Chromatin Dynamics during Lytic Infection with Herpes Simplex Virus 1

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. The Chromatinization of HSV-1 Genomes in Lytically Infected Cells, an Apparent Paradox
2.2. Chromatin and Histone Dynamics Are Altered in Cells Lytically Infected with HSV-1
2.2.1. HSV-1 DNA Is in Unstable Nucleosomes during Lytic Infections
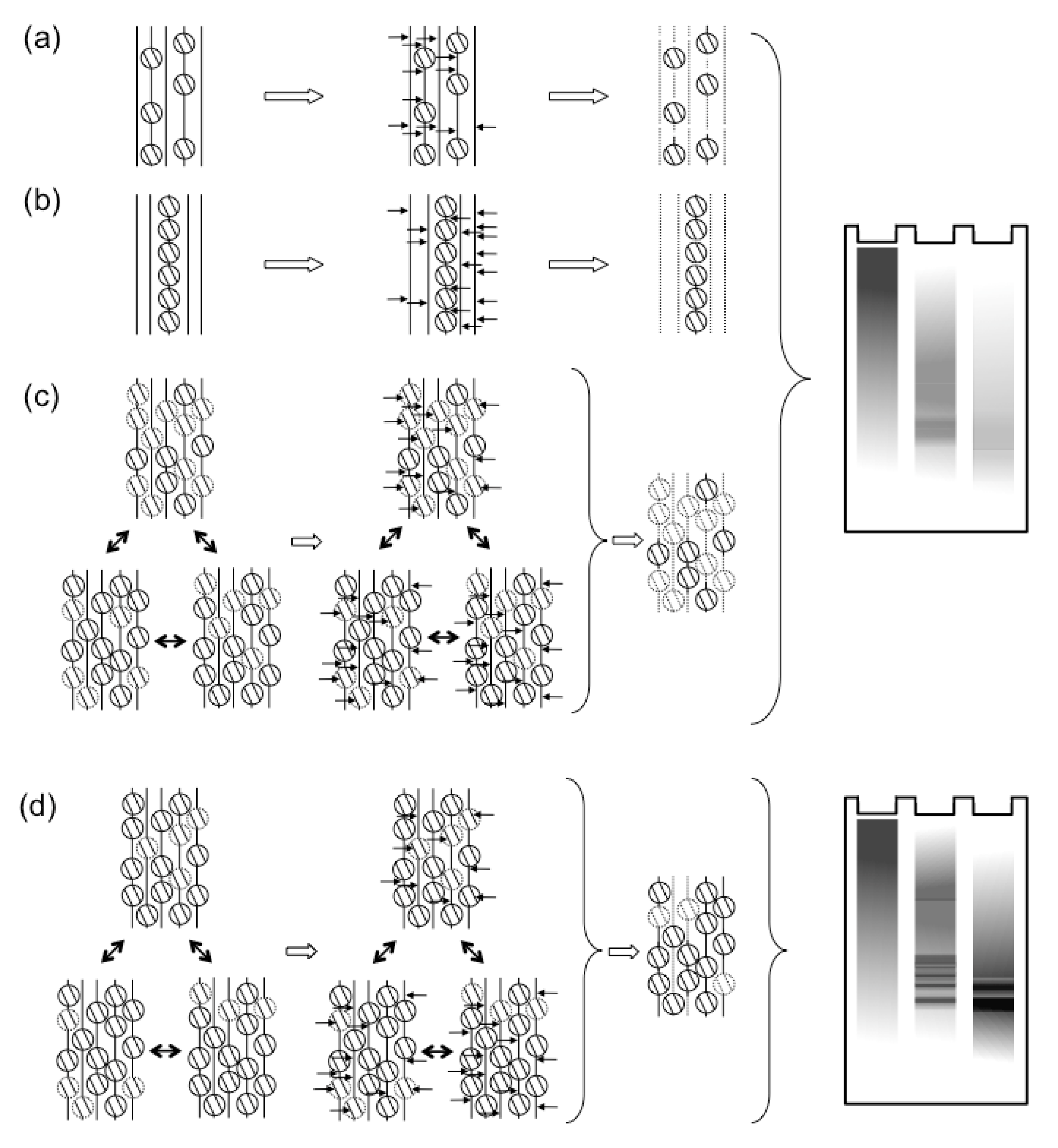
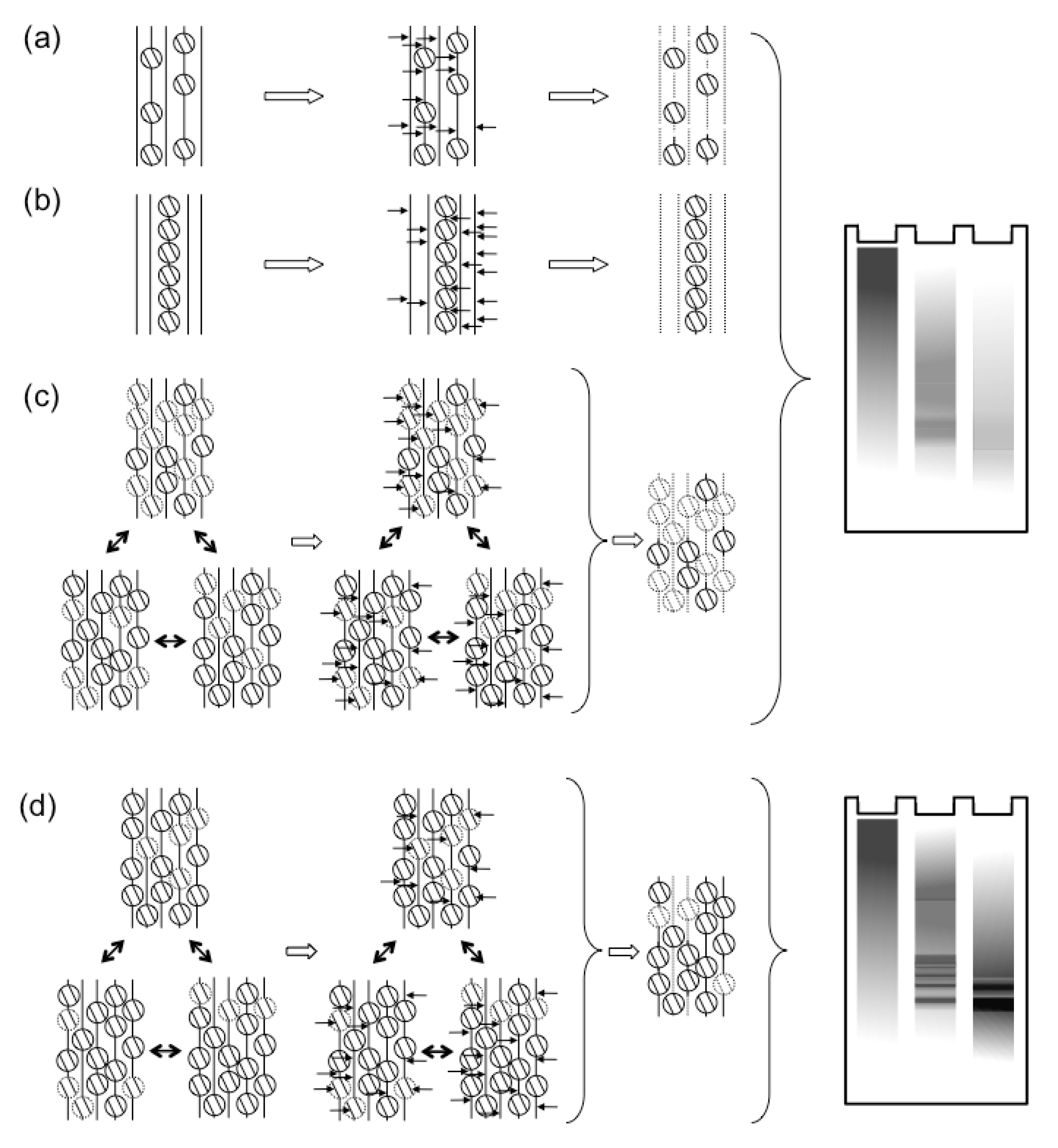
2.2.2. Histone Dynamics during Lytic Infections
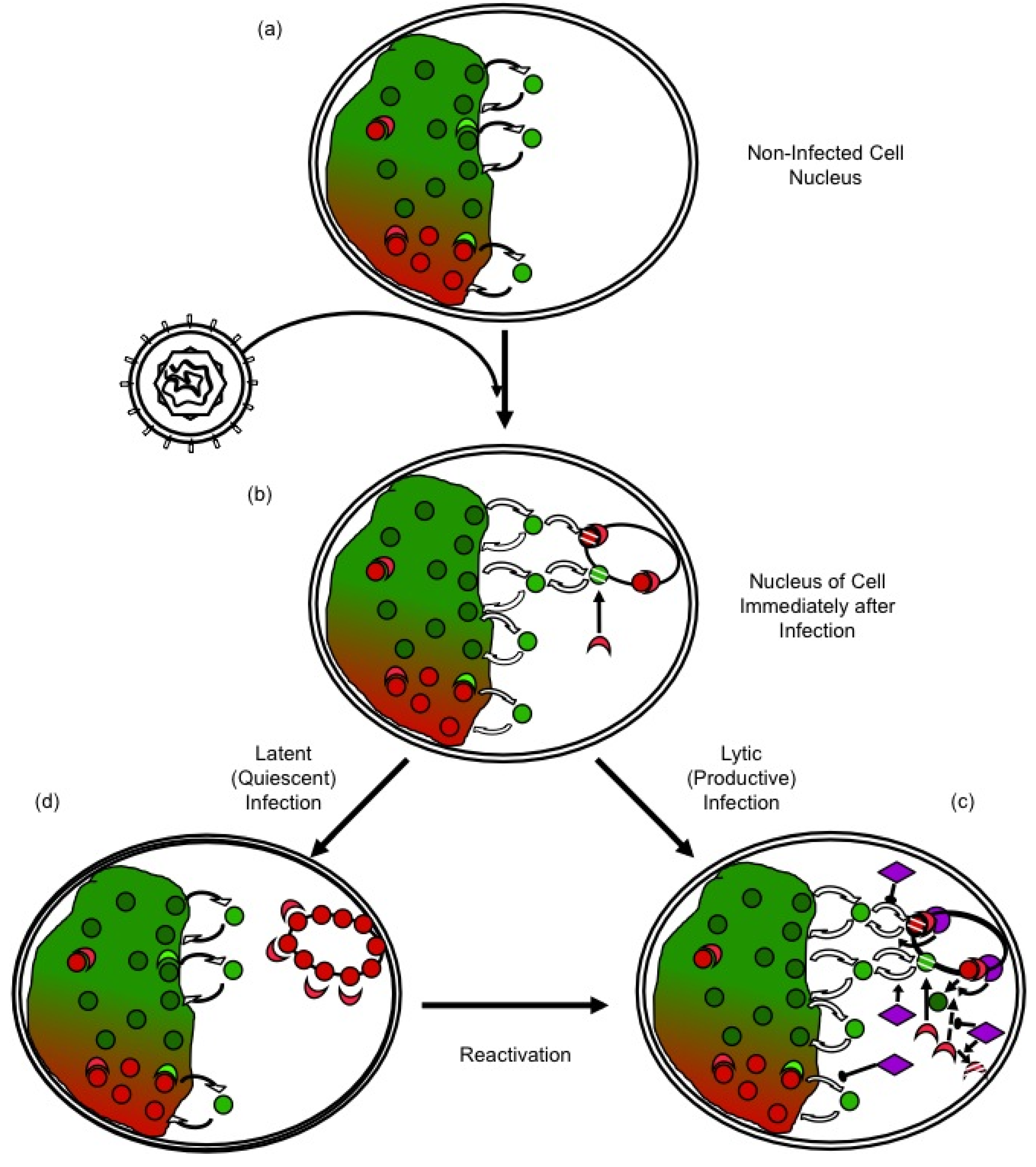
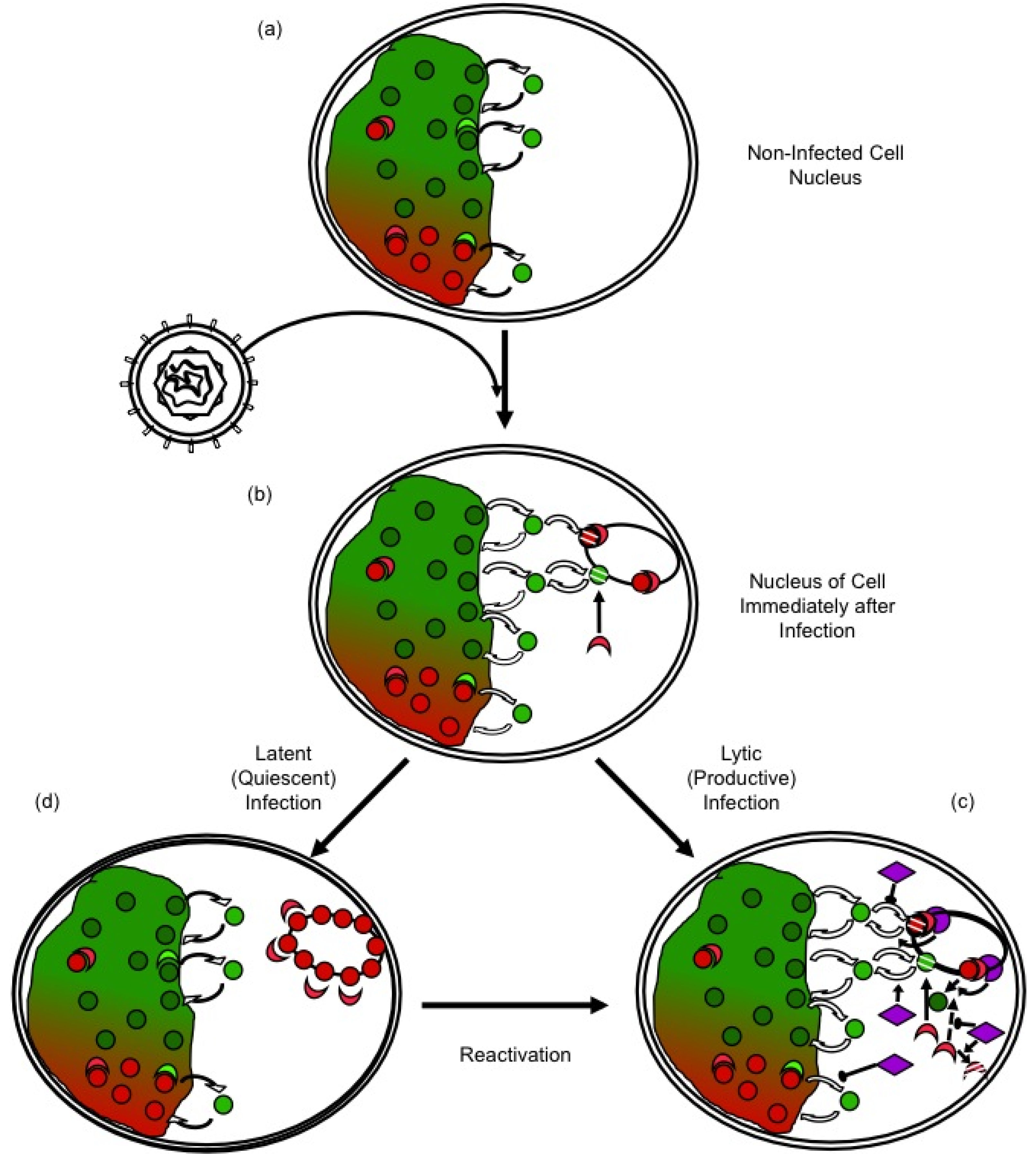
2.3. Potential Mechanisms of Regulation of Nucleosome Dynamics in HSV-1 Infected Cells
2.3.1. Mechanisms of Regulation of Nucleosome Dynamics
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Wilson, A.C.; LaMarco, K.; Peterson, M.G.; Herr, W. The vp16 accessory protein hcf is a family of polypeptides processed from a large precursor protein. Cell 1993, 74, 115–125. [Google Scholar] [CrossRef]
- Kristie, T.M.; Roizman, B. Host-cell proteins bind to the cis-acting site required for virion-mediated induction of herpes-simplex virus-1 alpha-genes. Proc. Natl. Acad. Sci. USA 1987, 84, 71–75. [Google Scholar] [CrossRef]
- Ohare, P.; Goding, C.R.; Haigh, A. Direct combinatorial interaction between a herpes-simplex virus regulatory protein and a cellular octamer-binding factor mediates specific induction of virus immediate-early gene-expression. EMBO J. 1988, 7, 4231–4238. [Google Scholar]
- Ohare, P.; Goding, C.R. Herpes-simplex virus regulatory elements and the immunoglobulin octamer domain bind a common factor and are both targets for virion transactivation. Cell 1988, 52, 435–445. [Google Scholar] [CrossRef]
- Preston, C.M.; Frame, M.C.; Campbell, M.E.M. A complex formed between cell components and an hsv structural polypeptide binds to a viral immediate early gene regulatory DNA-sequence. Cell 1988, 52, 425–434. [Google Scholar] [CrossRef]
- O'Hare, P. The virion transactivator of herpes simplex virus. Semin. Virol. 1993, 4, 145–156. [Google Scholar] [CrossRef]
- Smiley, J.R.; Smibert, C.; Everett, R.D. Expression of a cellular gene cloned in herpes simplex virus: Rabbit beta-globin is regulated as an early viral gene in infected fibroblasts. J. Virol. 1987, 61, 2368–2377. [Google Scholar]
- Smibert, C.A.; Smiley, J.R. Differential regulation of endogenous and transduced beta-globin genes during infection of erythroid cells with a herpes simplex virus type 1 recombinant. J. Virol. 1990, 64, 3882–3894. [Google Scholar]
- Boutell, C.; Everett, R.D. Regulation of alphaherpesvirus infections by the icp0 family of proteins. J. Gen. Virol. 2013, 94, 465–481. [Google Scholar] [CrossRef]
- Everett, R.D. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 2000, 22, 761–770. [Google Scholar] [CrossRef]
- Mossman, K.L.; Macgregor, P.F.; Rozmus, J.J.; Goryachev, A.B.; Edwards, A.M.; Smiley, J.R. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 2001, 75, 750–758. [Google Scholar] [CrossRef]
- Mossman, K.L.; Smiley, J.R. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J. Virol. 2002, 76, 1995–1998. [Google Scholar] [CrossRef]
- Coleman, H.M.; Connor, V.; Cheng, Z.S.; Grey, F.; Preston, C.M.; Efstathiou, S. Histone modifications associated with herpes simplex virus type 1 genomes during quiescence and following icp0-mediated de-repression. J. Gen. Virol. 2008, 89, 68–77. [Google Scholar] [CrossRef]
- Mossman, K.L.; Smiley, J.R. Truncation of the c-terminal acidic transcriptional activation domain of herpes simplex virus vp16 renders expression of the immediate-early genes almost entirely dependent on icp0. J. Virol. 1999, 73, 9726–9733. [Google Scholar]
- Preston, C.M.; Nicholl, M.J. Repression of gene expression upon infection of cells with herpes simplex virus type 1 mutants impaired for immediate-early protein synthesis. J. Virol. 1997, 71, 7807–7813. [Google Scholar]
- Harris, R.A.; Preston, C.M. Establishment of latency invitro by the herpes-simplex virus type-1 mutant in 1814. J. Gen. Virol. 1991, 72, 907–913. [Google Scholar] [CrossRef]
- Samaniego, L.A.; Neiderhiser, L.; DeLuca, N.A. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 1998, 72, 3307–3320. [Google Scholar]
- Ferenczy, M.W.; DeLuca, N.A. Epigenetic modulation of gene expression from quiescent herpes simplex virus genomes. J. Virol. 2009, 83, 8514–8524. [Google Scholar] [CrossRef]
- Danaher, R.J.; Jacob, R.J.; Steiner, M.R.; Allen, W.R.; Hill, J.M.; Miller, C.S. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J. Neurovirol. 2005, 11, 306–317. [Google Scholar] [CrossRef]
- Kuhn, M.A.; Nayak, S.; Camarena, V.; Gardner, J.; Wilson, A.; Mohr, I.; Chao, M.V.; Roehm, P.C. A cell culture model of facial palsy resulting from reactivation of latent herpes simplex type 1. Otol. Neurotol. 2012, 33, 87–92. [Google Scholar] [CrossRef]
- Du, T.; Zhou, G.Y.; Roizman, B. Induction of apoptosis accelerates reactivation of latent hsv-1 in ganglionic organ cultures and replication in cell cultures. Proc. Natl. Acad. Sci. USA 2012, 109, 14616–14621. [Google Scholar] [CrossRef]
- Du, T.; Zhou, G.Y.; Roizman, B. The activation of latent herpes simplex virus and suppression of lat and mi-rnas in trigeminal ganglia within the time-frame of a single cycle of viral replication. J. Neurovirol. 2012, 18, 32. [Google Scholar]
- Kim, J.Y.; Mandarino, A.; Chao, M.V.; Mohr, I.; Wilson, A.C. Transient reversal of episome silencing precedes vp16-dependent transcription during reactivation of latent hsv-1 in neurons. PLoS Pathog. 2012, 8, e1002540. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Dutnall, R.N. Cracking the histone code: One, two, three methyls, you're out! Mol. Cell 2003, 12, 3–4. [Google Scholar] [CrossRef]
- Turner, B.M. Histone acetylation and an epigenetic code. Bioessays 2000, 22, 836–845. [Google Scholar] [CrossRef]
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef]
- Cosgrove, M.S.; Wolberger, C. How does the histone code work? Biochem. Cell Biol. 2005, 83, 468–476. [Google Scholar] [CrossRef]
- Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 2004, 11, 1037–1043. [Google Scholar] [CrossRef]
- Hagglund, R.; Roizman, B. Role of icp0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 2004, 78, 2169–2178. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G.; Sakai, A.; Loeb, G.B.; Ahmad, K. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 2008, 19, 460–469. [Google Scholar] [CrossRef]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants h3.3 and h2a.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef]
- Jin, C.; Zang, C.; Wei, G.; Cui, K.; Peng, W.; Zhao, K.; Felsenfeld, G. H3.3/h2a.Z double variant-containing nucleosomes mark 'nucleosome-free regions' of active promoters and other regulatory regions. Nat. Genet. 2009, 41, 941–945. [Google Scholar] [CrossRef]
- Ausio, J. Histone variants—The structure behind the function. Brief. Funct. Genomic. Proteomic. 2006, 5, 228–243. [Google Scholar] [CrossRef]
- Ugrinova, I.; Pashev, I.G.; Pasheva, E.A. Nucleosome binding properties and co-remodeling activities of native and in vivo acetylated hmgb-1 and hmgb-2 proteins. Biochemistry 2009, 48, 6502–6507. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y. High mobility group proteins and their post-translational modifications. Biochim. Biophys. Acta 2008, 1784, 1159–1166. [Google Scholar] [CrossRef]
- Hock, R.; Furusawa, T.; Ueda, T.; Bustin, M. Hmg chromosomal proteins in development and disease. Trends Cell Biol. 2007, 17, 72–79. [Google Scholar] [CrossRef]
- Bonaldi, T.; Langst, G.; Strohner, R.; Becker, P.B.; Bianchi, M.E. The DNA chaperone hmgb1 facilitates acf/chrac-dependent nucleosome sliding. EMBO J. 2002, 21, 6865–6873. [Google Scholar] [CrossRef]
- Bustin, M. Chromatin unfolding and activation by hmgn(*) chromosomal proteins. Trends Biochem. Sci. 2001, 26, 431–437. [Google Scholar] [CrossRef]
- Parseghian, M.H.; Newcomb, R.L.; Hamkalo, B.A. Distribution of somatic h1 subtypes is non-random on active vs. Inactive chromatin ii: Distribution in human adult fibroblasts. J. Cell. Biochem. 2001, 83, 643–659. [Google Scholar] [CrossRef]
- Parseghian, M.H.; Newcomb, R.L.; Winokur, S.T.; Hamkalo, B.A. The distribution of somatic h1 subtypes is non-random on active vs. Inactive chromatin: Distribution in human fetal fibroblasts. Chromosome Res 2000, 8, 405–424. [Google Scholar] [CrossRef]
- Bhattacharjee, R.N.; Banks, G.C.; Trotter, K.W.; Lee, H.L.; Archer, T.K. Histone h1 phosphorylation by cdk2 selectively modulates mouse mammary tumor virus transcription through chromatin remodeling. Mol. Cell. Biol. 2001, 21, 5417–5425. [Google Scholar] [CrossRef]
- Gibson, W.; Roizman, B. Compartmentalization of spermine and spermidine in the herpes simplex virion. Proc. Natl. Acad. Sci. USA 1971, 68, 2818–2821. [Google Scholar] [CrossRef]
- Mouttet, M.E.; Guetard, D.; Bechet, J.M. Random cleavage of intranuclear herpes simplex virus DNA by micrococcal nuclease. FEBS Lett. 1979, 100, 107–109. [Google Scholar] [CrossRef]
- Lacasse, J.J.; Schang, L.M. Herpes simplex virus 1 DNA is in unstable nucleosomes throughout the lytic infection cycle, and the instability of the nucleosomes is independent of DNA replication. J. Virol. 2012, 86, 11287–11300. [Google Scholar] [CrossRef]
- Lacasse, J.J.; Schang, L.M. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J. Virol. 2010, 84, 1920–1933. [Google Scholar] [CrossRef]
- Leinbach, S.S.; Summers, W.C. The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J. Gen. Virol. 1980, 51, 45–59. [Google Scholar] [CrossRef]
- Lentine, A.F.; Bachenheimer, S.L. Intracellular organization of herpes-simplex virus type-1 dna assayed by staphylococcal nuclease sensitivity. Virus Res. 1990, 16, 275–292. [Google Scholar] [CrossRef]
- Kent, J.R.; Zeng, P.Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar]
- Muggeridge, M.I.; Fraser, N.W. Chromosomal organization of the herpes simplex virus genome during acute infection of the mouse central nervous system. J. Virol. 1986, 59, 764–767. [Google Scholar]
- Deshmane, S.L.; Fraser, N.W. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 1989, 63, 943–947. [Google Scholar]
- Ferenczy, M.W.; Deluca, N.A. Epigenetic modulation of gene expression from quiescent herpes simplex virus genomes. J. Virol. 2009, 83, 8514–8524. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; DeLuca, N.A. Reversal of heterochromatic silencing of quiescent herpes simplex virus type 1 by icp0. J. Virol. 2011, 85, 3424–3435. [Google Scholar] [CrossRef]
- Kubat, N.J.; Tran, R.K.; McAnany, P.; Bloom, D.C. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 2004, 78, 1139–1149. [Google Scholar] [CrossRef]
- Kubat, N.J.; Amelio, A.L.; Giordani, N.V.; Bloom, D.C. The herpes simplex virus type 1 latency-associated transcript (lat) enhancer/rcr is hyperacetylated during latency independently of lat transcription. J. Virol. 2004, 78, 12508–12518. [Google Scholar] [CrossRef]
- Amelio, A.L.; Giordani, N.V.; Kubat, N.J.; O'Neil, J.E.; Bloom, D.C. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (lat) enhancer and a decrease in lat abundance precede an increase in icp0 transcriptional permissiveness at early times postexplant. J. Virol. 2006, 80, 2063–2068. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Zhou, C.H.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar]
- Herrera, F.J.; Triezenberg, S.J. Vp16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 2004, 78, 9689–9696. [Google Scholar] [CrossRef]
- Huang, J.; Kent, J.R.; Placek, B.; Whelan, K.A.; Hollow, C.M.; Zeng, P.Y.; Fraser, N.W.; Berger, S.L. Trimethylation of histone h3 lysine 4 by set1 in the lytic infection of human herpes simplex virus 1. J. Virol. 2006, 80, 5740–5746. [Google Scholar] [CrossRef]
- Oh, J.; Fraser, N.W. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J. Virol. 2008, 82, 3530–3537. [Google Scholar] [CrossRef]
- Hancock, M.H.; Cliffe, A.R.; Knipe, D.M.; Smiley, J.R. Herpes simplex virus vp16, but not icp0, is required to reduce histone occupancy and enhance histone acetylation on viral genomes in u2os osteosarcoma cells. J. Virol. 2010, 84, 1366–1375. [Google Scholar] [CrossRef]
- Placek, B.J.; Huang, J.; Kent, J.R.; Dorsey, J.; Rice, L.; Fraser, N.W.; Berger, S.L. The histone variant h3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 2009, 83, 1416–1421. [Google Scholar] [CrossRef]
- Kristie, T.M.; Liang, Y.; Vogel, J.L. Control of alpha-herpesvirus ie gene expression by hcf-1 coupled chromatin modification activities. Biochim. Biophys. Acta 2010, 1799, 257–265. [Google Scholar] [CrossRef]
- Ng, H.H.; Robert, F.; Young, R.A.; Struhl, K. Targeted recruitment of set1 histone methylase by elongating pol ii provides a localized mark and memory of recent transcriptional activity. Mol. Cell 2003, 11, 709–719. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog lsd1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the histone demethylase lsd1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef]
- Kutluay, S.B.; DeVos, S.L.; Klomp, J.E.; Triezenberg, S.J. Transcriptional coactivators are not required for herpes simplex virus type 1 immediate-early gene expression in vitro. J. Virol. 2009, 83, 3436–3449. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus icp0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008, 82, 12030–12038. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; Ranayhossaini, D.J.; DeLuca, N.A. Activities of icp0 involved in the reversal of silencing of quiescent herpes simplex virus 1. J. Virol. 2011, 85, 4993–5002. [Google Scholar]
- Kristie, T.M.; Liang, Y.; Vogel, J.L. Control of alpha-herpesvirus ie gene expression by hcf-1 coupled chromatin modification activities. Biochim. Biophys. Acta 2010, 1799, 257–265. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Zhou, G.Y.; Te, D.; Roizman, B. The corest/rest repressor is both necessary and inimical for expression of herpes simplex virus genes. MBio 2011, 2, e00313-10. [Google Scholar]
- Kobiler, O.; Lipman, Y.; Therkelsen, K.; Daubechies, I.; Enquist, L.W. Herpesviruses carrying a brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat. Commun. 2010, 1, 146. [Google Scholar] [CrossRef]
- Hoshino, Y.; Dalai, S.K.; Wang, K.; Pesnicak, L.; Lau, T.Y.; Knipe, D.M.; Cohen, J.I.; Straus, S.E. Comparative efficacy and immunogenicity of replication-defective, recombinant glycoprotein, and DNA vaccines for herpes simplex virus 2 infections in mice and guinea pigs. J. Virol. 2005, 79, 4554. [Google Scholar] [CrossRef]
- Lomonte, P.; Thomas, J.; Texier, P.; Caron, C.; Khochbin, S.; Epstein, A.L. Functional interaction between class ii histone deacetylases and icp0 of herpes simplex virus type 1. J. Virol. 2004, 78, 6744–6757. [Google Scholar] [CrossRef]
- Poon, A.P.; Gu, H.; Roizman, B. Icp0 and the us3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 9993–9998. [Google Scholar] [CrossRef]
- Gu, H.D.; Liang, Y.; Mandel, G.; Roizman, B. Components of the rest/corest/histone deacetylase repressor complex are disrupted, modified, and translocated in hsv-1-infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 7571–7576. [Google Scholar] [CrossRef]
- Lomonte, P.; Sullivan, K.F.; Everett, R.D. Degradation of nucleosome-associated centromeric histone h3-like protein cenp-a induced by herpes simplex virus type 1 protein icp0. J. Biol. Chem. 2001, 276, 5829–5835. [Google Scholar] [CrossRef]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral e3 ligase targets rnf8 and rnf168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef]
- Smibert, C.A.; Smiley, J.R. Differential regulation of endogenous and transduced beta- globin genes during infection of erythroid-cells with a herpes- simplex virus type-1 recombinant. J. Virol. 1990, 64, 3882–3894. [Google Scholar]
- Smiley, J.R.; Johnson, D.C.; Pizer, L.I.; Everett, R.D. The icp4 binding sites in the herpes simplex virus type 1 glycoprotein d (gd) promoter are not essential for efficient gd transcription during virus infection. J. Virol. 1992, 66, 623–631. [Google Scholar]
- Carrozza, M.J.; DeLuca, N. The high mobility group protein 1 is a coactivator of herpes simplex virus ICP4 in vitro. J. Virol. 1998, 72, 6752–6757. [Google Scholar]
- Panagiotidis, C.A.; Silverstein, S.J. The host-cell architectural protein hmg i(y) modulates binding of herpes simplex virus type 1 icp4 to its cognate promoter. Virology 1999, 256, 64–74. [Google Scholar] [CrossRef]
- Tumbar, T.; Sudlow, G.; Belmont, A.S. Large-scale chromatin unfolding and remodeling induced by vp16 acidic activation domain. J. Cell Biol. 1999, 145, 1341–1354. [Google Scholar] [CrossRef]
- Belmont, A.; Hu, Y.; Sinclair, P.; Wu, W.; Bian, Q.; Kireev, I. Insights into interphase large-scale chromatin structure from analysis of engineered chromosome regions. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 453–460. [Google Scholar] [CrossRef]
- Zhou, G.Y.; Du, T.; Roizman, B. Hsv carrying wt rest establishes latency but reactivates only if the synthesis of rest is suppressed. Proc. Natl. Acad. Sci. USA 2013, 110, E498–E506. [Google Scholar] [CrossRef]
- Kutluay, S.B.; Triezenberg, S.J. Role of chromatin during herpesvirus infections. Biochim. Biophys. Acta 2009, 1790, 456–466. [Google Scholar] [CrossRef]
- Harley, C.A.; Dasgupta, A.; Wilson, D.W. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: Role for organelle acidification in assembly of infectious particles. J. Virol. 2001, 75, 1236–1251. [Google Scholar] [CrossRef]
- Schek, N.; Bachenheimer, S.L. Degradation of cellular messenger-rnas induced by a virion-associated factor during herpes-simplex virus-infection of vero cells. J. Virol. 1985, 55, 601–610. [Google Scholar]
- Sorenson, C.M.; Hart, P.A.; Ross, J. Analysis of herpes-simplex virus-induced messenger-rna destabilizing activity using an invitro messenger-rna decay system. Nucleic Acids Res. 1991, 19, 4459–4465. [Google Scholar] [CrossRef]
- Yager, D.R.; Bachenheimer, S.L. Synthesis and metabolism of cellular transcripts in HSV-1 infected cells. Virus Genes 1988, 1, 135–148. [Google Scholar] [CrossRef]
- Conn, K.L.; Hendzel, M.J.; Schang, L.M. Linker histones are mobilized during infection with herpes simplex virus type 1. J. Virol. 2008, 82, 8629–8646. [Google Scholar] [CrossRef]
- Higashi, T.; Matsunaga, S.; Isobe, K.; Morimoto, A.; Shimada, T.; Kataoka, S.; Watanabe, W.; Uchiyama, S.; Itoh, K.; Fukui, K. Histone h2a mobility is regulated by its tails and acetylation of core histone tails. Biochem. Biophys. Res. Commun. 2007, 357, 627–632. [Google Scholar] [CrossRef]
- Kimura, H. Histone dynamics in living cells revealed by photobleaching. DNA Repair 2005, 4, 939–950. [Google Scholar] [CrossRef]
- Gong, M.; Ni, J.H.; Jia, H.T. Increased exchange rate of histone h1 on chromatin by exogenous myogenin expression. Cell Res. 2002, 12, 395–400. [Google Scholar] [CrossRef]
- Conn, K.L.; Hendzel, M.J.; Schang, L.M. Core histones H2B and H4 are mobilized during infection with herpes simplex virus 1. J. Virol. 2011, 85, 13234–13252. [Google Scholar] [CrossRef]
- Conn, K.L.; Hendzel, M.J.; Schang, L.M.; University of Alberta, Edmonton, AB, Canada. The differential mobilization of histones h3.1 and h3.3 by herpes simplex virus 1 is consistent with their differential assembly in viral chromatin. Submitted for publication; 2013. [Google Scholar]
- Lukashchuk, V.; Everett, R. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by nd10 components atrx and hdaxx. J. Virol. 2010, 84, 4026–4040. [Google Scholar] [CrossRef]
- Glass, M.; Everett, R.D. Components of promyelocytic leukemia nuclear bodies (nd10) act cooperatively to repress herpesvirus infection. J. Virol. 2013, 87, 2174–2185. [Google Scholar]
- Narayanan, A.; Ruyechan, W.T.; Kristie, T.M. The coactivator host cell factor-1 mediates set1 and mll1 h3k4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc. Natl. Acad. Sci. USA 2007, 104, 10835–10840. [Google Scholar] [CrossRef]
- Liang, Y.; Quenelle, D.; Vogel, J.; Mascaro, C.; Ortega, A.; Kristie, T. A novel selective lsd1/kdm1a inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. MBio 2013, 4, e00558-12. [Google Scholar]
- Liang, Y.; Vogel, J.; Arbuckle, J.; Rai, G.; Jadhav, A.; Simeonov, A.; Maloney, D.; Kristie, T. Targeting the jmjd2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci. Transl. Med. 2013, 5, 167ra165. [Google Scholar]
- Bryant, K.; Colgrove, R.; Knipe, D. Cellular snf2h chromatin-remodeling factor promotes herpes simplex virus 1 immediate-early gene expression and replication. MBio 2011, 2, e00330-10. [Google Scholar]
- Memedula, S.; Belmont, A. Sequential recruitment of hat and swi/snf components to condensed chromatin by VP16. Curr. Biol. 2003, 13, 241–246. [Google Scholar] [CrossRef]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone h3.1 and h3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef]
- Lewis, P.; Elsaesser, S.; Noh, K.; Stadler, S.; Allis, C. 3-specific histone chaperone and cooperates with atrx in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar]
- Sawatsubashi, S.; Murata, T.; Lim, J.; Fujiki, R.; Ito, S.; Suzuki, E.; Tanabe, M.; Zhao, Y.; Kimura, S.; Fujiyama, S.; et al. A histone chaperone, dek, transcriptionally coactivates a nuclear receptor. Genes Dev. 2010, 24, 159–170. [Google Scholar] [CrossRef]
- Everett, R.; Murray, J.; Orr, A.; Preston, C. Herpes simplex virus type 1 genomes are associated with nd10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 2007, 81, 10991–11004. [Google Scholar] [CrossRef]
- Goldberg, A.; Banaszynski, L.; Noh, K.; Lewis, P.; Elsaesser, S.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant h3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef]
- Peng, H.; Vogel, J.L.; Nogueira, M.; Kristie, T.M. The transcriptional coactivator hcf-1 couples hsv DNA replication components to the histone chaperone asf1b. In Proceedings of the 34th International Herpesvirus Workshop, Ithaca, NY, USA, 26–30 July 2009; p. 5.15.
- Groth, A.; Corpet, A.; Cook, A.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of replication fork progression through histone supply and demand. Science 2007, 318, 1928–1931. [Google Scholar] [CrossRef]
- Lomonte, P.; Morency, E. Centromeric protein cenp-b proteasomal degradation induced by the viral protein icp0. FEBS Lett. 2007, 581, 658–662. [Google Scholar]
- Catez, F.; Gross, S.; Morency, E.; Sabra, M.; Texier, P.; Lomonte, P. The icp0 protein of hsv-1 induces centromere architecture breakdown, and triggers the interphase centromere damage response (icdr). In In Proceedings of the 33rd International Herpesvirus Workshop, Estoril, Portugal, 27 July–1 August 2008.
- Catez, F.; Crepin, S.; Gross, S.; Francelle, L.; Labetoulle, M.; Lomonte, P. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PLoS One 2012, 7, e44227. [Google Scholar]
- Bodor, D.; Valente, L.; Mata, J.; Black, B.; Jansen, L. Assembly in g1 phase and long-term stability are unique intrinsic features of cenp-a nucleosomes. Mol. Biol. Cell 2013, 24, 923–932. [Google Scholar] [CrossRef]
- Shuaib, M.; Ouararhni, K.; Dimitrov, S.; Hamiche, A. Hjurp binds cenp-a via a highly conserved n-terminal domain and mediates its deposition at centromeres. Proc. Natl. Acad. Sci. USA 2010, 107, 1349–1354. [Google Scholar] [CrossRef]
- Barnhart, M.; Kuich, P.; Stellfox, M.; Ward, J.; Bassett, E.; Black, B.; Foltz, D. Hjurp is a cenp-a chromatin assembly factor sufficient to form a functional de novo kinetochore. J. Cell Biol. 2011, 194, 229–243. [Google Scholar] [CrossRef]
- Foltz, D.; Jansen, L.; Bailey, A.; Yates, J.r.; Bassett, E.; Wood, S.; Black, B.; Cleveland, D. Centromere-specific assembly of cenp-a nucleosomes is mediated by hjurp. Cell 2009, 137, 472–484. [Google Scholar] [CrossRef]
- Li, W.; Cun, W.; Liu, L.; Hong, M.; Wang, L.; Wang, L.; Dong, C.; Li, Q. The transactivating effect of hsv-1 icp0 is enhanced by its interaction with the pcaf component of histone acetyltransferase. Arch. Virol. 2009, 154, 1755–1764. [Google Scholar] [CrossRef]
- Belmont, A.S.; Dietzel, S.; Nye, A.C.; Strukov, Y.G.; Tumbar, T. Large-scale chromatin structure and function. Curr. Opin. Cell Biol. 1999, 11, 307–311. [Google Scholar] [CrossRef]
- Gu, H.D.; Roizman, B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the corest-rest complex. Proc. Natl. Acad. Sci. USA 2007, 104, 17134–17139. [Google Scholar] [CrossRef]
- Melroe, G.; Silva, L.; Schaffer, P.; Knipe, D. Recruitment of activated irf-3 and cbp/p300 to herpes simplex virus icp0 nuclear foci: Potential role in blocking ifn-beta induction. Virology 2007, 360, 305–321. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. Circadian clock histone acetyl transferase localizes at nd10 nuclear bodies and enables herpes simplex virus gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17721–17726. [Google Scholar]
- Kutluay, S.B.; Triezenberg, S.J. Regulation of histone deposition on the herpes simplex virus type 1 genome during lytic infection. J. Virol. 2009, 83, 5835–5845. [Google Scholar] [CrossRef]
- Lee, J.; Lee, Y.; Lee, M.; Park, E.; Kang, S.; Chung, C.; Lee, K.; Kim, K. Dual modification of bmal1 by sumo2/3 and ubiquitin promotes circadian activation of the clock/bmal1 complex. Mol. Cell. Biol. 2008, 28, 6056–6065. [Google Scholar] [CrossRef]
- Doi, M.; Hirayama, J.; Sassone-Corsi, P. Circadian regulator clock is a histone acetyltransferase. Cell 2006, 125, 497–508. [Google Scholar]
- Wysocka, J.; Myers, M.; Laherty, C.; Eisenman, R.; Herr, W. Human sin3 deacetylase and trithorax-related set1/ash2 histone h3-k4 methyltransferase are tethered together selectively by the cell-proliferation factor hcf-1. Genes Dev. 2003, 17, 896–911. [Google Scholar] [CrossRef]
- Vogel, J.L.; Kristie, T.M. The dynamics of hcf-1 modulation of herpes simplex virus chromatin during initiation of infection. Viruses 2013, 5, 1272–1291. [Google Scholar] [CrossRef]
- Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Borchers, C.; Tempst, P.; Zhang, Y. Purification and functional characterization of a histone h3-lysine 4-specific methyltransferase. Mol. Cell 2001, 8, 1207–1217. [Google Scholar] [CrossRef]
- Nishioka, K.; Chuikov, S.; Sarma, K.; Erdjument-Bromage, H.; Allis, C.; Tempst, P.; Reinberg, D. Set9, a novel histone h3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002, 16, 479–489. [Google Scholar] [CrossRef]
- Wilkinson, D.; Weller, S. Herpes simplex virus type 1 disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 2006, 119, 2695–2703. [Google Scholar] [CrossRef]
- North, J.; Javaid, S.; Ferdinand, M.; Chatterjee, N.; Picking, J.; Shoffner, M.; Nakkula, R.; Bartholomew, B.; Ottesen, J.; Fishel, R.; et al. Phosphorylation of histone h3(t118) alters nucleosome dynamics and remodeling. Nucleic Acids Res. 2011, 39, 6465–6474. [Google Scholar]
- Kumar, A.; Kashyap, M.; Bhavesh, N.; Yogavel, M.; Sharma, A. Structural delineation of histone post-translation modifications in histone-nucleosome assembly protein complex. J. Struct. Biol. 2012, 180, 1–9. [Google Scholar] [CrossRef]
- Metzger, E.; Imhof, A.; Patel, D.; Kahl, P.; Hoffmeyer, K.; Friedrichs, N.; Muller, J.; Greschik, H.; Kirfel, J.; Ji, S.; et al. Phosphorylation of histone H3T6 by PKCbeta(i) controls demethylation at histone H3K4. Nature 2010, 464, 792–796. [Google Scholar] [CrossRef]
- Gehani, S.; Agrawal-Singh, S.; Dietrich, N.; Christophersen, N.; Helin, K.; Hansen, K. Polycomb group protein displacement and gene activation through msk-dependent h3k27me3s28 phosphorylation. Mol. Cell 2010, 39, 886–900. [Google Scholar] [CrossRef]
- Fischle, W.; Tseng, B.; Dormann, H.; Ueberheide, B.; Garcia, B.; Shabanowitz, J.; Hunt, D.; Funabiki, H.; Allis, C. Regulation of hp1-chromatin binding by histone h3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef]
- Hirota, T.; Lipp, J.; Toh, B.; Peters, J. Histone h3 serine 10 phosphorylation by aurora b causes hp1 dissociation from heterochromatin. Nature 2005, 438, 1176–1180. [Google Scholar] [CrossRef]
- Lo, W.; Trievel, R.; Rojas, J.; Duggan, L.; Hsu, J.; Allis, C.; Marmorstein, R.; Berger, S. Phosphorylation of serine 10 in histone h3 is functionally linked in vitro and in vivo to gcn5-mediated acetylation at lysine 14. Mol. Cell 2000, 5, 917–926. [Google Scholar] [CrossRef]
- Raghuram, N.; Carrero, G.; Th'ng, J.; Hendzel, M.J. Molecular dynamics of histone h1. Biochem. Cell Biol. 2009, 87, 189–206. [Google Scholar] [CrossRef]
- Herrera, J.E.; West, K.L.; Schiltz, R.L.; Nakatani, Y.; Bustin, M. Histone h1 is a specific repressor of core histone acetylation in chromatin. Mol. Cell. Biol. 2000, 20, 523–529. [Google Scholar] [CrossRef]
- Horn, P.; Carruthers, L.; Logie, C.; Hill, D.; Solomon, M.; Wade, P.; Imbalzano, A.; Hansen, J.; Peterson, C. Phosphorylation of linker histones regulates atp-dependent chromatin remodeling enzymes. Nat. Struct. Biol. 2002, 9, 263–267. [Google Scholar] [CrossRef]
- Hendzel, M.J.; Lever, M.A.; Crawford, E.; Th'ng, J.P. The c-terminal domain is the primary determinant of histone h1 binding to chromatin in vivo. J. Biol. Chem. 2004, 279, 20028–20034. [Google Scholar]
- Everett, R.D. Icp0 induces the accumulation of colocalizing conjugated ubiquitin. J. Virol. 2000, 74, 9994–10005. [Google Scholar] [CrossRef]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein icp0 and is isolated ring finger domain act as ubiquitin e3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef]
- Jason, L.; Finn, R.; Lindsey, G.; Ausio, J. Histone h2a ubiquitination does not preclude histone h1 binding, but it facilitates its association with the nucleosome. J. Biol. Chem. 2005, 280, 4975–4982. [Google Scholar]
- Trojer, P.; Cao, A.; Gao, Z.; Li, Y.; Zhang, J.; Xu, X.; Li, G.; Losson, R.; Erdjument-Bromage, H.; Tempst, P.; et al. L3mbtl2 protein acts in concert with pcg protein-mediated monoubiquitination of h2a to establish a repressive chromatin structure. Mol. Cell 2011, 42, 438–450. [Google Scholar] [CrossRef]
- Lilley, C.; Chaurushiya, M.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.; Stewart, G.; Durocher, D.; Weitzman, M. A viral e3 ligase targets rnf8 and rnf168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef]
- Conn, K.L.; Hendzel, M.J.; Schang, L.M.; University of Alberta, Edmonton, AB, Canada. Histone H2A and H2A.Z are mobilized in HSV-1 infected cells. Unpublished Work; 2013. [Google Scholar]
- Mattiroli, F.; Vissers, J.; van Dijk, W.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.; Sixma, T. Rnf168 ubiquitinates k13–15 on h2a/h2ax to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef]
- Thakar, A.; Parvin, J.; Zlatanova, J. Brca1/bard1 e3 ubiquitin ligase can modify histones h2a and h2b in the nucleosome particle. J. Biomol. Struct. Dyn. 2010, 27, 399–406. [Google Scholar] [CrossRef]
- Lilley, C.; Chaurushiya, M.; Boutell, C.; Everett, R.; Weitzman, M. The intrinsic antiviral defense to incoming hsv-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein icp0. PLoS Pathog. 2011, 7, e1002084. [Google Scholar] [CrossRef]
- Chandrasekharan, M.; Huang, F.; Sun, Z. Ubiquitination of histone h2b regulates chromatin dynamics by enhancing nucleosome stability. Proc. Natl. Acad. Sci. USA 2009, 106, 16686–16691. [Google Scholar] [CrossRef]
- Batta, K.; Zhang, Z.; Yen, K.; Goffman, D.; Pugh, B. Genome-wide function of h2b ubiquitylation in promoter and genic regions. Genes Dev. 2011, 25, 2254–2265. [Google Scholar] [CrossRef]
- Trujillo, K.; Osley, M. A role for H2B ubiquitylation in DNA replication. Mol. Cell 2012, 48, 734–746. [Google Scholar] [CrossRef]
- Briggs, S.; Xiao, T.; Sun, Z.; Caldwell, J.; Shabanowitz, J.; Hunt, D.; Allis, C.; Strahl, B. Gene silencing: Trans-histone regulatory pathway in chromatin. Nature 2002, 418, 498. [Google Scholar] [CrossRef]
- Venkatesh, S.; Smolle, M.; Li, H.; Gogol, M.; Saint, M.; Kumar, S.; Natarajan, K.; Workman, J. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 2012, 489, 452–455. [Google Scholar] [CrossRef]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of homologous recombination by MF20-dependent H2B ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef]
- Boutell, C.; Cuchet-Lourenco, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R. A viral ubiquitin ligase has substrate preferential sumo targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef]
- Shiio, Y.; Eisenman, R. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef]
- Nathan, D.; Ingvarsdottir, K.; Sterner, D.; Bylebyl, G.; Dokmanovic, M.; Dorsey, J.; Whelan, K.; Krsmanovic, M.; Lane, W.; Meluh, P.; et al. Histone sumoylation is a negative regulator in saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev. 2006, 20, 966–976. [Google Scholar] [CrossRef]
- Kalocsay, M.; Hiller, N.; Jentsch, S. Chromosome-wide rad51 spreading and sumo-h2a.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cel 2009, 33, 335–343. [Google Scholar] [CrossRef]
- Messner, S.; Hottiger, M. Histone adp-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011, 21, 534–542. [Google Scholar]
- Petesch, S.; Lis, J. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at hsp70 loci. Cell 2008, 134, 74–84. [Google Scholar] [CrossRef]
- Grady, S.; Hwang, J.; Vastag, L.; Rabinowitz, J.; Shenk, T. Herpes simplex virus 1 infection activates poly(adp-ribose) polymerase and triggers the degradation of poly(adp-ribose) glycohydrolase. J. Virol. 2012, 86, 8259–8268. [Google Scholar] [CrossRef]
- Taylor, T.; Knipe, D. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar] [CrossRef]
- Li, Z.; Yamauchi, Y.; Kamakura, M.; Murayama, T.; Goshima, F.; Kimura, H.; Nishiyama, Y. Herpes simplex virus requires poly(adp-ribose) polymerase activity for efficient replication and induces extracellular signal-related kinase-dependent phosphorylation and ICP0-dependent nuclear localization of tankyrase 1. J. Virol. 2012, 86, 492–503. [Google Scholar] [CrossRef]
- Dechassa, M.; Sabri, A.; Pondugula, S.; Kassabov, S.; Chatterjee, N.; Kladde, M.; Bartholomew, B. Swi/snf has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol. Cell 2010, 38, 590–602. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Xu, Y.; Ayrapetov, M.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B. Histone h2a.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 2012, 48, 723–733. [Google Scholar]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.; Hu, S.; Woodard, C.; Lin, J.; Taverna, S.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and tip60 histone acetyltransferase to promote virus replication. Cell Host Microbe 2011, 10, 390–400. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Conn, K.L.; Schang, L.M. Chromatin Dynamics during Lytic Infection with Herpes Simplex Virus 1. Viruses 2013, 5, 1758-1786. https://doi.org/10.3390/v5071758
Conn KL, Schang LM. Chromatin Dynamics during Lytic Infection with Herpes Simplex Virus 1. Viruses. 2013; 5(7):1758-1786. https://doi.org/10.3390/v5071758
Chicago/Turabian StyleConn, Kristen L., and Luis M. Schang. 2013. "Chromatin Dynamics during Lytic Infection with Herpes Simplex Virus 1" Viruses 5, no. 7: 1758-1786. https://doi.org/10.3390/v5071758
APA StyleConn, K. L., & Schang, L. M. (2013). Chromatin Dynamics during Lytic Infection with Herpes Simplex Virus 1. Viruses, 5(7), 1758-1786. https://doi.org/10.3390/v5071758