The Lysine 65 Residue in HIV-1 Reverse Transcriptase Function and in Nucleoside Analog Drug Resistance

{kind=link}
{kind=link}
Abstract
:1. Introduction
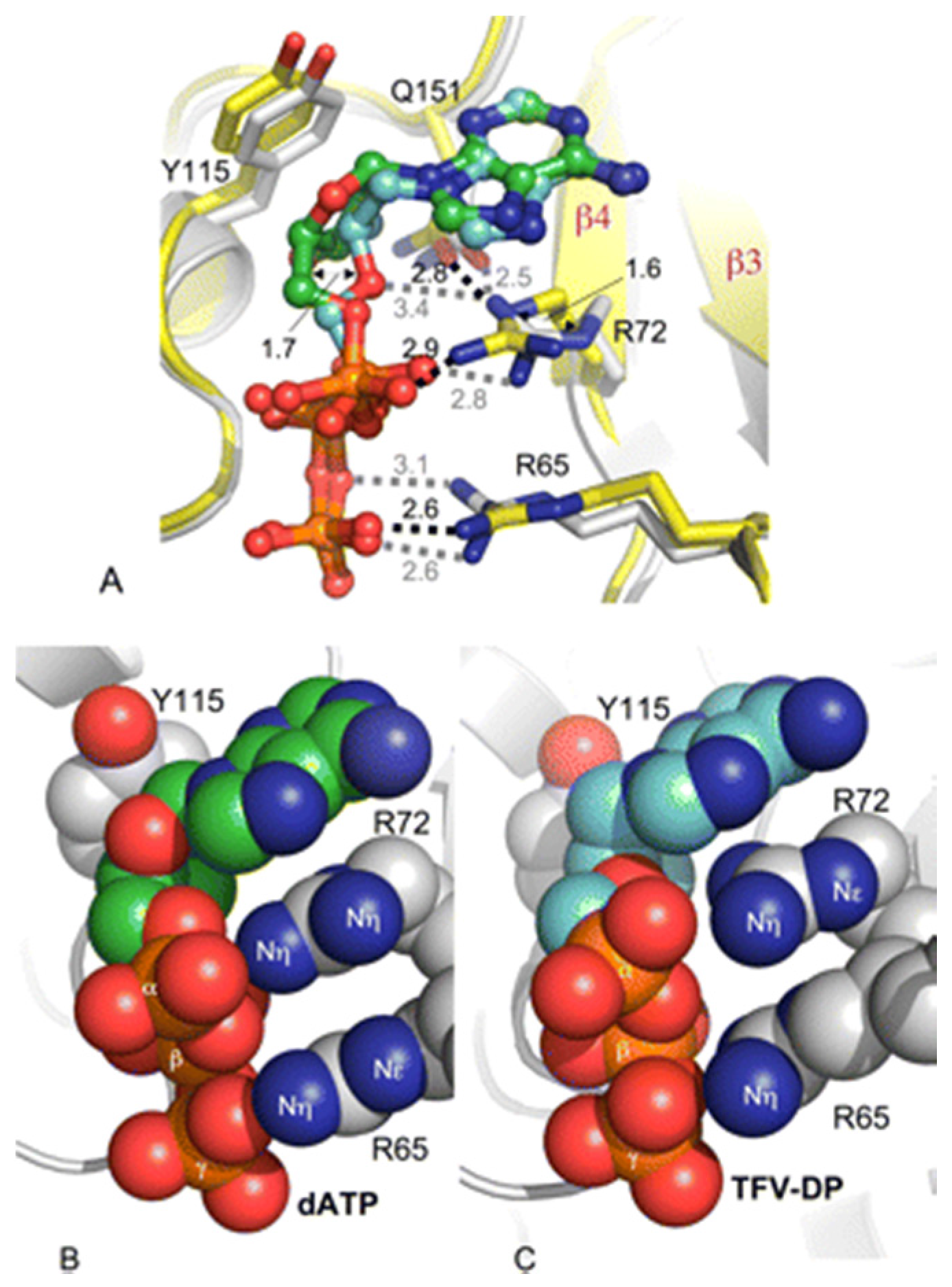
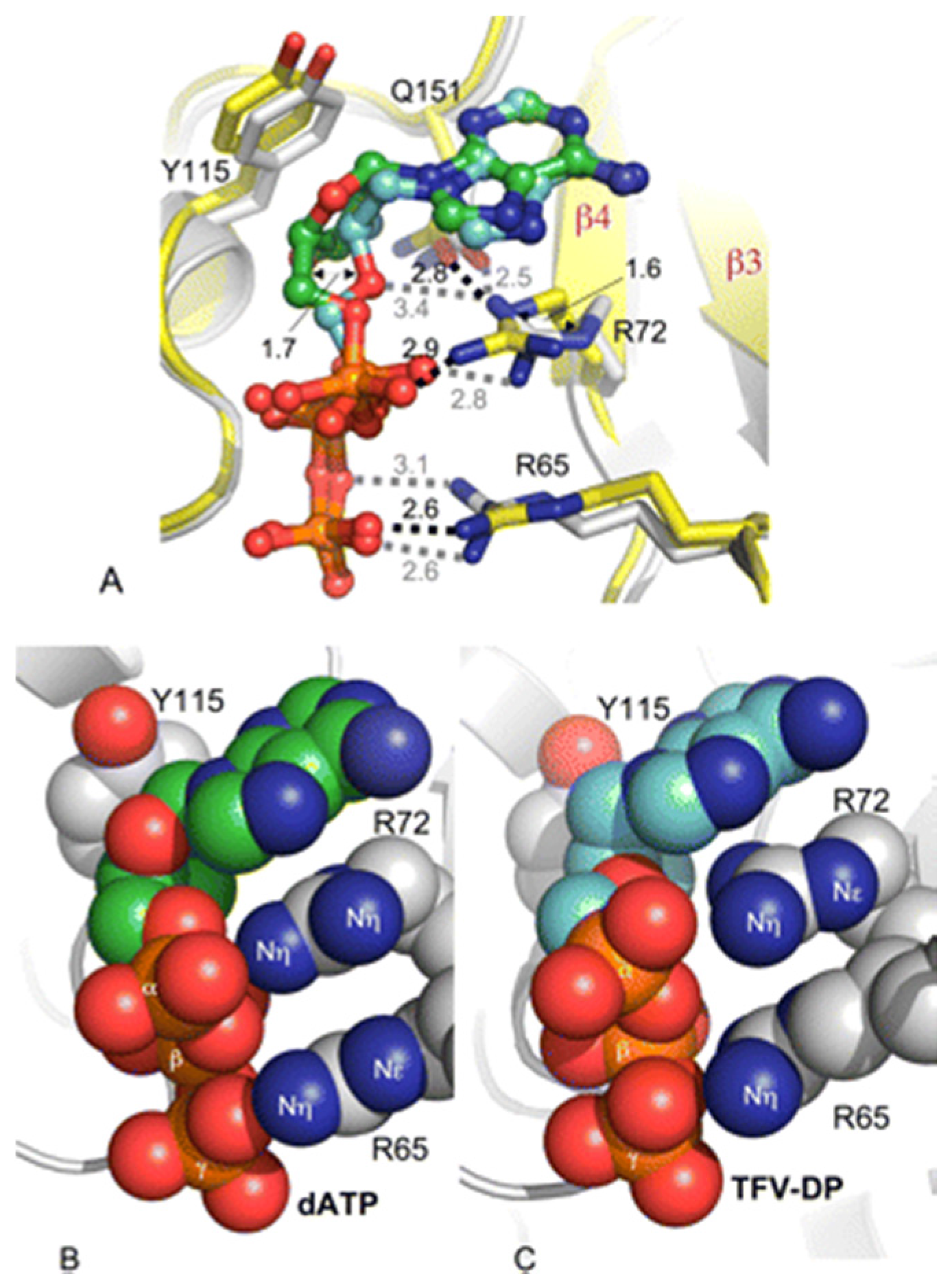
2. K65 in HIV-1 RT Function
3. K65 in Nucleoside Analog-Resistance
4. Antagonism of K65R with other NRTI-Resistance Mutations
5. K65R Confers Hypersusceptibility to other NRTIs
6. Clinical Prevalence of K65R
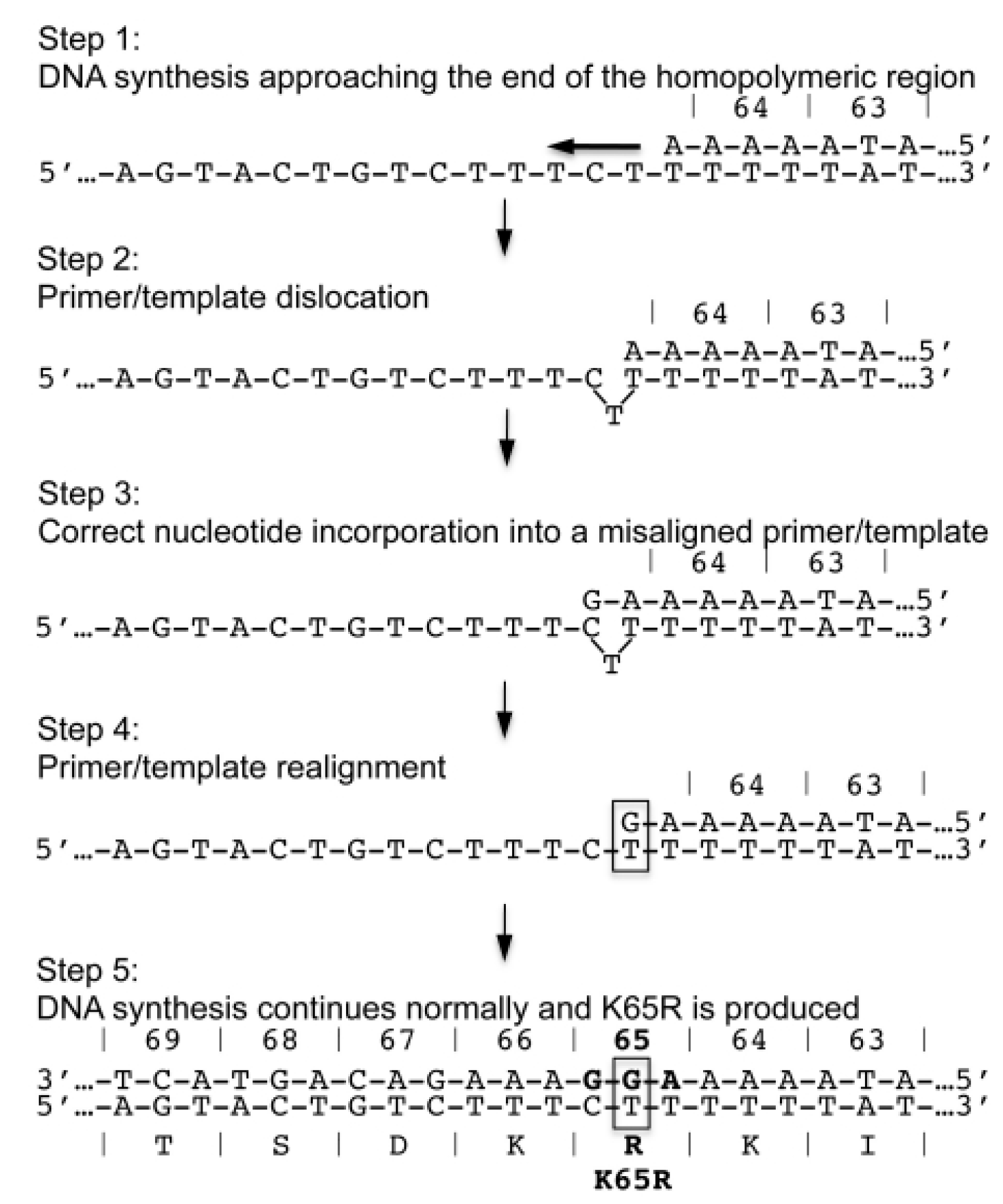
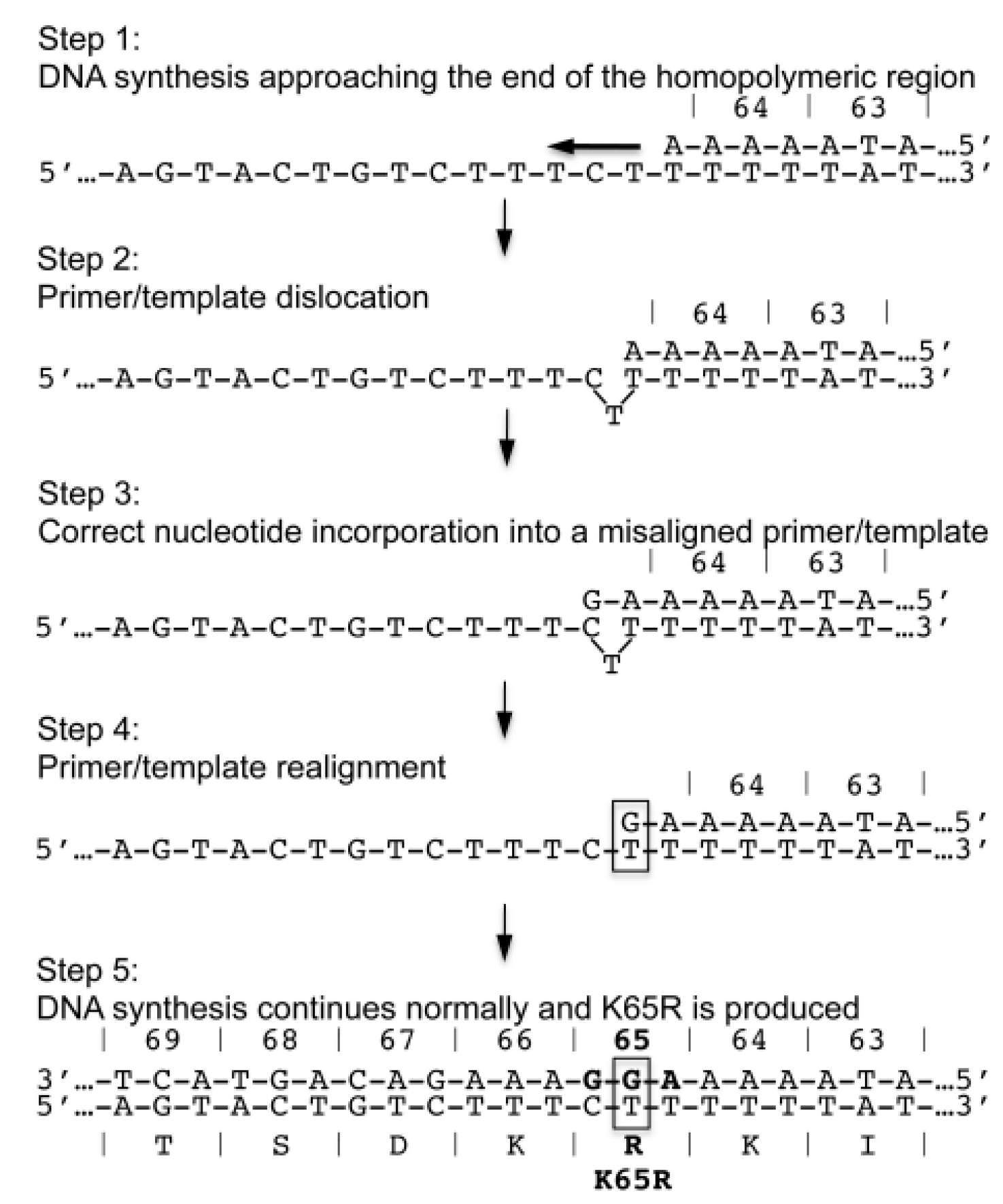
7. Subtype Differences
8. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Meyer, P.R.; Matsuura, S.E.; Mian, A.M.; So, A.G.; Scott, W.A. A mechanism of AZT resistance: An increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell 1999, 4, 35–43. [Google Scholar]
- Arion, D.; Kaushik, N.; McCormick, S.; Borkow, G.; Parniak, M.A. Phenotypic mechanism of HIV-1 resistance to 3'-azido-3'-deoxythymidine (AZT): Increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 1998, 37, 15908–15917. [Google Scholar]
- Tu, X.; Das, K.; Han, Q.; Bauman, J.D.; Clark, A.D.; Hou, X.; Frenkel, Y.V.; Gaffney, B.L.; Jones, R.A.; Boyer, P.L.; et al. Structural basis of HIV-1 resistance to AZT by excision. Nat. Struct. Mol. Biol. 2010, 17, 1202–1209. [Google Scholar]
- Gu, Z.; Gao, Q.; Li, X.; Parniak, M.A.; Wainberg, M.A. Novel mutation in the human immunodeficiency virus type 1 reverse transcriptase gene that encodes cross-resistance to 2',3'-dideoxyinosine and 2',3'-dideoxycytidine. J. Virol. 1992, 66, 7128–7135. [Google Scholar]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark, A.D.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark, P.; et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6320–6324. [Google Scholar]
- Gao, H.Q.; Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. The role of steric hindrance in 3TC resistance of human immunodeficiency virus type-1 reverse transcriptase. J. Mol. Biol. 2000, 300, 403–418. [Google Scholar]
- Shirasaka, T.; Kavlick, M.F.; Ueno, T.; Gao, W.Y.; Kojima, E.; Alcaide, M.L.; Chokekijchai, S.; Roy, B.M.; Arnold, E.; Yarchoan, R. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. USA 1995, 92, 2398–2402. [Google Scholar]
- Hachiya, A.; Kodama, E.N.; Schuckmann, M.M.; Kirby, K.A.; Michailidis, E.; Sakagami, Y.; Oka, S.; Singh, K.; Sarafianos, S.G. K70Q Adds High-Level Tenofovir Resistance to "Q151M Complex" HIV Reverse Transcriptase through the Enhanced Discrimination Mechanism. PLoS One 2011, 6, e16242. [Google Scholar]
- Deval, J.; Selmi, B.; Boretto, J.; Egloff, M.P.; Guerreiro, C.; Sarfati, S.; Canard, B. The molecular mechanism of multidrug resistance by the Q151M human immunodeficiency virus type 1 reverse transcriptase and its suppression using alpha-boranophosphate nucleotide analogues. J. Biol. Chem. 2002, 277, 42097–42104. [Google Scholar]
- Feng, J.Y.; Myrick, F.; Selmi, B.; Deval, J.; Canard, B.; Borroto-Esoda, K. Effects of HIV Q151M-associated multi-drug resistance mutations on the activities of (-)-beta-D-1',3'-dioxolan guanine. Antivir. Res. 2005, 66, 153–158. [Google Scholar]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar]
- Garforth, S.J.; Domaoal, R.A.; Lwatula, C.; Landau, M.J.; Meyer, A.J.; Anderson, K.S.; Prasad, V.R. K65R and K65A substitutions in HIV-1 reverse transcriptase enhance polymerase fidelity by decreasing both dNTP misinsertion and mispaired primer extension efficiencies. J. Mol. Biol. 2010, 401, 33–44. [Google Scholar]
- Garforth, S.J.; Kim, T.W.; Parniak, M.A.; Kool, E.T.; Prasad, V.R. Site-directed mutagenesis in the fingers subdomain of HIV-1 reverse transcriptase reveals a specific role for the beta3-beta4 hairpin loop in dNTP selection. J. Mol. Biol. 2007, 365, 38–49. [Google Scholar]
- Gu, Z.; Gao, Q.; Fang, H.; Salomon, H.; Parniak, M.A.; Goldberg, E.; Cameron, J.; Wainberg, M.A. Identification of a mutation at codon 65 in the IKKK motif of reverse transcriptase that encodes human immunodeficiency virus resistance to 2',3'-dideoxycytidine and 2',3'-dideoxy-3'-thiacytidine. Antimicrob. Agents Chemother. 1994, 38, 275–281. [Google Scholar]
- Zhang, D.; Caliendo, A.M.; Eron, J.J.; DeVore, K.M.; Kaplan, J.C.; Hirsch, M.S.; D'Aquila, R.T. Resistance to 2',3'-dideoxycytidine conferred by a mutation in codon 65 of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 1994, 38, 282–287. [Google Scholar]
- Johnson, V.A.; Calvez, V.; Günthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.; Wensing, A.M.; Richman, D.D. 2011 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2011, 19, 156–164. [Google Scholar]
- Stone, C.; Ait-Khaled, M.; Craig, C.; Griffin, P.; Tisdale, M. Human immunodeficiency virus type 1 reverse transcriptase mutation selection during in vitro exposure to tenofovir alone or combined with abacavir or lamivudine. Antimicrob. Agents Chemother. 2004, 48, 1413–1415. [Google Scholar]
- Harrigan, P.R.; Stone, C.; Griffin, P.; Nájera, I.; Bloor, S.; Kemp, S.; Tisdale, M.; Larder, B. Resistance profile of the human immunodeficiency virus type 1 reverse transcriptase inhibitor abacavir (1592U89) after monotherapy and combination therapy. CNA2001 Investigative Group. J. Infect. Dis. 2000, 181, 912–920. [Google Scholar]
- Winters, M.A.; Shafer, R.W.; Jellinger, R.A.; Mamtora, G.; Gingeras, T.; Merigan, T.C. Human immunodeficiency virus type 1 reverse transcriptase genotype and drug susceptibility changes in infected individuals receiving dideoxyinosine monotherapy for 1 to 2 years. Antimicrob. Agents Chemother. 1997, 41, 757–762. [Google Scholar]
- Gu, Z.; Arts, E.J.; Parniak, M.A.; Wainberg, M.A. Mutated K65R recombinant reverse transcriptase of human immunodeficiency virus type 1 shows diminished chain termination in the presence of 2',3'-dideoxycytidine 5'-triphosphate and other drugs. Proc. Natl. Acad. Sci. USA 1995, 92, 2760–2764. [Google Scholar]
- Gu, Z.; Fletcher, R.S.; Arts, E.J.; Wainberg, M.A.; Parniak, M.A. The K65R mutant reverse transcriptase of HIV-1 cross-resistant to 2', 3'-dideoxycytidine, 2',3'-dideoxy-3'-thiacytidine, and 2',3'-dideoxyinosine shows reduced sensitivity to specific dideoxynucleoside triphosphate inhibitors in vitro. J. Biol. Chem. 1994, 269, 28118–28122. [Google Scholar]
- Sluis-Cremer, N.; Arion, D.; Kaushik, N.; Lim, H.; Parniak, M.A. Mutational analysis of Lys65 of HIV-1 reverse transcriptase. Biochem. J. 2000, 348 Pt. 1, 77–82. [Google Scholar]
- Larder, B.A.; Chesebro, B.; Richman, D.D. Susceptibilities of zidovudine-susceptible and -resistant human immunodeficiency virus isolates to antiviral agents determined by using a quantitative plaque reduction assay. Antimicrob. Agents Chemother. 1990, 34, 436–441. [Google Scholar]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 2001, 75, 4832–4842. [Google Scholar]
- Larder, B.A.; Kemp, S.D. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science 1989, 246, 1155–1158. [Google Scholar]
- White, K.L.; Chen, J.M.; Feng, J.Y.; Margot, N.A.; Ly, J.K.; Ray, A.S.; Macarthur, H.L.; McDermott, M.J.; Swaminathan, S.; Miller, M.D. The K65R reverse transcriptase mutation in HIV-1 reverses the excision phenotype of zidovudine resistance mutations. Antivir. Ther. 2006, 11, 155–163. [Google Scholar]
- Meyer, P.R.; Matsuura, S.E.; So, A.G.; Scott, W.A. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. USA 1998, 95, 13471–13476. [Google Scholar]
- Garforth, S.J.; Parniak, M.A.; Prasad, V.R. Utilization of a deoxynucleoside diphosphate substrate by HIV reverse transcriptase. PLoS One 2008, 3, e2074. [Google Scholar]
- Das, K.; Bandwar, R.P.; White, K.L.; Feng, J.Y.; Sarafianos, S.G.; Tuske, S.; Tu, X.; Clark, A.D.; Boyer, P.L.; Hou, X.; et al. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J. Biol. Chem. 2009, 284, 35092–35100. [Google Scholar]
- Parikh, U.M.; Bacheler, L.; Koontz, D.; Mellors, J.W. The K65R mutation in human immunodeficiency virus type 1 reverse transcriptase exhibits bidirectional phenotypic antagonism with thymidine analog mutations. J. Virol. 2006, 80, 4971–4977. [Google Scholar]
- Wilson, J.E.; Aulabaugh, A.; Caligan, B.; McPherson, S.; Wakefield, J.K.; Jablonski, S.; Morrow, C.D.; Reardon, J.E.; Furman, P.A. Human immunodeficiency virus type-1 reverse transcriptase. Contribution of Met-184 to binding of nucleoside 5'-triphosphate. J. Biol Chem. 1996, 271, 13656–13662. [Google Scholar]
- Parikh, U.M.; Barnas, D.C.; Faruki, H.; Mellors, J.W. Antagonism between the HIV-1 Reverse-Transcriptase Mutation K65R and Thymidine-Analogue Mutations at the Genomic Level. J. Infect. Dis. 2006, 194, 651–660. [Google Scholar]
- Michailidis, E.; Huber, A.D.; Ryan, E.M.; Ong, Y.T.; Leslie, M.D.; Matzek, K.B.; Singh, K.; Marchand, B.; Hagedorn, A.N.; Kirby, K.A.; et al. 4'-ethynyl-2-fluoro-2'-deoxyadenosine (EFdA) inhibits HIV-1 reverse transcriptase with multiple mechanisms. J. Biol .Chem. 2014. [Google Scholar] [CrossRef]
- Michailidis, E.; Ryan, E.M.; Hachiya, A.; Kirby, K.A.; Marchand, B.; Leslie, M.D.; Huber, A.D.; Ong, Y.T.; Jackson, J.C.; Singh, K.; et al. Hypersusceptibility mechanism of Tenofovir-resistant HIV to EFdA. Retrovirology 2013, 10, 65. [Google Scholar]
- Rhee, S.-Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar]
- Shafer, R.W. Rationale and uses of a public HIV drug-resistance database. J. Infect. Dis. 2006, 194 (Suppl 1), S51–S58. [Google Scholar]
- Wolf, K.; Walter, H.; Beerenwinkel, N.; Keulen, W.; Kaiser, R.; Hoffmann, D.; Lengauer, T.; Selbig, J.; Vandamme, A.M.; Korn, K.; et al. Tenofovir resistance and resensitization. Antimicrob Agents Chemother. 2003, 47, 3478–3484. [Google Scholar]
- Margot, N.A.; Isaacson, E.; McGowan, I.; Cheng, A.K.; Schooley, R.T.; Miller, M.D. Genotypic and phenotypic analyses of HIV-1 in antiretroviral-experienced patients treated with tenofovir DF. AIDS 2002, 16, 1227–1235. [Google Scholar]
- Miller, M.D. K65R, TAMs and tenofovir. AIDS Rev. 2004, 6, 22–33. [Google Scholar]
- Deval, J.; White, K.L.; Miller, M.D.; Parkin, N.T.; Courcambeck, J.; Halfon, P.; Selmi, B.; Boretto, J.; Canard, B. Mechanistic basis for reduced viral and enzymatic fitness of HIV-1 reverse transcriptase containing both K65R and M184V mutations. J. Biol. Chem. 2004, 279, 509–516. [Google Scholar]
- Deval, J.; Navarro, J.-M.; Selmi, B.; Courcambeck, J.; Boretto, J.; Halfon, P.; Garrido-Urbani, S.; Sire, J.; Canard, B. A loss of viral replicative capacity correlates with altered DNA polymerization kinetics by the human immunodeficiency virus reverse transcriptase bearing the K65R and L74V dideoxynucleoside resistance substitutions. J. Biol. Chem. 2004, 279, 25489–25496. [Google Scholar]
- Sluis-Cremer, N.; Sheen, C.W.; Zelina, S.; Argoti Torres, P.S.; Parikh, U.M.; Mellors, J.W. Molecular Mechanism by which K70E in HIV-1 Reverse Transcriptase Confers Resistance to Nucleoside Reverse Transcriptase Inhibitors. Antimicrob Agents Chemother. 2007, 51, 48–53. [Google Scholar]
- White, K.L.; Margot, N.A.; Wrin, T.; Petropoulos, C.J.; Miller, M.D.; Naeger, L.K. Molecular mechanisms of resistance to human immunodeficiency virus type 1 with reverse transcriptase mutations K65R and K65R+M184V and their effects on enzyme function and viral replication capacity. Antimicrob Agents Chemother. 2002, 46, 3437–3446. [Google Scholar]
- Shah, F.S.; Curr, K.A.; Hamburgh, M.E.; Parniak, M.; Mitsuya, H.; Arnez, J.G.; Prasad, V.R. Differential influence of nucleoside analog-resistance mutations K65R and L74V on the overall mutation rate and error specificity of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2000, 275, 27037–27044. [Google Scholar]
- Lwatula, C.; Garforth, S.J.; Prasad, V.R. Lys66 residue as a determinant of high mismatch extension and misinsertion rates of HIV-1 reverse transcriptase. FEBS J. 2012, 279, 4010–4024. [Google Scholar]
- Fourati, S.; Visseaux, B.; Armenia, D.; Morand-Joubert, L.; Artese, A.; Charpentier, C.; van Den Eede, P.; Costa, G.; Alcaro, S.; Wirden, M.; et al. Identification of a rare mutation at reverse transcriptase Lys65 (K65E) in HIV-1-infected patients failing on nucleos(t)ide reverse transcriptase inhibitors. J. Antimicrob Chemother. 2013, 68, 2199–2204. [Google Scholar]
- Svarovskaia, E.S.; Feng, J.Y.; Margot, N.A.; Myrick, F.; Goodman, D.; Ly, J.K.; White, K.L.; Kutty, N.; Wang, R.; Borroto-Esoda, K.; et al. The A62V and S68G mutations in HIV-1 reverse transcriptase partially restore the replication defect associated with the K65R mutation. J. Acquir. Immune Defic. Syndr. 2008, 48, 428–436. [Google Scholar]
- Margot, N.A.; Lu, B.; Cheng, A.; Miller, M.D. Study 903 Team Resistance development over 144 weeks in treatment-naive patients receiving tenofovir disoproxil fumarate or stavudine with lamivudine and efavirenz in Study 903. HIV Med. 2006, 7, 442–450. [Google Scholar]
- Tebit, D.M.; Arts, E.J. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect. Dis. 2011, 11, 45–56. [Google Scholar]
- Gao, F.; Robertson, D.L.; Carruthers, C.D.; Morrison, S.G.; Jian, B.; Chen, Y.; Barré-Sinoussi, F.; Girard, M.; Srinivasan, A.; Abimiku, A.G.; et al. A comprehensive panel of near-full-length clones and reference sequences for non-subtype B isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 5680–5698. [Google Scholar]
- Brenner, B.G.; Oliveira, M.; Doualla-Bell, F.; Moisi, D.D.; Ntemgwa, M.; Frankel, F.; Essex, M.; Wainberg, M.A. HIV-1 subtype C viruses rapidly develop K65R resistance to tenofovir in cell culture. AIDS 2006, 20, F9–F13. [Google Scholar]
- Xu, H.-T.; Martinez-Cajas, J.L.; Ntemgwa, M.L.; Coutsinos, D.; Frankel, F.A.; Brenner, B.G.; Wainberg, M.A. Effects of the K65R and K65R/M184V reverse transcriptase mutations in subtype C HIV on enzyme function and drug resistance. Retrovirology 2009, 6, 14. [Google Scholar]
- Invernizzi, C.F.; Coutsinos, D.; Oliveira, M.; Moisi, D.; Brenner, B.G.; Wainberg, M.A. Signature nucleotide polymorphisms at positions 64 and 65 in reverse transcriptase favor the selection of the K65R resistance mutation in HIV-1 subtype C. J. Infect. Dis 2009, 200, 1202–1206. [Google Scholar]
- Coutsinos, D.; Invernizzi, C.F.; Xu, H.; Brenner, B.G.; Wainberg, M.A. Factors affecting template usage in the development of K65R resistance in subtype C variants of HIV type-1. Antivir. Chem. Chemother. 2010, 20, 117–131. [Google Scholar]
- Coutsinos, D.; Invernizzi, C.F.; Moisi, D.; Oliveira, M.; Martinez-Cajas, J.L.; Brenner, B.G.; Wainberg, M.A. A template-dependent dislocation mechanism potentiates K65R reverse transcriptase mutation development in subtype C variants of HIV-1. PLoS One 2011, 6, e20208. [Google Scholar]
- Invernizzi, C.F.; Coutsinos, D.; Oliveira, M.; Schildknecht, R.S.; Xu, H.; Gaseitsiwe, S.; Moisi, D.; Brenner, B.G.; Wainberg, M.A. The preferential selection of K65R in HIV-1 subtype C is attenuated by nucleotide polymorphisms at thymidine analogue mutation sites. J. Antimicrob Chemother. 2013, 68, 2192–2196. [Google Scholar]
- Doualla-Bell, F.; Avalos, A.; Brenner, B.; Gaolathe, T.; Mine, M.; Gaseitsiwe, S.; Oliveira, M.; Moisi, D.; Ndwapi, N.; Moffat, H.; et al. High prevalence of the K65R mutation in human immunodeficiency virus type 1 subtype C isolates from infected patients in Botswana treated with didanosine-based regimens. Antimicrob Agents Chemother. 2006, 50, 4182–4185. [Google Scholar]
- Hosseinipour, M.C.; van Oosterhout, J.J.G.; Weigel, R.; Phiri, S.; Kamwendo, D.; Parkin, N.; Fiscus, S.A.; Nelson, J.A.E.; Eron, J.J.; Kumwenda, J. The public health approach to identify antiretroviral therapy failure: High-level nucleoside reverse transcriptase inhibitor resistance among Malawians failing first-line antiretroviral therapy. AIDS 2009, 23, 1127–1134. [Google Scholar]
- Sungkanuparph, S.; Manosuthi, W.; Kiertiburanakul, S.; Saekang, N.; Pairoj, W.; Chantratita, W. Prevalence and risk factors for developing K65R mutations among HIV-1 infected patients who fail an initial regimen of fixed-dose combination of stavudine, lamivudine, and nevirapine. J. Clin. Virol. 2008, 41, 310–313. [Google Scholar]
- Tolle, M.; Howard, L.; Kirk, B.; Gomila, A.; Schwarzwald, H.; Anabwani, G. Reverse transcriptase genotypes in pediatric patients failing initial antiretroviral therapy in Gaborone, Botswana. J. Int. Assoc. Physicians AIDS Care (Chic.) 2012, 11, 260–268. [Google Scholar]
- Li, J.F.; Lipscomb, J.; Wei, X. Detection of minority K65R variants in NRTI-naive subtype B and C HIV-1-infected individuals. J. Infect. Dis. 2011, 203, 798–802. [Google Scholar]
- Kozal, M.J.; Chiarella, J.; St John, E.P.; Moreno, E.A.; Simen, B.B.; Arnold, T.E.; Lataillade, M. Prevalence of low-level HIV-1 variants with reverse transcriptase mutation K65R and the effect of antiretroviral drug exposure on variant levels. Antivir. Ther. 2011, 16, 925–929. [Google Scholar]
- Chan, P.A.; Huang, A.; Kantor, R. Low prevalence of transmitted K65R and other tenofovir resistance mutations across different HIV-1 subtypes: Implications for pre-exposure prophylaxis. J. Int. AIDS Soc. 2012, 15, 17701. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garforth, S.J.; Lwatula, C.; Prasad, V.R. The Lysine 65 Residue in HIV-1 Reverse Transcriptase Function and in Nucleoside Analog Drug Resistance. Viruses 2014, 6, 4080-4094. https://doi.org/10.3390/v6104080
Garforth SJ, Lwatula C, Prasad VR. The Lysine 65 Residue in HIV-1 Reverse Transcriptase Function and in Nucleoside Analog Drug Resistance. Viruses. 2014; 6(10):4080-4094. https://doi.org/10.3390/v6104080
Chicago/Turabian StyleGarforth, Scott J., Chisanga Lwatula, and Vinayaka R. Prasad. 2014. "The Lysine 65 Residue in HIV-1 Reverse Transcriptase Function and in Nucleoside Analog Drug Resistance" Viruses 6, no. 10: 4080-4094. https://doi.org/10.3390/v6104080
APA StyleGarforth, S. J., Lwatula, C., & Prasad, V. R. (2014). The Lysine 65 Residue in HIV-1 Reverse Transcriptase Function and in Nucleoside Analog Drug Resistance. Viruses, 6(10), 4080-4094. https://doi.org/10.3390/v6104080