y.C. Impact of novel human immunodeficiency virus type 1 reverse transcriptase mutations p119s and t165a on 4'-ethynylthymidine analog resistance profile. Antimicrob. Agents Chemother. 2009, 53, 4640–4646. [Google Scholar] [CrossRef]
- Das, K.; Arnold, E. Hiv-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013, 3, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Spence, R.A.; Kati, W.M.; Anderson, K.S.; Johnson, K.A. Mechanism of inhibition of hiv-1 reverse transcriptase by nonnucleoside inhibitors. Science 1995, 267, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Rittinger, K.; Divita, G.; Goody, R.S. Human immunodeficiency virus reverse transcriptase substrate-induced conformational changes and the mechanism of inhibition by nonnucleoside inhibitors. Proc. Natl. Acad. Sci. USA 1995, 92, 8046–8049. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Radzio, J.; Anderson, K.S.; Sluis-Cremer, N. Probing nonnucleoside inhibitor-induced active-site distortion in hiv-1 reverse transcriptase by transient kinetic analyses. Protein Sci. 2007, 16, 1728–1737. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Milton, J.; Weaver, K.L.; Short, S.A.; Stuart, D.I.; Stammers, D.K. Structural basis for the resilience of efavirenz (dmp-266) to drug resistance mutations in hiv-1 reverse transcriptase. Struct. Fold Des. 2000, 8, 1089–1094. [Google Scholar] [CrossRef]
- Martins, S.; Ramos, M.J.; Fernandes, P.A. The current status of the nnrti family of antiretrovirals used in the haart regime against hiv infection. Curr. Med. Chem. 2008, 15, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Kuritzkes, D.R. Altered viral fitness and drug susceptibility in human inmmunodeficiency virus type 1 (hiv-1) carrying mutations that confer resistance to non-nucleoside reverse transcriptase and integrase strand-transfer inhibitors. J. Virol. 2014, 88, 9268–9276. [Google Scholar] [CrossRef] [PubMed]
- Merluzzi, V.J.; Hargrave, K.D.; Labadia, M.; Grozinger, K.; Skoog, M.; Wu, J.C.; Shih, C.K.; Eckner, K.; Hattox, S.; Adams, J.; et al. Inhibition of hiv-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science 1990, 250, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Dueweke, T.J.; Poppe, S.M.; Romero, D.L.; Swaney, S.M.; So, A.G.; Downey, K.M.; Althaus, I.W.; Reusser, F.; Busso, M.; Resnick, L.; et al. U-90152, a potent inhibitor of human immunodeficiency virus type 1 replication. Antimicrob. Agents Chemother. 1993, 37, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Young, S.D.; Britcher, S.F.; Tran, L.O.; Payne, L.S.; Lumma, W.C.; Lyle, T.A.; Huff, J.R.; Anderson, P.S.; Olsen, D.B.; Carroll, S.S.; et al. L-743, 726 (dmp-266): A novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1995, 39, 2602–2605. [Google Scholar] [CrossRef]
- Andries, K.; Azijn, H.; Thielemans, T.; Ludovici, D.; Kukla, M.; Heeres, J.; Janssen, P.; de Corte, B.; Vingerhoets, J.; Pauwels, R.; et al. Tmc125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2004, 48, 4680–4686. [Google Scholar] [CrossRef]
- Janssen, P.A.; Lewi, P.J.; Arnold, E.; Daeyaert, F.; de Jonge, M.; Heeres, J.; Koymans, L.; Vinkers, M.; Guillemont, J.; Pasquier, E.; et al. In search of a novel anti-hiv drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1e)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (r278474, rilpivirine). J. Med. Chem. 2005, 48, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; de Mendoza, C. Genetic mechanisms of resistance to nrti and nnrti. HIV Clin. Trials 2002, 3, 237–248. [Google Scholar] [CrossRef]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 a resolution of hiv-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef]
- Esnouf, R.; Ren, J.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. Mechanism of inhibition of hiv-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 303–308. [Google Scholar] [CrossRef]
- Department of Health and Human Services. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents; Office of Aids Research Advisory Council: Bethesda, USA, 2011; pp. 1–174.
- Deparment of Health and Human Services - A working group of the office of AIDS research advisory council. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents; 2014; pp. 1–284. Available online: http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf (accessed on 15 October 2014).
- Tambuyzer, L.; Azijn, H.; Rimsky, L.T.; Vingerhoets, J.; Lecocq, P.; Kraus, G.; Picchio, G.; de Bethune, M.P. Compilation and prevalence of mutations associated with resistance to non-nucleoside reverse transcriptase inhibitors. Antivir. Therapy 2009, 14, 103–109. [Google Scholar]
- Hsiou, Y.; Ding, J.; Das, K.; Clark, A.D., Jr.; Boyer, P.L.; Lewi, P.; Janssen, P.A.; Kleim, J.P.; Rosner, M.; Hughes, S.H.; et al. The lys103asn mutation of hiv-1 rt: A novel mechanism of drug resistance. J. Mol. Biol. 2001, 309, 437–445. [Google Scholar] [CrossRef]
- Lindberg, J.; Sigurdsson, S.; Lowgren, S.; Andersson, H.O.; Sahlberg, C.; Noreen, R.; Fridborg, K.; Zhang, H.; Unge, T. Structural basis for the inhibitory efficacy of efavirenz (dmp-266), msc194 and pnu142721 towards the hiv-1 rt k103n mutant. Eur. J. Biochem./FEBS 2002, 269, 1670–1677. [Google Scholar] [CrossRef]
- Harrigan, P.R.; Mo, T.; Wynhoven, B.; Hirsch, J.; Brumme, Z.; McKenna, P.; Pattery, T.; Vingerhoets, J.; Bacheler, L.T. Rare mutations at codon 103 of hiv-1 reverse transcriptase can confer resistance to non-nucleoside reverse transcriptase inhibitors. AIDS 2005, 19, 549–554. [Google Scholar] [CrossRef]
- Parkin, N.T.; Gupta, S.; Chappey, C.; Petropoulos, C.J. The k101p and k103r/v179d mutations in human immunodeficiency virus type 1 reverse transcriptase confer resistance to nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2006, 50, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Keeling, J.; Darby, G.; Jones, Y.; Stuart, D.; et al. High resolution structures of hiv-1 rt from four rt-inhibitor complexes. Nat. Struct. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Non-nucleoside reverse transcriptase inhibitors (nnrtis): Past, present, and future. Chem. Biodivers. 2004, 1, 44–64. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, C.J.; Parkin, N.T.; Limoli, K.L.; Lie, Y.S.; Wrin, T.; Huang, W.; Tian, H.; Smith, D.; Winslow, G.A.; Capon, D.J.; et al. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2000, 44, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, S.L.; Kelly, D.V.; Tseng, A.L.; Humar, A.; Harrigan, P.R. Non-nucleoside reverse transcriptase inhibitor failure impairs hiv-rna responses to efavirenz-containing salvage antiretroviral therapy. AIDS 2001, 15, 1581–1584. [Google Scholar] [CrossRef] [PubMed]
- Delaugerre, C.; Rohban, R.; Simon, A.; Mouroux, M.; Tricot, C.; Agher, R.; Huraux, J.M.; Katlama, C.; Calvez, V. Resistance profile and cross-resistance of hiv-1 among patients failing a non-nucleoside reverse transcriptase inhibitor-containing regimen. J. Med. Virol. 2001, 65, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Bacheler, L.; Jeffrey, S.; Hanna, G.; D’Aquila, R.; Wallace, L.; Logue, K.; Cordova, B.; Hertogs, K.; Larder, B.; Buckery, R.; et al. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J. Virol. 2001, 75, 4999–5008. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Gamarnik, A.; Limoli, K.; Petropoulos, C.J.; Whitcomb, J.M. Amino acid substitutions at position 190 of human immunodeficiency virus type 1 reverse transcriptase increase susceptibility to delavirdine and impair virus replication. J. Virol. 2003, 77, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Kleim, J.P.; Bender, R.; Kirsch, R.; Meichsner, C.; Paessens, A.; Riess, G. Mutational analysis of residue 190 of human immunodeficiency virus type 1 reverse transcriptase. Virology 1994, 200, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Ludovici, D.W.; de Corte, B.L.; Kukla, M.J.; Ye, H.; Ho, C.Y.; Lichtenstein, M.A.; Kavash, R.W.; Andries, K.; de Bethune, M.P.; Azijn, H.; et al. Evolution of anti-hiv drug candidates. Part 3: Diarylpyrimidine (dapy) analogues. Bioorganic Med. Chem. Lett. 2001, 11, 2235–2239. [Google Scholar] [CrossRef]
- Vingerhoets, J.; Azijn, H.; Fransen, E.; de Baere, I.; Smeulders, L.; Jochmans, D.; Andries, K.; Pauwels, R.; de Bethune, M.P. Tmc125 displays a high genetic barrier to the development of resistance: Evidence from in vitro selection experiments. J. Virol. 2005, 79, 12773–12782. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Clark, A.D., Jr.; Lewi, P.J.; Heeres, J.; de Jonge, M.R.; Koymans, L.M.; Vinkers, H.M.; Daeyaert, F.; Ludovici, D.W.; Kukla, M.J.; et al. Roles of conformational and positional adaptability in structure-based design of tmc125-r165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant hiv-1 variants. J. Med. Chem. 2004, 47, 2550–2560. [Google Scholar] [CrossRef]
- Das, K.; Bauman, J.D.; Clark, A.D., Jr.; Frenkel, Y.V.; Lewi, P.J.; Shatkin, A.J.; Hughes, S.H.; Arnold, E. High-resolution structures of hiv-1 reverse transcriptase/tmc278 complexes: Strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 1466–1471. [Google Scholar] [CrossRef]
- Lansdon, E.B.; Brendza, K.M.; Hung, M.; Wang, R.; Mukund, S.; Jin, D.; Birkus, G.; Kutty, N.; Liu, X. Crystal structures of hiv-1 reverse transcriptase with etravirine (tmc125) and rilpivirine (tmc278): Implications for drug design. J. Med. Chem. 2010, 53, 4295–4299. [Google Scholar] [CrossRef]
- Xu, H.T.; Quan, Y.; Schader, S.M.; Oliveira, M.; Bar-Magen, T.; Wainberg, M.A. The m230l nonnucleoside reverse transcriptase inhibitor resistance mutation in hiv-1 reverse transcriptase impairs enzymatic function and viral replicative capacity. Antimicrob. Agents Chemother. 2010, 54, 2401–2408. [Google Scholar] [CrossRef]
- Madruga, J.V.; Cahn, P.; Grinsztejn, B.; Haubrich, R.; Lalezari, J.; Mills, A.; Pialoux, G.; Wilkin, T.; Peeters, M.; Vingerhoets, J.; et al. Efficacy and safety of tmc125 (etravirine) in treatment-experienced hiv-1-infected patients in duet-1: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 2007, 370, 29–38. [Google Scholar] [CrossRef]
- Lazzarin, A.; Campbell, T.; Clotet, B.; Johnson, M.; Katlama, C.; Moll, A.; Towner, W.; Trottier, B.; Peeters, M.; Vingerhoets, J.; et al. Efficacy and safety of tmc125 (etravirine) in treatment-experienced hiv-1-infected patients in duet-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 2007, 370, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Vingerhoets, J.; Tambuyzer, L.; Azijn, H.; Hoogstoel, A.; Nijs, S.; Peeters, M.; de Bethune, M.P.; de Smedt, G.; Woodfall, B.; Picchio, G. Resistance profile of etravirine: Combined analysis of baseline genotypic and phenotypic data from the randomized, controlled phase iii clinical studies. AIDS 2010, 24, 503–514. [Google Scholar] [CrossRef]
- Towner, W.J.; Cassetti, I.; Domingo, P.; Nijs, S.; Kakuda, T.N.; Vingerhoets, J.; Woodfall, B. Etravirine: Clinical review of a treatment option for hiv type-1-infected patients with non-nucleoside reverse transcriptase inhibitor resistance. Antivir. Therapy 2010, 15, 803–816. [Google Scholar] [CrossRef]
- Marcelin, A.G.; Flandre, P.; Descamps, D.; Morand-Joubert, L.; Charpentier, C.; Izopet, J.; Trabaud, M.A.; Saoudin, H.; Delaugerre, C.; Tamalet, C.; et al. Factors associated with virological response to etravirine in nonnucleoside reverse transcriptase inhibitor-experienced hiv-1-infected patients. Antimicrob. Agents Chemother. 2010, 54, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.T.; Colby-Germinario, S.P.; Asahchop, E.L.; Oliveira, M.; McCallum, M.; Schader, S.M.; Han, Y.; Quan, Y.; Sarafianos, S.G.; Wainberg, M.A. Effect of mutations at position e138 in hiv-1 reverse transcriptase and their interactions with the m184i mutation on defining patterns of resistance to nonnucleoside reverse transcriptase inhibitors rilpivirine and etravirine. Antimicrob. Agents Chemother. 2013, 57, 3100–3109. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, H.T.; Oliveira, M.; Asahchop, E.L.; McCallum, M.; Quashie, P.K.; Han, Y.; Quan, Y.; Wainberg, M.A. Molecular mechanism of antagonism between the y181c and e138k mutations in hiv-1 reverse transcriptase. J. Virol. 2012, 86, 12983–12990. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; de Mendoza, C.; Pattery, T.; Gonzalez Mdel, M.; Villacian, J.; Soriano, V. Phenotypic impact of resistance mutations on etravirine susceptibility in hiv patients with prior failure to nonnucleoside analogues. AIDS 2008, 22, 2395–2398. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Vingerhoets, J.; Fransen, S.; Tambuyzer, L.; Azijn, H.; Frantzell, A.; Paredes, R.; Coakley, E.; Nijs, S.; Clotet, B.; et al. Connection domain mutations in hiv-1 reverse transcriptase do not impact etravirine susceptibility and virologic responses to etravirine-containing regimens. Antimicrob. Agents Chemother. 2011, 55, 2872–2879. [Google Scholar] [CrossRef]
- Xu, H.T.; Colby-Germinario, S.P.; Oliveira, M.; Han, Y.; Quan, Y.; Zanichelli, V.; Wainberg, M.A. The connection domain mutation n348i in hiv-1 reverse transcriptase enhances resistance to etravirine and rilpivirine but restricts the emergence of the e138k resistance mutation by diminishing viral replication capacity. J. Virol. 2014, 88, 1536–1547. [Google Scholar] [CrossRef]
- Sharma, M.; Saravolatz, L.D. Rilpivirine: A new non-nucleoside reverse transcriptase inhibitor. J. Antimicrob. Chemother. 2013, 68, 250–256. [Google Scholar] [CrossRef]
- Johnson, B.C.; Pauly, G.T.; Rai, G.; Patel, D.; Bauman, J.D.; Baker, H.L.; Das, K.; Schneider, J.P.; Maloney, D.J.; Arnold, E.; et al. A comparison of the ability of rilpivirine (tmc278) and selected analogues to inhibit clinically relevant hiv-1 reverse transcriptase mutants. Retrovirology 2012, 9, 99. [Google Scholar] [CrossRef]
- Asahchop, E.L.; Wainberg, M.A.; Oliveira, M.; Xu, H.; Brenner, B.G.; Moisi, D.; Ibanescu, I.R.; Tremblay, C. Distinct resistance patterns to etravirine and rilpivirine in viruses containing nonnucleoside reverse transcriptase inhibitor mutations at baseline. AIDS 2013, 27, 879–887. [Google Scholar] [CrossRef]
- Xu, H.T.; Colby-Germinario, S.P.; Huang, W.; Oliveira, M.; Han, Y.; Quan, Y.; Petropoulos, C.J.; Wainberg, M.A. Role of the k101e substitution in hiv-1 reverse transcriptase in resistance to rilpivirine and other nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5649–5657. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, K.; Marchand, B.; Rai, D.K.; Sharma, B.; Michailidis, E.; Ryan, E.M.; Matzek, K.B.; Leslie, M.D.; Hagedorn, A.N.; Li, Z.; et al. Biochemical mechanism of hiv-1 resistance to rilpivirine. J. Biol. Chem. 2012, 287, 38110–38123. [Google Scholar] [CrossRef]
- Rimsky, L.; Van Eygen, V.; Hoogstoel, A.; Stevens, M.; Boven, K.; Picchio, G.; Vingerhoets, J. 96-week resistance analyses of rilpivirine in treatment-naive, hiv-1-infected adults from the echo and thrive phase iii trials. Antivir. Therapy 2013, 18, 967–977. [Google Scholar] [CrossRef]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4690. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Krohn, A.; et al. Rational design of peptide-based hiv proteinase inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Huff, J.R.; Kahn, J. Discovery and clinical development of hiv-1 protease inhibitors. Adv. Protein Chem. 2001, 56, 213–251. [Google Scholar] [PubMed]
- Louis, J.M.; Ishima, R.; Torchia, D.A.; Weber, I.T. Hiv-1 protease: Structure, dynamics, and inhibition. Adv. Pharmacol. 2007, 55, 261–298. [Google Scholar] [PubMed]
- Hull, M.W.; Montaner, J.S. Ritonavir-boosted protease inhibitors in hiv therapy. Ann. Med. 2011, 43, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Kempf, D.J.; Marsh, K.C.; Kumar, G.; Rodrigues, A.D.; Denissen, J.F.; McDonald, E.; Kukulka, M.J.; Hsu, A.; Granneman, G.R.; Baroldi, P.A.; et al. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. 1997, 41, 654–660. [Google Scholar] [PubMed]
- Stanford HIV Drug Resistance Database. Major HIV-1 drug resistance mutations. Available online: http://hivdb.Stanford.edu (accessed on 16 June 2014).
- Swain, A.L.; Miller, M.M.; Green, J.; Rich, D.H.; Schneider, J.; Kent, S.B.; Wlodawer, A. X-ray crystallographic structure of a complex between a synthetic protease of human immunodeficiency virus 1 and a substrate-based hydroxyethylamine inhibitor. Proc. Natl. Acad. Sci. USA 1990, 87, 8805–8809. [Google Scholar] [CrossRef] [PubMed]
- Gulnik, S.V.; Suvorov, L.I.; Liu, B.; Yu, B.; Anderson, B.; Mitsuya, H.; Erickson, J.W. Kinetic characterization and cross-resistance patterns of hiv-1 protease mutants selected under drug pressure. Biochemistry 1995, 34, 9282–9287. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.A.; Calvez, V.; Gunthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.; Wensing, A.M.; Richman, D.D. 2011 update of the drug resistance mutations in hiv-1. Top. Antivir. Med. 2011, 19, 156–164. [Google Scholar] [PubMed]
- Markowitz, M.; Conant, M.; Hurley, A.; Schluger, R.; Duran, M.; Peterkin, J.; Chapman, S.; Patick, A.; Hendricks, A.; Yuen, G.J.; et al. A preliminary evaluation of nelfinavir mesylate, an inhibitor of human immunodeficiency virus (hiv)-1 protease, to treat hiv infection. J. Infect. Dis. 1998, 177, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Taylor, J.; Fessel, W.J.; Kaufman, D.; Towner, W.; Troia, P.; Ruane, P.; Hellinger, J.; Shirvani, V.; Zolopa, A.; et al. Hiv-1 protease mutations and protease inhibitor cross-resistance. Antimicrob. Agents Chemother. 2010, 54, 4253–4261. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, B.; Louis, J.M.; Reed, C.C.; Adomat, J.M.; Krouse, J.; Wang, Y.F.; Harrison, R.W.; Weber, I.T. Structural and kinetic analysis of drug resistant mutants of hiv-1 protease. Eur. J. Biochem./FEBS 1999, 263, 238–245. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for hiv-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef] [PubMed]
- King, N.M.; Prabu-Jeyabalan, M.; Nalivaika, E.A.; Schiffer, C.A. Combating susceptibility to drug resistance: Lessons from hiv-1 protease. Chem. Biol. 2004, 11, 1333–1338. [Google Scholar] [PubMed]
- King, N.M.; Melnick, L.; Prabu-Jeyabalan, M.; Nalivaika, E.A.; Yang, S.S.; Gao, Y.; Nie, X.; Zepp, C.; Heefner, D.L.; Schiffer, C.A. Lack of synergy for inhibitors targeting a multi-drug-resistant hiv-1 protease. Protein Sci. 2002, 11, 418–429. [Google Scholar] [CrossRef]
- Munshi, S.; Chen, Z.; Yan, Y.; Li, Y.; Olsen, D.B.; Schock, H.B.; Galvin, B.B.; Dorsey, B.; Kuo, L.C. An alternate binding site for the p1-p3 group of a class of potent hiv-1 protease inhibitors as a result of concerted structural change in the 80s loop of the protease. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 381–388. [Google Scholar] [CrossRef]
- Johnson, V.A.; Calvez, V.; Gunthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Richman, D.D. Update of the drug resistance mutations in hiv-1: March 2013. Top. Antivir. Med. 2013, 21, 6–14. [Google Scholar]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F., 2nd; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; et al. Viracept (nelfinavir mesylate, ag1343): A potent, orally bioavailable inhibitor of hiv-1 protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef]
- Sugiura, W.; Matsuda, Z.; Yokomaku, Y.; Hertogs, K.; Larder, B.; Oishi, T.; Okano, A.; Shiino, T.; Tatsumi, M.; Matsuda, M.; et al. Interference between d30n and l90m in selection and development of protease inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2002, 46, 708–715. [Google Scholar] [CrossRef]
- Menendez-Arias, L.; Martinez, M.A.; Quinones-Mateu, M.E.; Martinez-Picado, J. Fitness variations and their impact on the evolution of antiretroviral drug resistance. Curr. Drug Targets 2003, 3, 355–371. [Google Scholar] [CrossRef]
- Chang, M.W.; Torbett, B.E. Accessory mutations maintain stability in drug-resistant hiv-1 protease. J. Mol. Biol. 2011, 410, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Imamichi, H.; Imamichi, T.; Lane, H.C.; Falloon, J.; Vasudevachari, M.B.; Salzman, N.P. Drug resistance during indinavir therapy is caused by mutations in the protease gene and in its gag substrate cleavage sites. J. Virol. 1997, 71, 6662–6670. [Google Scholar] [PubMed]
- Doyon, L.; Croteau, G.; Thibeault, D.; Poulin, F.; Pilote, L.; Lamarre, D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 1996, 70, 3763–3769. [Google Scholar] [PubMed]
- Robinson, L.H.; Myers, R.E.; Snowden, B.W.; Tisdale, M.; Blair, E.D. Hiv type 1 protease cleavage site mutations and viral fitness: Implications for drug susceptibility phenotyping assays. AIDS Res. Hum. Retrovir. 2000, 16, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Pettit, S.C.; Henderson, G.J.; Schiffer, C.A.; Swanstrom, R. Replacement of the p1 amino acid of human immunodeficiency virus type 1 gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J. Virol. 2002, 76, 10226–10233. [Google Scholar] [CrossRef] [PubMed]
- Feher, A.; Weber, I.T.; Bagossi, P.; Boross, P.; Mahalingam, B.; Louis, J.M.; Copeland, T.D.; Torshin, I.Y.; Harrison, R.W.; Tozser, J. Effect of sequence polymorphism and drug resistance on two hiv-1 gag processing sites. Eur. J. Biochem./FEBS 2002, 269, 4114–4120. [Google Scholar] [CrossRef]
- Doyon, L.; Payant, C.; Brakier-Gingras, L.; Lamarre, D. Novel gag-pol frameshift site in human immunodeficiency virus type 1 variants resistant to protease inhibitors. J. Virol. 1998, 72, 6146–6150. [Google Scholar] [PubMed]
- Gatanaga, H.; Suzuki, Y.; Tsang, H.; Yoshimura, K.; Kavlick, M.F.; Nagashima, K.; Gorelick, R.J.; Mardy, S.; Tang, C.; Summers, M.F.; et al. Amino acid substitutions in gag protein at non-cleavage sites are indispensable for the development of a high multitude of hiv-1 resistance against protease inhibitors. J. Biol. Chem. 2002, 277, 5952–5961. [Google Scholar] [CrossRef] [PubMed]
- Dam, E.; Quercia, R.; Glass, B.; Descamps, D.; Launay, O.; Duval, X.; Krausslich, H.G.; Hance, A.J.; Clavel, F.; Group, A.S. Gag mutations strongly contribute to hiv-1 resistance to protease inhibitors in highly drug-experienced patients besides compensating for fitness loss. PLoS Pathog. 2009, 5, e1000345. [Google Scholar] [CrossRef] [PubMed]
- Myint, L.; Matsuda, M.; Matsuda, Z.; Yokomaku, Y.; Chiba, T.; Okano, A.; Yamada, K.; Sugiura, W. Gag non-cleavage site mutations contribute to full recovery of viral fitness in protease inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2004, 48, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.; Wong, E.C.; Rode, R.; Shafer, R.; de Luca, A.; Nadler, J.; Hawkins, T.; Cohen, C.; Harrington, R.; Kempf, D.; et al. Virologic response to lopinavir-ritonavir-based antiretroviral regimens in a multicenter international clinical cohort: Comparison of genotypic interpretation scores. Antimicrob. Agents Chemother. 2008, 52, 4050–4056. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Thornton, L. Fosamprenavir: Advancing hiv protease inhibitor treatment options. Expert Opin. Pharmacother. 2004, 5, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, A.G.; Masquelier, B.; Descamps, D.; Izopet, J.; Charpentier, C.; Alloui, C.; Bouvier-Alias, M.; Signori-Schmuck, A.; Montes, B.; Chaix, M.L.; et al. Tipranavir-ritonavir genotypic resistance score in protease inhibitor-experienced patients. Antimicrob. Agents Chemother. 2008, 52, 3237–3243. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, S.; Hill, A.; Picchio, G.; DeMasi, R.; de Paepe, E.; de Bethune, M.P. Influence of baseline protease inhibitor resistance on the efficacy of darunavir/ritonavir or lopinavir/ritonavir in the titan trial. J. Acquir. Immune Defic. Syndr. 2008, 49, 563–564. [Google Scholar] [CrossRef]
- Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.; Nalivaika, E.A.; Ozen, A.; et al. Molecular basis for drug resistance in hiv-1 protease. Viruses 2010, 2, 2509–2535. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Parham, G.L.; Martyr, C.D.; Nyalapatla, P.R.; Osswald, H.L.; Agniswamy, J.; Wang, Y.F.; Amano, M.; Weber, I.T.; Mitsuya, H. Highly potent hiv-1 protease inhibitors with novel tricyclic p2 ligands: Design, synthesis, and protein-ligand x-ray studies. J. Med. Chem. 2013, 56, 6792–6802. [Google Scholar] [CrossRef]
- Chow, S.A.; Vincent, K.A.; Ellison, V.; Brown, P.O. Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science 1992, 255, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Bushman, F.D.; Craigie, R. Identification of discrete functional domains of hiv-1 integrase and their organization within an active multimeric complex. EMBO J. 1993, 12, 3269–3275. [Google Scholar] [PubMed]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; et al. Inhibitors of strand transfer that prevent integration and inhibit hiv-1 replication in cells. Science 2000, 287, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Carayon, K.; Saib, A.; Deprez, E.; Mouscadet, J.F. Integrase and integration: Biochemical activities of hiv-1 integrase. Retrovirology 2008, 5, 114. [Google Scholar] [CrossRef]
- Craigie, R. The molecular biology of hiv integrase. Future Virol. 2012, 7, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Metifiot, M.; Marchand, C.; Pommier, Y. Hiv integrase inhibitors: 20-year landmark and challenges. Adv. Pharmacol. 2013, 67, 75–105. [Google Scholar]
- Hazuda, D.J.; Felock, P.J.; Hastings, J.C.; Pramanik, B.; Wolfe, A.L. Differential divalent cation requirements uncouple the assembly and catalytic reactions of human immunodeficiency virus type 1 integrase. J. Virol. 1997, 71, 7005–7011. [Google Scholar] [PubMed]
- Hicks, C.; Gulick, R.M. Raltegravir: The first hiv type 1 integrase inhibitor. Clin. Infect. Dis. 2009, 48, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.A.; Steigbigel, R.T.; Gatell, J.M.; Rockstroh, J.K.; Katlama, C.; Yeni, P.; Lazzarin, A.; Clotet, B.; Kumar, P.N.; Eron, J.E.; et al. Subgroup and resistance analyses of raltegravir for resistant hiv-1 infection. N. Engl. J. Med. 2008, 359, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Canducci, F.; Sampaolo, M.; Marinozzi, M.C.; Boeri, E.; Spagnuolo, V.; Galli, A.; Castagna, A.; Lazzarin, A.; Clementi, M.; Gianotti, N. Dynamic patterns of human immunodeficiency virus type 1 integrase gene evolution in patients failing raltegravir-based salvage therapies. AIDS 2009, 23, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Hurt, C.B.; Sebastian, J.; Hicks, C.B.; Eron, J.J. Resistance to hiv integrase strand transfer inhibitors among clinical specimens in the united states, 2009–2012. Clin. Infect. Dis. 2014, 58, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Malet, I.; Delelis, O.; Valantin, M.A.; Montes, B.; Soulie, C.; Wirden, M.; Tchertanov, L.; Peytavin, G.; Reynes, J.; Mouscadet, J.F.; et al. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob. Agents Chemother. 2008, 52, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, K.; Wakasa-Morimoto, C.; Kobayashi, M.; Miki, S.; Noshi, T.; Seki, T.; Kanamori-Koyama, M.; Kawauchi, S.; Suyama, A.; Fujishita, T.; et al. Secondary mutations in viruses resistant to hiv-1 integrase inhibitors that restore viral infectivity and replication kinetics. Antivir. Res. 2009, 81, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Malet, I.; Na, L.; Tchertanov, L.; Calvez, V.; Marcelin, A.G.; Subra, F.; Deprez, E.; Mouscadet, J.F. The g140s mutation in hiv integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance q148h mutation. Nucleic Acids Res. 2009, 37, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Marchand, C. The elvitegravir quad pill: The first once-daily dual-target anti-hiv tablet. Expert Opin. Investig. Drugs 2012, 21, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.L.; Varghese, V.; Rhee, S.Y.; Gatell, J.M.; Shafer, R.W. Hiv-1 integrase inhibitor resistance and its clinical implications. J. Infect. Dis. 2011, 203, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Goethals, O.; Clayton, R.; van Ginderen, M.; Vereycken, I.; Wagemans, E.; Geluykens, P.; Dockx, K.; Strijbos, R.; Smits, V.; Vos, A.; et al. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J. Virol. 2008, 82, 10366–10374. [Google Scholar] [CrossRef] [PubMed]
- Shimura, K.; Kodama, E.; Sakagami, Y.; Matsuzaki, Y.; Watanabe, W.; Yamataka, K.; Watanabe, Y.; Ohata, Y.; Doi, S.; Sato, M.; et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (jtk-303/gs-9137). J. Virol. 2008, 82, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yoshinaga, T.; Seki, T.; Wakasa-Morimoto, C.; Brown, K.W.; Ferris, R.; Foster, S.A.; Hazen, R.J.; Miki, S.; Suyama-Kagitani, A.; et al. In vitro antiretroviral properties of s/gsk1349572, a next-generation hiv integrase inhibitor. Antimicrob. Agents Chemother. 2011, 55, 813–821. [Google Scholar]
- Hightower, K.E.; Wang, R.; Deanda, F.; Johns, B.A.; Weaver, K.; Shen, Y.; Tomberlin, G.H.; Carter, H.L., 3rd; Broderick, T.; Sigethy, S.; et al. Dolutegravir (s/gsk1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant hiv-1 integrase-DNA complexes. Antimicrob. Agents Chemother. 2011, 55, 4552–4559. [Google Scholar] [CrossRef] [PubMed]
- Eron, J.J.; Clotet, B.; Durant, J.; Katlama, C.; Kumar, P.; Lazzarin, A.; Poizot-Martin, I.; Richmond, G.; Soriano, V.; Ait-Khaled, M.; et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant hiv type 1 infection: 24-week results of the viking study. J. Infect. Dis. 2013, 207, 740–748. [Google Scholar] [CrossRef]
- Quashie, P.K.; Mesplede, T.; Han, Y.S.; Oliveira, M.; Singhroy, D.N.; Fujiwara, T.; Underwood, M.R.; Wainberg, M.A. Characterization of the r263k mutation in hiv-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J. Virol. 2012, 86, 2696–2705. [Google Scholar] [CrossRef]
- Cahn, P.; Pozniak, A.L.; Mingrone, H.; Shuldyakov, A.; Brites, C.; Andrade-Villanueva, J.F.; Richmond, G.; Buendia, C.B.; Fourie, J.; Ramgopal, M.; et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with hiv: Week 48 results from the randomised, double-blind, non-inferiority sailing study. Lancet 2013, 382, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Mesplede, T.; Quashie, P.K.; Osman, N.; Han, Y.; Singhroy, D.N.; Lie, Y.; Petropoulos, C.J.; Huang, W.; Wainberg, M.A. Viral fitness cost prevents hiv-1 from evading dolutegravir drug pressure. Retrovirology 2013, 10, 22. [Google Scholar]
- Quashie, P.K.; Mesplede, T.; Han, Y.S.; Veres, T.; Osman, N.; Hassounah, S.; Sloan, R.D.; Xu, H.T.; Wainberg, M.A. Biochemical analysis of the role of g118r-linked dolutegravir drug resistance substitutions in hiv-1 integrase. Antimicrob. Agents Chemother. 2014, 58, 3580. [Google Scholar] [CrossRef]
- Oliveira, M.; Mesplede, T.; Quashie, P.K.; Moisi, D.; Wainberg, M.A. Resistance mutations against dolutegravir in hiv integrase impair the emergence of resistance against reverse transcriptase inhibitors. AIDS 2014, 28, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Haqqani, A.A.; Tilton, J.C. Entry inhibitors and their use in the treatment of hiv-1 infection. Antivir. Res. 2013, 98, 158–170. [Google Scholar] [CrossRef] [PubMed]
- De Feo, C.J.; Weiss, C.D. Escape from human immunodeficiency virus type 1 (hiv-1) entry inhibitors. Viruses 2012, 4, 3859–3911. [Google Scholar] [CrossRef] [PubMed]
- Cheng-Mayer, C.; Quiroga, M.; Tung, J.W.; Dina, D.; Levy, J.A. Viral determinants of human immunodeficiency virus type 1 t-cell or macrophage tropism, cytopathogenicity, and cd4 antigen modulation. J. Virol. 1990, 64, 4390–4398. [Google Scholar] [PubMed]
- Berger, E.A. Hiv entry and tropism: The chemokine receptor connection. AIDS 1997, 11 Suppl A, S3–S16. [Google Scholar] [PubMed]
- Picchio, G.R.; Gulizia, R.J.; Wehrly, K.; Chesebro, B.; Mosier, D.E. The cell tropism of human immunodeficiency virus type 1 determines the kinetics of plasma viremia in scid mice reconstituted with human peripheral blood leukocytes. J. Virol. 1998, 72, 2002–2009. [Google Scholar] [PubMed]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; di Marzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of hiv-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. Hiv-1 entry cofactor: Functional cdna cloning of a seven-transmembrane, g protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (uk-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor ccr5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [PubMed]
- Dragic, T.; Trkola, A.; Thompson, D.A.; Cormier, E.G.; Kajumo, F.A.; Maxwell, E.; Lin, S.W.; Ying, W.; Smith, S.O.; Sakmar, T.P.; et al. A binding pocket for a small molecule inhibitor of hiv-1 entry within the transmembrane helices of ccr5. Proc. Natl. Acad. Sci. USA 2000, 97, 5639–5644. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.; Jakobsen, M.R.; Sterjovski, J.; Ellett, A.; Posta, F.; Lee, B.; Jubb, B.; Westby, M.; Lewin, S.R.; Ramsland, P.A.; et al. Hiv-1 escape from the ccr5 antagonist maraviroc associated with an altered and less-efficient mechanism of gp120-ccr5 engagement that attenuates macrophage tropism. J. Virol. 2011, 85, 4330–4342. [Google Scholar] [CrossRef]
- Fatkenheuer, G.; Nelson, M.; Lazzarin, A.; Konourina, I.; Hoepelman, A.I.; Lampiris, H.; Hirschel, B.; Tebas, P.; Raffi, F.; Trottier, B.; et al. Subgroup analyses of maraviroc in previously treated r5 hiv-1 infection. N. Engl. J. Med. 2008, 359, 1442–1455. [Google Scholar] [CrossRef]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; et al. Maraviroc for previously treated patients with r5 hiv-1 infection. N. Engl. J. Med. 2008, 359, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Kuritzkes, D.R. A piece de resistance: How hiv-1 escapes small molecule ccr5 inhibitors. Curr. Opin. HIV AIDS 2009, 4, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Pastore, C.; Ramos, A.; Mosier, D.E. Intrinsic obstacles to human immunodeficiency virus type 1 coreceptor switching. J. Virol. 2004, 78, 7565–7574. [Google Scholar] [CrossRef]
- Pastore, C.; Nedellec, R.; Ramos, A.; Pontow, S.; Ratner, L.; Mosier, D.E. Human immunodeficiency virus type 1 coreceptor switching: V1/v2 gain-of-fitness mutations compensate for v3 loss-of-fitness mutations. J. Virol. 2006, 80, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Westby, M. Resistance to ccr5 antagonists. Curr. Opin. HIV AIDS 2007, 2, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Westby, M.; Smith-Burchnell, C.; Mori, J.; Lewis, M.; Mosley, M.; Stockdale, M.; Dorr, P.; Ciaramella, G.; Perros, M. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the ccr5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 2007, 81, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, C.G.; Ketas, T.J.; Klasse, P.J.; Moore, J.P. Resistance to ccr5 inhibitors caused by sequence changes in the fusion peptide of hiv-1 gp41. Proc. Natl. Acad. Sci. USA 2009, 106, 5318–5323. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Maeda, Y.; Terasawa, H.; Monde, K.; Harada, S.; Yusa, K. A combination of polymorphic mutations in v3 loop of hiv-1 gp120 can confer noncompetitive resistance to maraviroc. Virology 2011, 413, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Berro, R.; Klasse, P.J.; Jakobsen, M.R.; Gorry, P.R.; Moore, J.P.; Sanders, R.W. V3 determinants of hiv-1 escape from the ccr5 inhibitors maraviroc and vicriviroc. Virology 2012, 427, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of hiv-1 replication in humans by t-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of hiv-1 into host cd4 lymphocytes. Nat. Rev. 2004, 3, 215–225. [Google Scholar]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 1998, 72, 986–993. [Google Scholar] [PubMed]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (t-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; Rodes, B.; Toro, C.; Martin-Carbonero, L.; Gonzalez-Lahoz, J.; Soriano, V. Evolution of the gp41 env region in hiv-infected patients receiving t-20, a fusion inhibitor. AIDS 2002, 16, 1959–1961. [Google Scholar] [CrossRef] [PubMed]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment hiv-1 isolates. AIDS 2004, 18, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; Rodes, B.; Lebel-Binay, S.; Faudon, J.L.; Jimenez, V.; Soriano, V. Dynamics of enfuvirtide resistance in hiv-infected patients during and after long-term enfuvirtide salvage therapy. J. Clin. Virol. 2005, 34, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, C.; Marfil, S.; Garcia, E.; Martinez-Picado, J.; Bonjoch, A.; Bofill, M.; Moreno, S.; Ribera, E.; Domingo, P.; Clotet, B.; et al. Genetic evolution of gp41 reveals a highly exclusive relationship between codons 36, 38 and 43 in gp41 under long-term enfuvirtide-containing salvage regimen. AIDS 2006, 20, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.; Harrison, J.E.; Blackburn, L.A.; Martin, J.N.; Deeks, S.G.; Doms, R.W. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J. Virol. 2007, 81, 3240–3250. [Google Scholar] [CrossRef]
- Xu, L.; Pozniak, A.; Wildfire, A.; Stanfield-Oakley, S.A.; Mosier, S.M.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; et al. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar] [CrossRef]
- Cilliers, T.; Moore, P.; Coetzer, M.; Morris, L. In vitro generation of hiv type 1 subtype c isolates resistant to enfuvirtide. AIDS Res. Hum. Retrovir. 2005, 21, 776–783. [Google Scholar] [CrossRef]
- Mink, M.; Mosier, S.M.; Janumpalli, S.; Davison, D.; Jin, L.; Melby, T.; Sista, P.; Erickson, J.; Lambert, D.; Stanfield-Oakley, S.A.; et al. Impact of human immunodeficiency virus type 1 gp41 amino acid substitutions selected during enfuvirtide treatment on gp41 binding and antiviral potency of enfuvirtide in vitro. J. Virol. 2005, 79, 12447–12454. [Google Scholar] [CrossRef]
- Menzo, S.; Castagna, A.; Monachetti, A.; Hasson, H.; Danise, A.; Carini, E.; Bagnarelli, P.; Lazzarin, A.; Clementi, M. Genotype and phenotype patterns of human immunodeficiency virus type 1 resistance to enfuvirtide during long-term treatment. Antimicrob. Agents Chemother. 2004, 48, 3253–3259. [Google Scholar] [CrossRef] [PubMed]
- Nameki, D.; Kodama, E.; Ikeuchi, M.; Mabuchi, N.; Otaka, A.; Tamamura, H.; Ohno, M.; Fujii, N.; Matsuoka, M. Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp41 and rev-responsive element functions. J. Virol. 2005, 79, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.L.; Cammack, N. Resistance to enfuvirtide, the first hiv fusion inhibitor. J. Antimicrob. Chemother. 2004, 54, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, V.D.; Cheng, S.F.; Wu, C.W.; Karthikeyan, R.; Chen, C.J.; Chang, D.K. The llsgiv stretch of the n-terminal region of hiv-1 gp41 is critical for binding to a model peptide, t20. Protein Eng. 2003, 16, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Sista, P.; Giguel, F.; Greenberg, M.; Kuritzkes, D.R. Relative replicative fitness of human immunodeficiency virus type 1 mutants resistant to enfuvirtide (t-20). J. Virol. 2004, 78, 4628–4637. [Google Scholar] [CrossRef] [PubMed]
- Marconi, V.; Bonhoeffer, S.; Paredes, R.; Lu, J.; Hoh, R.; Martin, J.N.; Deeks, S.G.; Kuritzkes, D.R. Viral dynamics and in vivo fitness of hiv-1 in the presence and absence of enfuvirtide. J. Acquir. Immune Defic. Syndr. 2008, 48, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wilson, K.L.; Seedorff, J.E.; Ahrens, D.; Green, J.; Davison, D.K.; Jin, L.; Stanfield-Oakley, S.A.; Mosier, S.M.; Melby, T.E.; et al. Impact of the enfuvirtide resistance mutation n43d and the associated baseline polymorphism e137k on peptide sensitivity and six-helix bundle structure. Biochemistry 2008, 47, 6662–6670. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Terakawa, Y.; Watanabe, K.; Ohno, H.; Nakano, H.; Nakatsu, T.; Kato, H.; Izumi, K.; Kodama, E.; Matsuoka, M.; et al. X-ray crystallographic study of an hiv-1 fusion inhibitor with the gp41 s138a substitution. J. Mol. Biol. 2009, 392, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Haleyur Giri Setty, M.K.; Hewlett, I.K. Point of care technologies for hiv. AIDS Res. Treat. 2014, 2014, 497046. [Google Scholar] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Recent trends in hiv-1 drug resistance. Curr. Opin. Virol. 2013, 3, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Dineva, M.A.; Chua, Y.L.; Ritchie, A.V.; Ushiro-Lumb, I.; Wisniewski, C.A. Simple amplification-based assay: A nucleic acid-based point-of-care platform for hiv-1 testing. J. Infect. Dis. 2010, 201 Suppl 1, S65–72. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.R.; Bennett, D.E.; Bertagnolio, S.; Gilks, C.F.; Sutherland, D. World health organization surveys to monitor hiv drug resistance prevention and associated factors in sentinel antiretroviral treatment sites. Antivir. Therapy 2008, 13 Suppl 2, 15–23. [Google Scholar]
- Van den Eede, P.; van Wesenbeeck, L.; Verlinden, Y.; Feyaerts, M.; Smits, V.; Verheyen, A.; Vanhooren, L.; Deloof, A.; Villacian, J.; Pattery, T. Hiv-1 genotyping of the protease-reverse transcriptase and integrase genes to detect mutations that confer antiretroviral resistance. Methods Mol. Biol. 2013, 1030, 37–55. [Google Scholar] [PubMed]
- Asahchop, E.L.; Wainberg, M.A.; Sloan, R.D.; Tremblay, C.L. Antiviral drug resistance and the need for development of new hiv-1 reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2012, 56, 5000–5008. [Google Scholar] [CrossRef] [PubMed]
- Pirrone, V.; Thakkar, N.; Jacobson, J.M.; Wigdahl, B.; Krebs, F.C. Combinatorial approaches to the prevention and treatment of hiv-1 infection. Antimicrob. Agents Chemother. 2011, 55, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyidogan, P.; Anderson, K.S. Current Perspectives on HIV-1 Antiretroviral Drug Resistance. Viruses 2014, 6, 4095-4139. https://doi.org/10.3390/v6104095
Iyidogan P, Anderson KS. Current Perspectives on HIV-1 Antiretroviral Drug Resistance. Viruses. 2014; 6(10):4095-4139. https://doi.org/10.3390/v6104095
Chicago/Turabian StyleIyidogan, Pinar, and Karen S. Anderson. 2014. "Current Perspectives on HIV-1 Antiretroviral Drug Resistance" Viruses 6, no. 10: 4095-4139. https://doi.org/10.3390/v6104095
APA StyleIyidogan, P., & Anderson, K. S. (2014). Current Perspectives on HIV-1 Antiretroviral Drug Resistance. Viruses, 6(10), 4095-4139. https://doi.org/10.3390/v6104095