West Nile Virus Drug Discovery

Abstract
:1. Introduction
3. Inhibitors of Viral Targets
3.1. Viral Entry Inhibitors
3.2. Therapeutic Antibody
3.3. NS3
3.3.1. NS3 Protease
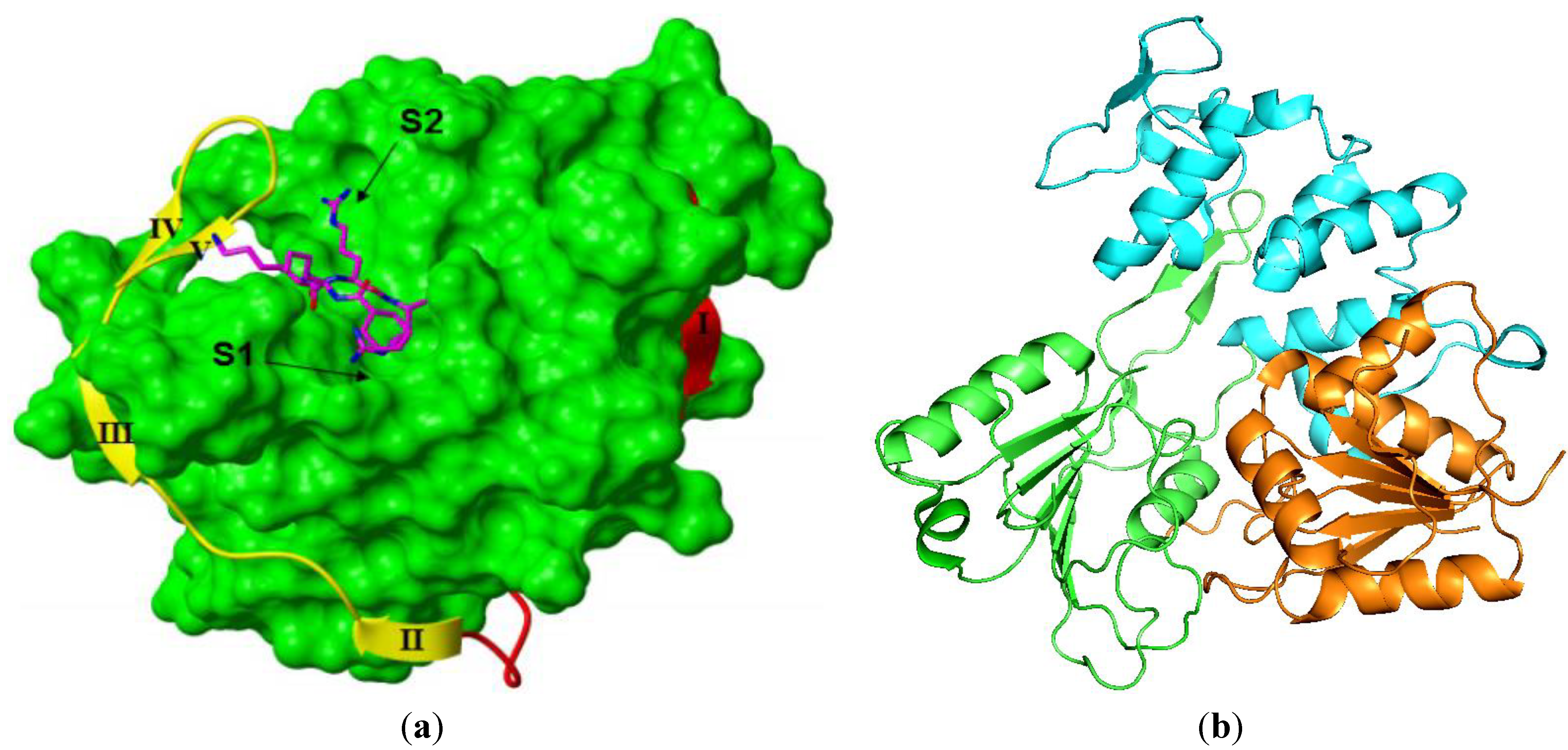

{kind=link}
{kind=link}
| Compound | WNV Ki (µM) | Binding Mode | Co-crystallised with WNV NS2B/3pro | Anti-WNV cellular activity | Ref |
|---|---|---|---|---|---|
| Aprotinin | 0.026; 0.09 ± 0.02 | non-covalent | 34 | [73,74] | |
| Bz-Nle-KRR-H | 4.1 | covalent; warhead | 31 | [70] | |
| Bz-Nle-KKR-H | 1.9 | covalent; warhead | [70] | ||
| Bz-Nle-KR(p-guanidinyl)F-H | 12 | covalent; warhead | [70] | ||
| rrrrrr-NH2 (hexa-D-R-NH2) | 0.478 | non-covalent | [75] | ||
| rrrrrrr-NH2 (hepta-D-R-NH2) | 0.041 | non-covalent | [75] | ||
| rrrrrrrr-NH2 (octa-D-R-NH2) | 0.017 | non-covalent | [75] | ||
| rrrrrrrrr-NH2 (nona-D-R-NH2) | 0.006 | non-covalent | yes | [75] | |
| rrrrrrrrrr-NH2 (deca-D-R-NH2) | 0.002 | non-covalent | [75] | ||
| rrrrrrrrrrr-NH2 (undeca-D-R-NH2) | 0.001 | non-covalent | [75] | ||
| rrrrrrrrrrrr-NH2 (dodeca-D-R-NH2) | 0.001 | non-covalent | [75] | ||
| 2-naphthoyl-KKR-H | 0.041 | covalent; warhead | 33 | [71] | |
| phenylacetyl-KKR-H | 0.009; 0.70 ± 0.04 | covalent; warhead | yes | [71,72] | |
| 4-phenylphenylacetyl-KKR-H | 0.006; 0.056 ± 0.004 | covalent; warhead | [71,72] | ||
| acetyl-KKR-H | 0.49 ± 0.32 | covalent; warhead | [72] | ||
| propionyl-KKR-H | 0.43 ± 0.06 | covalent; warhead | [72] | ||
| cyclopropionyl-KKR-H | 0.19 ± 0.01 | covalent; warhead | [72] | ||
| benzoyl-KKR-H | 0.21 ± 0.09 | covalent; warhead | [72] | ||
| acetyl-KR-H | 0.09 ± 0.02 | covalent; warhead | [76] | ||
| propionyl-KR-H | 0.17 ± 0.06 | covalent; warhead | [76] | ||
| cyclopropionyl-KR-H | 0.22 ± 0.05 | covalent; warhead | [76] | ||
| benzoyl-KR-H | 0.92 ± 0.09 | covalent; warhead | [76] | ||
| acetyl-Lys-Lys-agmatine | 9.1 ± 2.1 | non-covalent | [77] | ||
| 4-phenylphenylacetyl-Lys-Lys-agmatine | 2.05 ± 0.13 | non-covalent | [77] | ||
| 2-chloro-4-phenyl-phenacetyl-L-Lys-Lys-agmatine | 1.3 ± 0.2 | non-covalent | [78] | ||
| 4-chloro-4-phenyl-phenacetyl-L-Lys-Lys-agmatine | 2.4 ± 0.5 | non-covalent | [78] | ||
| 2-methyl-4-phenyl-phenacetyl-L-Lys-Lys-agmatine | 3.4 ± 0.6 | non-covalent | [78] | ||
| 4-methyl-4-phenyl-phenacetyl-L-Lys-Lys-agmatine4 | 3.5 ± 0.7 | non-covalent | [78] |
| Compound (Substrate) | Core | Method | WNV IC50 (µM) | Biophysical method (Kd, µM) | Anti-WNV cellular activity EC50 [CC50] in µM | Ref |
|---|---|---|---|---|---|---|
| cpd A (Boc-GKR-AMC) | 8-hydroxyquinoline | Diverse library screening | 6.4 ± 0.6 | [82] | ||
| cpd B (Boc-GKR-AMC) | 8-hydroxyquinoline | Diverse library screening | 6.8 ± 1.2; 3.6 ± 2.0 | 1.4 ± 0.4 [140 ± 1.98] | [82,83] | |
| Compound 14 | 8-hydroxyquinoline | Derivatization of cpd B | 1.0 ± 0.08 (GKR-AMC); 2.01 ± 0.08 (nkRR-AMC) | [83] | ||
| Compound 12j (Boc-GKR-AMC) | 1-oxo-1,2-dihydroisoquinoline | Focus library design and screening | 30 | [84] | ||
| Palmatine (pERTKR-AMC) | natural product | unknown | 96 | 3.6 [1031] | [85] | |
| cpd 1 (nKRR-AMC) | Carbamimidoylsulfanyl-methyl | In silico FBS | 178 | NMR (40) | [86] | |
| cpd 1 (nKRR-AMC) | In silico FBS | 2.8 ± 0.1 | NMR (90 ± 40) | [87] | ||
| cpd 1 (nKRR-AMC) | In silico FBS | 34.2 ± 0.1 | [87] | |||
| SID-852843 (Pyr-RTKR-AMC) | pyrazolyl benzoic acid ester | Diverse library screening | 0.105 | [88] | ||
| SID-4245669 (Pyr-RTKR-AMC) | pyrazolyl benzoic acid ester | Diverse library screening | 0.11 | [88] | ||
| SID-3717586 (Pyr-RTKR-AMC) | pyrazolyl benzoic acid ester | Diverse library screening | 1.353 | [88] | ||
| cpd 7a | Derivatization of pyrazole ester | 1.96 | [89] | |||
| cpd 10a | Derivatization of pyrazole ester | 4.03 | [89] | |||
| cpd 4; 166347 (FRET) | guanidinylated 2,5-dideoxystreptamine | Diverse library screening | 1.2 ± 0.3 | [74] | ||
| cpd 9; 166550 (FRET) | guanidinylated 2,5-dideoxystreptamine | Diverse library screening | 4 ± 2 | [74] | ||
| cpd 7; 166346 (FRET) | guanidinylated 2,5-dideoxystreptamine | Diverse library screening | 6 ± 1 | [74] | ||
| cpd 2; 166750 (FRET) | guanidinylated 2,5-dideoxystreptamine | Diverse library screening | 8 ± 1 | [74] | ||
| cpd 6; 166631 (FRET) | guanidinylated 2,5-dideoxystreptamine | Diverse library screening | 8 ± 1 | [74] | ||
| cpd 1a24 (Pyr-RTKR-AMC) | 2-{6-[2-(5-phenyl-4H-[1,2,4]triazol-3-ylsulfanyl)acetylamino]-2-{6-[2-(5-phenyl-4H-[1,2,4]triazol-3-ylsulfanyl)acetylamino]-benzothiazol-2-ylsulfanyl}acetamide | Diverse library screening | 3.4 ± 0.2 | [90] | ||
| cpd 1a16 (Pyr-RTKR-AMC) | 1,3,4,5-tetrasubstituted 1H-pyrrol-2(5H)-one | Diverse library screening | 2.2 ± 0.7 | [91] | ||
| cpd 1a40 (Pyr-RTKR-AMC) | 9,10-dihydro-3H,4aH-1,3,9,10a-tetraazaphenanthren-4-one | Diverse library screening | 2.2 ± 0.7 | [92] | ||
| Tyrothricin (M23) | decapolypeptide antibiotic | Diverse library screening | 2 ± 0.2 | [93] | ||
| Cpd 1; NSC86314 (Pyr-RTKR-AMC) | In silico docking | 0.26 | 42.77 [212.5] | [94] | ||
| Cpd 2; NSC16898 (Pyr-RTKR-AMC) | In silico docking | 0.44 | 17.01 [235.8] | [94] |
3.3.2. NS3 Helicase
3.4. NS4B
3.5. NS5
3.5.1. MTase and GTase
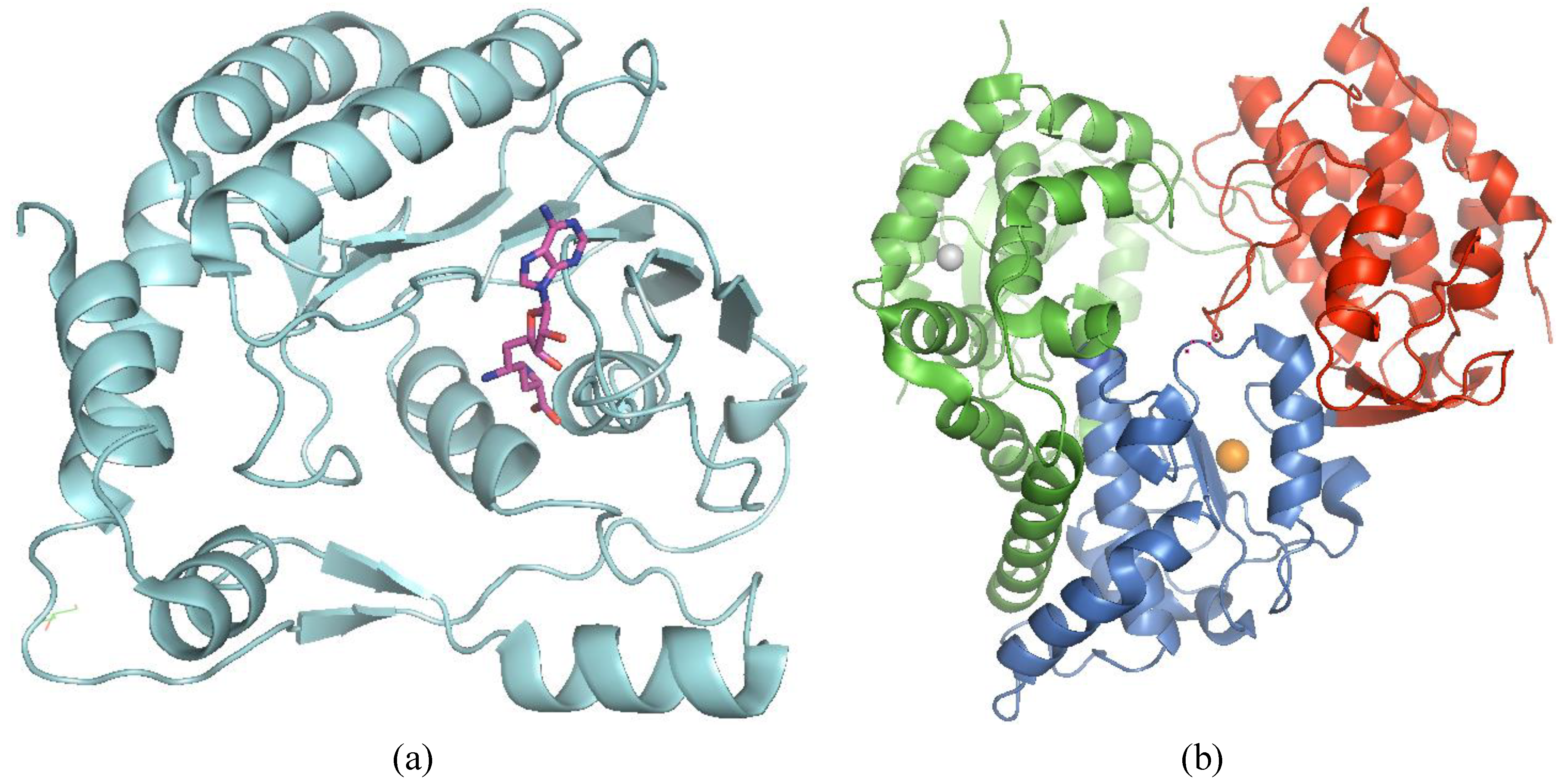
3.5.2. RdRp
4. Inhibitors of Host Targets
4.1. Inhibitors of Viral Replication and Translation
4.2. Inhibitors of Host Pyrimidine or Purine Biosynthesis
4.3. Inhibitors to Virus Assembly and Maturation
5. Discussion and Perspectives
Abbreviations
| DENV | dengue virus |
| WNV | West Nile virus |
| YFV | yellow fever virus |
| TBEV | tick-borne encephalitis virus |
| HCV | hepatitis C virus |
| WEEV | Western equine encephalitis virus |
| VSV | vesicular stomatitis virus |
| HBV | hepatitis B virus |
| HIV | human immunodeficiency virus |
| DAA | direct antiviral agent |
| GTase | guanylyltransferase |
| PI | protease inhibitors |
| HTS | high-throughput screening |
| MTase | methyltransferase |
| RdRp | RNA-dependent RNA polymerase |
| NI | nucleoside analog inhibitor |
| NNI | non-nucleoside inhibitor |
| TI | therapeutic index |
| SAR | structure-activity relationship |
| ER | endoplasmic reticulum |
Acknowledgments
Conflicts of Interest
References
- Shi, P.Y.; Brinton, M.A.; Veal, J.M.; Zhong, Y.Y.; Wilson, W. D. Evidence for the existence of a pseudoknot structure at the 3’ terminus of the flavivirus genomic RNA. Biochemistry 1996, 35, 4222–4230. [Google Scholar]
- Khromykh, A.A.; Meka, H.; Guyatt, K.J.; Westaway, E.G. Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 2001, 75, 6719–6728. [Google Scholar] [CrossRef]
- Friebe, P.; Harris, E. Interplay of RNA elements in the dengue virus 5’and 3’ ends required for viral RNA replication. J. Virol. 2010, 84, 6103–6118. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Thiel, H.J.; Rice, C.M. Flaviviridae: The Virus and Their Replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2007; Volume 1, p. 1101. [Google Scholar]
- Westaway, E.G.; Mackenzie, J.M.; Khromykh, A.A. Replication and gene function in Kunjin virus. Curr. Top. Microbiol. Immunol. 2002, 267, 323–351. [Google Scholar]
- Melian, E.B.; Hinzman, E.; Nagasaki, T.; Firth, A.E.; Wills, N.M.; Nouwens, A.S.; Blitvich, B.J.; Leung, J.; Funk, A.; Atkins, J.F.; et al. NS1’ of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J. Virol. 2010, 84, 1641–1647. [Google Scholar]
- Roosendaal, J.; Westaway, E.G.; Khromykh, A.; Mackenzie, J.M. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 2006, 80, 4623–4632. [Google Scholar] [CrossRef]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Conway, M.J.; Montgomery, R.R.; Fikrig, E. West Nile Virus: Biology, transmission, and human infection. Clin. Microbiol. Rev. 2012, 25, 635–648. [Google Scholar] [CrossRef]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R., II; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef]
- Perera, R.; Kuhn, R.J. Structural proteomics of dengue virus. Curr. Opin. Microbiol. 2008, 11, 369–377. [Google Scholar] [CrossRef]
- Perera, R.; Khaliq, M.; Kuhn, R.J. Closing the door on Flaviviruses: Entry as a target for antiviral drug design. Antivir. Res. 2008, 80, 11–22. [Google Scholar] [CrossRef]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2003, 100, 6986–6991. [Google Scholar] [CrossRef]
- Zhou, Z.; Khaliq, M.; Suk, J.E.; Patkar, C.; Li, L.; Kuhn, R.J.; Post, C.B. Antiviral compounds discovered by virtual screening of small-molecule libraries against dengue virus E protein. ACS Chem. Biol. 2008, 3, 765–775. [Google Scholar] [CrossRef]
- Poh, M.K.; Yip, A.; Zhang, S.; Priestle, J.P.; Ma, N.L.; Smit, J.M.; Wilschut, J.; Shi, P.Y.; Wenk, M.R.; Schul, W. A small molecule fusion inhibitor of dengue virus. Antivir. Res. 2009, 84, 260–266. [Google Scholar] [CrossRef]
- Yennamalli, R.; Subbarao, N.; Kampmann, T.; McGeary, R.P.; Young, P.R.; Kobe, B. Identification of novel target sites and an inhibitor of the dengue virus E protein. J. Comput. Aided Mol. Des. 2009, 23, 333–341. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Patel, S.J.; Vangrevelinghe, E.; Xu, H.Y.; Rao, R.; Jaber, D.; Schul, W.; Gu, F.; Heudi, O.; Ma, N.L.; et al. A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1823–1831. [Google Scholar] [CrossRef]
- Kampmann, T.; Yennamalli, R.; Campbell, P.; Stoermer, M.J.; Fairlie, D.P.; Kobe, B.; Young, P.R. In silico screening of small molecule libraries using the dengue virus envelope E protein has identified compounds with antiviral activity against multiple Flaviviruses. Antivir. Res. 2009, 84, 234–241. [Google Scholar] [CrossRef]
- Li, Z.; Khaliq, M.; Zhou, Z.; Post, C.B.; Kuhn, R.J.; Cushman, M. Design, synthesis, and biological evaluation of antiviral agents targeting flavivirus envelope proteins. J. Med. Chem. 2008, 51, 4660–4671. [Google Scholar] [CrossRef]
- Mayhoub, A.S.; Khaliqm, M.; Botting, C.; Li, Z.; Kuhn, R.J.; Cushman, M. An investigation of phenylthiazole anti-flaviviral agents. Bioorg. Med. Chem. 2011, 19, 3845–3854. [Google Scholar] [CrossRef]
- Mayhoub, A.S.; Khaliq, M.; Kuhn, R.J.; Cushman, M. Design, synthesis, and biological evaluation of thiazoles targeting flavivirus envelope proteins. J. Med. Chem. 2011, 54, 1704–1714. [Google Scholar] [CrossRef]
- Chu, J.J.; Rajamanonmani, R.; Li, J.; Bhuvanakantham, R.; Lescar, J.; Ng, M.L. Inhibition of West Nile virus entry by using a recombinant domain III from the envelope glycoprotein. J. Gen. Virol. 2005, 86, 405–412. [Google Scholar] [CrossRef]
- Hrobowski, Y.M.; Garry, R.F.; Michael, S.F. Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol. J. 2005, 2. [Google Scholar] [CrossRef]
- Diamond, M.S.; Pierson, T.C.; Roehrig, J.T. Molecular Virology and Control of Flaviviruses; Shi, P., Ed.; Caister Academic Press: Norfolk, UK, 2012; Chapter 11; p. 231. [Google Scholar]
- Haley, M.; Retter, A.S.; Fowler, D.; Gea-Banacloche, J.; O’Grady, N.P. The role for intravenous immunoglobulin in the treatment of West Nile virus encephalitis. Clin. Infect. Dis. 2003, 37, e88–e90. [Google Scholar] [CrossRef]
- Hamdan, A.; Green, P.; Mendelson, E.; Kramer, M.R.; Pitlik, S.; Weinberger, M. Possible benefit of intravenous immunoglobulin therapy in a lung transplant recipient with West Nile virus encephalitis. Transpl. Infect. Dis. 2002, 4, 160–162. [Google Scholar] [CrossRef]
- Saquib, R.; Randall, H.; Chandrakantan, A.; Spak, C.W.; Barri, Y.M. West Nile virus encephalitis in a renal transplant recipient: the role of intravenous immunoglobulin. Am. J. Kidney Dis. 2008, 52, e19–e21. [Google Scholar] [CrossRef]
- Shimoni, Z.; Niven, M.J.; Pitlick, S.; Bulvik, S. Treatment of West Nile virus encephalitis with intravenous immunoglobulin. Emerg. Infect. Dis. 2001, 7, 759. [Google Scholar]
- Oliphant, T.; Engle, M.; Nybakken, G.E.; Doane, C.; Johnson, S.; Huang, L.; Gorlatov, S.; Mehlhop, E.; Marri, A.; Chung, K.M.; et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 2005, 11, 522–530. [Google Scholar] [CrossRef]
- Morrey, J.D.; Siddharthan, V.; Olsen, A.L.; Roper, G.Y.; Wang, H.; Baldwin, T.J.; Koenig, S.; Johnson, S.; Nordstrom, J.L.; Diamond, M.S. Humanized monoclonal antibody against West Nile virus envelope protein administered after neuronal infection protects against lethal encephalitis in hamsters. J. Infect. Dis. 2006, 194, 1300–1308. [Google Scholar] [CrossRef]
- Morrey, J.D.; Siddharthan, V.; Olsen, A.L.; Wang, H.; Julander, J.G.; Hall, J.O.; Li, H.; Nordstrom, J.L.; Koenig, S.; Johnson, S.; et al. Defining limits of treatment with humanized neutralizing monoclonal antibody for West Nile virus neurological infection in a hamster model. Antimicrob. Agents Chemother. 2007, 51, 2396–2402. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Donchenko, A.P.; Koonin, E.V.; Blinov, V.M. N-terminal domains of putative helicases of flavi- and pestiviruses may be serine proteases. Nucleic Acids Res. 1989, 17, 3889–3897. [Google Scholar] [CrossRef]
- Wengler, G.; Wengler, G. The carboxy-terminal part of the NS 3 protein of the West Nile flavivirus can be isolated as a soluble protein after proteolytic cleavage and represents an RNA-stimulated NTPase. Virology 1991, 184, 707–715. [Google Scholar] [CrossRef]
- Wengler, G.; Czaya, G.; Färber, P.M.; Hegemann, J.H. In vitro synthesis of West Nile virus proteins indicates that the amino-terminal segment of the NS3 protein contains the active centre of the protease which cleaves the viral polyprotein after multiple basic amino acids. J. Gen. Virol. 1991, 72, 851–858. [Google Scholar] [CrossRef]
- Luo, D.; Xu, T.; Hunke, C.; Gruber, G.; Vasudevan, S.G.; Lescar, J. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 2008, 82, 173–183. [Google Scholar] [CrossRef]
- Assenberg, R.; Mastrangelo, E.; Walter, T.S.; Verma, A.; Milani, M.; Owens, R.J.; Stuart, D.I.; Grimes, J.M.; Mancini, E.J. Crystal structure of a novel conformational state of the flavivirus NS3 protein: Implications for polyprotein processing and viral replication. J. Virol. 2009, 83, 12895–12906. [Google Scholar] [CrossRef]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef]
- Mastrangelo, E.; Milani, M.; Bollati, M.; Selisko, B.; Peyrane, F.; Pandini, V.; Sorrentino, G.; Canard, B.; Konarev, P.V.; Svergun, D.I.; et al. Cystal structure and activity of Kunjin virus NS3 helicase; protease and helicase domain assembly in the full length NS3 protein. J. Mol. Biol. 2007, 372, 444–455. [Google Scholar] [CrossRef]
- Robin, G.; Chappell, K.; Stoermer, M.J.; Hu, S.H.; Young, P.R.; Fairlie, D.P.; Martin, J.L. Structure of West Nile virus NS3 protease: Ligand stabilization of the catalytic conformation. J. Mol. Biol. 2009, 385, 1568–1577. [Google Scholar] [CrossRef]
- Aleshin, A.E.; Shiryaev, S.A.; Strongin, A.Y.; Liddington, R.C. Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein Sci. 2007, 16, 795–806. [Google Scholar] [CrossRef]
- Hammamy, M.Z.; Haase, C.; Hammami, M.; Hilgenfeld, R.; Steinmetzer, T. Development and characterization of new peptidomimetic inhibitors of the West Nile virus NS2B-NS3 protease. ChemMedChem 2013, 8, 231–241. [Google Scholar] [CrossRef]
- Chernov, A.V.; Shiryaev, S.A.; Aleshin, A.E.; Ratnikov, B.I.; Smith, J.W.; Liddington, R.C.; Strongin, A.Y. The two-component NS2B-NS3 proteinase represses DNA unwinding activity of the West Nile virus NS3 helicase. J. Biol. Chem. 2008, 283, 17270–17278. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Chernov, A.V.; Aleshin, A.E.; Shiryaev, T.N.; Strongin, A.Y. NS4A regulates the ATPase activity of the NS3 helicase: A novel cofactor role of the non-structural protein NS4A from West Nile virus. J. Gen. Virol. 2009, 90, 2081–2085. [Google Scholar] [CrossRef]
- Borowski, P.; Niebuhr, A.; Mueller, O.; Bretner, M.; Felczak, K.; Kulikowski, T.; Schmitz, H. Purification and characterization of West Nile virus nucleoside triphosphatase (NTPase)/helicase: Evidence for dissociation of the NTPase and helicase activities of the enzyme. J. Virol. 2001, 75, 3220–3229. [Google Scholar] [CrossRef]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar]
- Bazan, J.F.; Fletterick, R.J. Detection of a trypsin-like serine protease domain in flaviviruses and pestiviruses. Virology 1989, 171, 637–639. [Google Scholar] [CrossRef]
- Falgout, B.; Pethel, M.; Zhang, Y.M.; Lai, C.J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 1991, 65, 2467–2475. [Google Scholar]
- McMinn, P.C. The molecular basis of virulence of the encephalitogenic flaviviruses. J. Gen. Virol. 1997, 78, 2711–2722. [Google Scholar]
- Chiou, S.S.; Chen, W.J. Mutations in the NS3 gene and 3'-NCR of Japanese encephalitis virus isolated from an unconventional ecosystem and implications for natural attenuation of the virus. Virology 2001, 289, 129–136. [Google Scholar] [CrossRef]
- Ramanathan, M.P.; Chambers, J.A.; Pankhong, P.; Chattergoon, M.; Attatippaholkun, W.; Dang, K.; Shah, N.; Weiner, D.B. Host cell killing by the West Nile Virus NS2B-NS3 proteolytic complex: NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway. Virology 2006, 345, 56–72. [Google Scholar] [CrossRef]
- De Clercq, E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob. Agents 2009, 33, 307–320. [Google Scholar] [CrossRef]
- Wyles, D.L. Antiviral resistance and the future landscape of hepatitis C virus infection therapy. J. Infect. Dis. 2013, 207, S33–S39. [Google Scholar] [CrossRef]
- Halfon, P.; Locarnini, S. Hepatitis C virus resistance to protease inhibitors. J. Hepatol. 2011, 55, 192–206. [Google Scholar] [CrossRef]
- Paolucci, S.; Fiorina, L.; Piralla, A.; Gulminetti, R.; Novati, S.; Barbarini, G.; Sacchi, P.; Gatti, M.; Dossena, L.; Baldanti, F. Naturally occurring mutations to HCV protease inhibitors in treatment-naïve patients. Virol. J. 2012, 9. [Google Scholar] [CrossRef]
- Cento, V.; Mirabelli, C.; Salpini, R.; Dimonte, S.; Artese, A.; Costa, G.; Mercurio, F.; Svicher, V.; Parrotta, L.; Bertoli, A.; et al. HCV genotypes are differently prone to the development of resistance to linear and macrocyclic protease inhibitors. PLoS One 2012, 7, e39652. [Google Scholar] [CrossRef]
- Murray, K.; Walker, C.; Herrington, E.; Lewis, J.A.; McCormick, J.; Beasley, D.W.; Tesh, R.B.; Fisher-Hoch, S. Persistent infection with West Nile virus years after initial infection. J. Infect. Dis. 2010, 201, 2–4. [Google Scholar] [CrossRef]
- Sejvar, J.J. The long-term outcomes of human West Nile virus infection. Clin. Infect. Dis. 2007, 44, 1617–1624. [Google Scholar] [CrossRef]
- Cook, R.L.; Xu, X.; Yablonsky, E.J.; Sakata, N.; Tripp, J.H.; Hess, R.; Piazza, P.; Rinaldo, C.R. Demographic and clinical factors associated with persistent symptoms after West Nile virus infection. Am. J. Trop. Med. Hyg. 2010, 83, 1133–1136. [Google Scholar] [CrossRef]
- Nolan, M.S.; Podoll, A.S.; Hause, A.M.; Akers, K.M.; Finkel, K.W.; Murray, K.O. Prevalence of chronic kidney disease and progression of disease over time among patients enrolled in the Houston West Nile virus cohort. PLoS One 2012, 7, e40374. [Google Scholar]
- Chappell, K.J.; Stoermer, M.J.; Fairlie, D.P.; Young, P.R. West Nile Virus NS2B/NS3 protease as an antiviral target. Curr. Med. Chem. 2008, 15, 2771–2784. [Google Scholar] [CrossRef]
- Bollati, M.; Alvarez, K.; Assenberg, R.; Baronti, C.; Canard, B.; Cook, S.; Coutard, B.; Decroly, E.; de Lamballerie, X.; Gould, E.A.; et al. Structure and functionality in flavivirus NS-proteins: perspectives for drug design. Antivir. Res. 2010, 87, 125–148. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Strongin, A.Y. Structural and functional parameters of the flaviviral protease: A promising antiviral drug target. Future Virol. 2010, 5, 593–606. [Google Scholar] [CrossRef]
- Steuber, H.; Hilgenfeld, R. Recent advances in targeting viral proteases for the discovery of novel antivirals. Curr. Top. Med. Chem. 2010, 10, 323–345. [Google Scholar] [CrossRef]
- Noble, C.G.; She, C.C.; Chao, A.T.; Shi, P.Y. Ligand-bound structures of the dengue virus protease reveal the active conformation. J. Virol. 2012, 86, 438–446. [Google Scholar] [CrossRef]
- Su, X.C.; Ozawa, K.; Yagi, H.; Lim, S.P.; Wen, D.; Ekonomiuk, D.; Huang, D.; Keller, T.H.; Sonntag, S.; Caflisch, A.; et al. NMR study of complexes between low molecular mass inhibitors and the West Nile virus NS2B-NS3 protease. FEBS J. 2009, 276, 4244–4255. [Google Scholar] [CrossRef]
- Su, X.C.; Ozawa, K.; Qi, R.; Vasudevan, S.G.; Lim, S.P.; Otting, G. NMR analysis of the dynamic exchange of the NS2B cofactor between open and closed conformations of the West Nile virus NS2B-NS3 protease. PLoS Negl. Trop. Dis. 2009, 3, e561. [Google Scholar] [CrossRef]
- Kim, Y.M.; Gayen, S.; Kang, C.; Joy, J.; Huang, Q.; Chen, A.S.; Wee, J.L.; Ang, M.J.; Lim, H.A.; Hung, A.W.; et al. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. J. Biol. Chem. 2013, 288, 12891–12900. [Google Scholar] [CrossRef]
- Knox, J.E.; Ma, N.L.; Yin, Z.; Patel, S.J.; Wang, W.L.; Chan, W.L.; Ranga Rao, K.R.; Wang, G.; Ngew, X.; Patel, V.; et al. Peptide inhibitors of West Nile NS3 protease: SAR study of tetrapeptide aldehyde inhibitors. J. Med. Chem. 2006, 49, 6585–6890. [Google Scholar] [CrossRef]
- Stoermer, M.J.; Chappell, K.J.; Liebscher, S.; Jensen, C.M.; Gan, C.H.; Gupta, P.K.; Xu, W.J.; Young, P.R.; Fairlie, D.P. Potent cationic inhibitors of West Nile virus NS2B/NS3 protease with serum stability, cell permeability and antiviral activity. J. Med. Chem. 2008, 51, 5714–5721. [Google Scholar] [CrossRef]
- Schüller, A.; Yin, Z.; Brian Chia, C.S.; Doan, D.N.; Kim, H.K.; Shang, L.; Loh, T.P.; Hill, J.; Vasudevan, S.G. Tripeptide inhibitors of dengue and West Nile virus NS2B-NS3 protease. Antivir. Res. 2011, 92, 96–101. [Google Scholar] [CrossRef]
- Mueller, N.H.; Yon, C.; Ganesh, V.K.; Padmanabhan, R. Characterization of the West Nile virus protease substrate specificity and inhibitors. Int. J. Biochem. Cell Biol. 2007, 39, 606–614. [Google Scholar] [CrossRef]
- Cregar-Hernandez, L.; Jiao, G.S.; Johnson, A.T.; Lehrer, A.T.; Wong, T.A.; Margosiak, S.A. Small molecule pan-dengue and West Nile virus NS3 protease inhibitors. Antivir. Chem. Chemother. 2011, 21, 209–217. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Ratnikov, B.I.; Chekanov, A.V.; Sikora, S.; Rozanov, D.V.; Godzik, A.; Wang, J.; Smith, J.W.; Huang, Z.; Lindberg, I.; et al. Cleavage targets and the D-arginine-based inhibitors of the West Nile virus NS3 processing proteinase. Biochem. J. 2006, 393, 503–511. [Google Scholar] [CrossRef]
- Kang, C.; Gayen, S.; Wang, W.; Severin, R.; Chen, A.S.; Lim, H.A.; Chia, C.S.; Schüller, A.; Doan, D.N.; Poulsen, A.; et al. Exploring the binding of peptidic West Nile virus NS2B-NS3 protease inhibitors by NMR. Antivir. Res. 2013, 97, 137–144. [Google Scholar] [CrossRef]
- Lim, H.A.; Joy, J.; Hill, J.; San Brian Chia, C. Novel agmatine and agmatine-like peptidomimetic inhibitors of the West Nile virus NS2B/NS3 serine protease. Eur. J. Med. Chem. 2011, 46, 3130–3134. [Google Scholar] [CrossRef]
- Lim, H.A.; Ang, M.J.; Joy, J.; Poulsen, A.; Wu, W.; Ching, S.C.; Hill, J.; Chia, C.S. Novel agmatine dipeptide inhibitors against the West Nile virus NS2B/NS3 protease: A P3 and N-cap optimization study. Eur. J. Med. Chem. 2013, 62, 199–205. [Google Scholar] [CrossRef]
- Nitsche, C.; Steuer, C.; Klein, C.D. Arylcyanoacrylamides as inhibitors of the Dengue and West Nile virus proteases. Bioorg. Med. Chem. 2011, 19, 7318–7337. [Google Scholar] [CrossRef]
- Eisert, W.G.; Hauel, N.; Stangier, J.; Wienen, W.; Clemens, A.; van Ryn, J. Dabigatran: An oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1885–1889. [Google Scholar] [CrossRef]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Misselwitz, F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat. Rev. Drug Discov. 2011, 10, 61–75. [Google Scholar] [CrossRef]
- Mueller, N.H.; Pattabiraman, N.; Ansarah-Sobrinho, C.; Viswanathan, P.; Pierson, T.C.; Padmanabhan, R. Identification and biochemical characterization of small-molecule inhibitors of west nile virus serine protease by a high-throughput screen. Antimicrob. Agents Chemother. 2008, 52, 3385–3393. [Google Scholar] [CrossRef]
- Ezgimen, M.; Lai, H.; Mueller, N.H.; Lee, K.; Cuny, G.; Ostrov, D.A.; Padmanabhan, R. Characterization of the 8-hydroxyquinoline scaffold for inhibitors of West Nile virus serine protease. Antivir. Res. 2012, 94, 18–24. [Google Scholar] [CrossRef]
- Dou, D.; Viwanathan, P.; Li, Y.; He, G.; Alliston, K.R.; Lushington, G.H.; Brown-Clay, J.D.; Padmanabhan, R.; Groutas, W.C. Design, synthesis, and in vitro evaluation of potential West Nile virus protease inhibitors based on the 1-oxo-1,2,3,4-tetrahydroisoquinoline and 1-oxo-1,2-dihydroisoquinoline scaffolds. J. Comb Chem. 2010, 12, 836–843. [Google Scholar] [CrossRef]
- Jia, F.; Zou, G.; Fan, J.; Yuan, Z. Identification of palmatine as an inhibitor of West Nile virus. Arch. Virol. 2010, 155, 1325–1329. [Google Scholar] [CrossRef]
- Ekonomiuk, D.; Su, X.C.; Ozawa, K.; Bodenreider, C.; Lim, S.P.; Yin, Z.; Keller, T.H.; Beer, D.; Patel, V.; Otting, G.; et al. Discovery of a non-peptidic inhibitor of west nile virus NS3 protease by high-throughput docking. PLoS Negl. Trop. Dis. 2009, 3, e356. [Google Scholar] [CrossRef]
- Ekonomiuk, D.; Su, X.C.; Ozawa, K.; Bodenreider, C.; Lim, S.P.; Otting, G.; Huang, D.; Caflisch, A. Flaviviral protease inhibitors identified by fragment-based library docking into a structure generated by molecular dynamics. J. Med Chem. 2009, 52, 4860–4868. [Google Scholar] [CrossRef]
- Johnston, P.A.; Phillips, J.; Shun, T.Y.; Shinde, S.; Lazo, J.S.; Huryn, D.M.; Myers, M.C.; Ratnikov, B.; Smith, J.W.; et al. HTS identifies novel and specific uncompetitive inhibitors of the two-component NS2B-NS3 proteinase of WestNile virus. Assay. Drug Dev. Technol. 2007, 5, 737–750. [Google Scholar] [CrossRef]
- Sidique, S.; Shiryaev, S.A.; Ratnikov, B.I.; Herath, A.; Su, Y.; Strongin, A.Y.; Cosford, N.D. Structure-activity relationship and improved hydrolytic stability of pyrazole derivatives that are allosteric inhibitors of West Nile Virus NS2B-NS3 proteinase. Bioorg. Med. Chem. Lett. 2009, 19, 5773–5777. [Google Scholar] [CrossRef]
- Samanta, S.; Lim, T.L.; Lam, Y. Synthesis and in vitro evaluation of West Nile virus protease inhibitors based on the 2-{6-[2-(5-phenyl-4H-{1,2,4]triazol-3-ylsulfanyl)acetylamino]benzothiazol-2-ylsulfanyl}acetamide scaffold. ChemMedChem 2013, 8, 994–1001. [Google Scholar] [CrossRef]
- Gao, Y.; Samanta, S.; Cui, T.; Lam, Y. Synthesis and in vitro evaluation of West Nile Virus Protease inhibitors based on the 1,3,4,5-Tetrasubstituted 1H-Pyrrol-2(5H)-one Scaffold. ChemMedChem 2013, in press. [Google Scholar]
- Samanta, S.; Cui, T.; Lam, Y. Discovery, synthesis, and in vitro evaluation of West Nile virus protease inhibitors based on the 9,10-dihydro-3H,4aH-1,3,9,10a-tetraazaphenanthren-4-one scaffold. ChemMedChem 2012, 7, 1210–1216. [Google Scholar] [CrossRef]
- Tomlinson, S.M.; Watowich, S.J. Use of parallel validation high-throughput screens to reduce false positives and identify novel dengue NS2B-NS3 protease inhibitors. Antivir. Res. 2012, 93, 245–252. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Cheltsov, A.V.; Gawlik, K.; Ratnikov, B.I.; Strongin, AY. Virtual ligand screening of the National Cancer Institute (NCI) compound library leads to the allosteric inhibitory scaffolds of the West Nile Virus NS3 proteinase. Assay Drug Dev. Technol. 2011, 9, 69–78. [Google Scholar] [CrossRef]
- Brault, A.C.; Huang, C.Y.; Langevin, S.A.; Kinney, R.M.; Bowen, R.A.; Ramey, W.N.; Panella, N.A.; Holmes, E.C.; Powers, A.M.; Miller, B.R. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat. Genet. 2007, 39, 1162–1166. [Google Scholar] [CrossRef]
- Mertens, E.; Kajaste-Rudnitski, A.; Torres, S.; Funk, A.; Frenkiel, M.P.; Iteman, I.; Khromykh, A.A.; Desprès, P. Viral determinants in the NS3 helicase and 2K peptide that promote West Nile virus resistance to antiviral action of 2',5'-oligoadenylate synthetase 1b. Virology 2010, 399, 176–185. [Google Scholar] [CrossRef]
- Mastrangelo, E.; Pezzullo, M.; de Burghgraeve, T.; Kaptein, S.; Pastorino, B.; Dallmeier, K.; de Lamballerie, X.; Neyts, J.; Hanson, A.M.; Frick, D.N.; et al. Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: New prospects for an old drug. J. Antimicrob. Chemother. 2012, 67, 1884–1894. [Google Scholar] [CrossRef]
- Kline, K.; McCarthy, J.S.; Pearson, M.; Loukas, A.; Hotez, P.J. Neglected tropical diseases of Oceania: Review of their prevalence, distribution, and opportunities for control. PLoS Negl. Trop. Dis. 2013, 7, e1755. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Sivakumaran, H.; Heaton, S.M.; Harrich, D.; Jans, D.A. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem. J. 2012, 443, 851–856. [Google Scholar] [CrossRef]
- Tay, M.Y.; Fraser, J.E.; Chan, W.K.; Moreland, N.J.; Rathore, A.P.; Wang, C.; Vasudevan, S.G.; Jans, D.A. Nuclear localization of dengue virus (DENV) 1–4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor Ivermectin. Antivir. Res. 2013, 99, 301–306. [Google Scholar]
- Borowski, P.; Heising, M.V.; Miranda, I.B.; Liao, C.L.; Choe, J.; Baier, A. Viral NS3 helicase activity is inhibited by peptides reproducing the Arg-rich conserved motif of the enzyme (motif VI). Biochem. Pharmacol. 2008, 76, 28–38. [Google Scholar] [CrossRef]
- Bretner, M.; Baier, A.; Kopańska, K.; Najda, A.; Schoof, A.; Reinholz, M.; Lipniacki, A.; Piasek, A.; Kulikowski, T.; Borowski, P. Synthesis and biological activity of 1H-benzotriazole and 1H-benzimidazole analogues—Inhibitors of the NTpase/helicase of HCV and of some related Flaviviridae. Antivir. Chem. Chemother. 2005, 16, 315–326. [Google Scholar]
- Ujjinamatada, R.K.; Baier, A.; Borowski, P.; Hosmane, R.S. An analogue of AICAR with dual inhibitory activity against WNV and HCV NTPase/helicase: Synthesis and in vitro screening of 4-carbamoyl-5-(4,6-diamino-2,5-dihydro-1,3,5-triazin-2-yl)imidazole-1-beta-D-ribofuranoside. Bioorg. Med. Chem. Lett. 2007, 17, 2285–2288. [Google Scholar] [CrossRef]
- Borowski, P.; Deinert, J.; Schalinski, S.; Bretner, M.; Ginalski, K.; Kulikowski, T.; Shugar, D. Halogenated benzimidazoles and benzotriazoles as inhibitors of the NTPase/helicase activities of hepatitis C and related viruses. Eur. J. Biochem. 2003, 270, 1645–1653. [Google Scholar] [CrossRef]
- Bretner, M.; Schalinski, S.; Haag, A.; Lang, M.; Schmitz, H.; Baier, A.; Behrens, S.E.; Kulikowski, T.; Borowski, P. Synthesis and evaluation of ATP-binding site directed potential inhibitors of nucleoside triphosphatases/helicases and polymerases of hepatitis C and other selected Flaviviridae viruses. Antivir. Chem. Chemother. 2004, 15, 35–42. [Google Scholar]
- Zhang, N.; Chen, H.M.; Koch, V.; Schmitz, H.; Minczuk, M.; Stepien, P.; Fattom, A.I.; Naso, R.B.; Kalicharran, K.; Borowski, P.; et al. Potent inhibition of NTPase/helicase of the West Nile Virus by ring-expanded (“fat”) nucleoside analogues. J. Med Chem. 2003, 46, 4776–4789. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, H.M.; Koch, V.; Schmitz, H.; Liao, C.L.; Bretner, M.; Bhadti, V.S.; Fattom, A.I.; Naso, R.B.; Hosmane, R.S.; et al. Ring-expanded (“fat”) nucleoside and nucleotide analogues exhibit potent in vitro activity against flaviviridae NTPases/helicases, including those of the West Nile virus, hepatitis C virus, and Japanese encephalitis virus. J. Med. Chem. 2003, 46, 4149–4164. [Google Scholar] [CrossRef]
- Borowski, P.; Lang, M.; Haag, A.; Schmitz, H.; Choe, J.; Chen, H.M.; Hosmane, R.S. Characterization of imidazo[4,5-d]pyridazine nucleosides as modulators of unwinding reaction mediated by West Nile virus nucleoside triphosphatase/helicase: Evidence for activity on the level of substrate and/or enzyme. Antimicrob. Agents Chemother. 2002, 46, 1231–1239. [Google Scholar] [CrossRef]
- Munoz-Jordan, J.L.; Laurent-Rolle, M.; Ashour, J.; Martinez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; Garcia-Sastre, A. Inhibition of Alpha/Beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef]
- Puig-Basagoiti, F.; Tilgner, M.; Bennett, C.J.; Zhou, Y.; Muñoz-Jordán, J.L.; García-Sastre, A.; Bernard, K.A.; Shi, P.Y. A mouse cell-adapted NS4B mutation attenuates West Nile virus RNA synthesis. Virology 2007, 361, 229–341. [Google Scholar] [CrossRef]
- Wicker, J.A.; Whiteman, M.C.; Beasley, D.W.; Davis, C.T.; Zhang, S.; Schneider, B.S.; Higgs, S.; Kinney, R.M.; Barrett, A.D. A single amino acid substitution in the central portion of the West Nile virus NS4B protein confers a highly attenuated phenotype in mice. Virology 2006, 349, 245–253. [Google Scholar] [CrossRef]
- Welte, T.; Xie, G.; Wicker, J.A.; Whiteman, M.C.; Li, L.; Rachamallu, A.; Barrett, A.; Wang, T. Immune responses to an attenuated West Nile virus NS4B-P38G mutant strain. Vaccine 2011, 29, 4853–4861. [Google Scholar] [CrossRef]
- Xie, X.; Wang, Q.Y.; Xu, H.Y.; Qing, M.; Kramer, L.; Yuan, Z.; Shi, P.Y. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 2011, 85, 11183–11195. [Google Scholar] [CrossRef]
- Van Cleef, K.W.; Overheul, G.J.; Thomassen, M.C.; Kaptein, S.J.; Davidson, A.D.; Jacobs, M.; Neyts, J.; van Kuppeveld, F.J.; van Rij, R.P. Identification of a new dengue virus inhibitor that targets the viral NS4B protein and restricts genomic RNA replication. Antivir. Res. 2013, 99, 165–171. [Google Scholar] [CrossRef]
- Zou, G.; Puig-Basagoiti, F.; Zhang, B.; Qing, M.; Chen, L.; Pankiewicz, K.W.; Felczak, K.; Yuan, Z. A single-amino acid substitution in West Nile virus 2K peptide between NS4A and NS4B confers resistance to lycorine, a flavivirus inhibitor. Virology 2009, 384, 242–252. [Google Scholar] [CrossRef]
- Patkar, C.G.; Larsen, M.; Owston, M.; Smith, J.L.; Kuhn, R.J. Identification of inhibitors of yellow fever virus replication using a replicon-based high-throughput assay. Antimicrob. Agents Chemother. 2009, 53, 4103–4114. [Google Scholar] [CrossRef]
- Bollati, M.; Alvarez, K.; Assenberg, R.; Baronti, C.; Canard, B.; Cook, S.; Coutard, B.; Decroly, E.; de Lamballerie, X.; Gould, E.A.; et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antivir. Res. 2010, 87, 125–148. [Google Scholar] [CrossRef]
- Noble, C.G.; Shi, P.Y. Structural biology of dengue virus enzymes: Towards rational design of therapeutics. Antivir. Res. 2012, 96, 115–126. [Google Scholar] [CrossRef]
- Lescar, J.; Lim, S.P.; Shi, P. Molecular Virology and Control of Flaviviruses; Shi, P., Ed.; Caister Academic Press: Norfolk, UK, 2012; Chapter 6; p. 101. [Google Scholar]
- Davidson, A.D. New insights into Flavivirus nonstructural protein 5. Adv. Virus Res. 2009, 74, 41–101. [Google Scholar]
- Dong, H.; Zhang, B.; Shi, P.Y. Flavivirus methyltransferase: A novel antiviral target. Antivir. Res. 2008, 80, 1–10. [Google Scholar] [CrossRef]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.; Shi, P.Y.; Li, H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2'-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Dong, H.; Chang, D.C.; Hua, M.H.; Lim, S.P.; Chionh, Y.H.; Hia, F.; Lee, Y.H.; Kukkaro, P.; Lok, S.-M.; Dedon, P.C.; et al. 2’-O Methylation of Internal Adenosine by Flavivirus NS5 Methyltransferase. PLoS Patho. 2012, 8, 1–12. [Google Scholar]
- Issur, M.; Geiss, B.J.; Bougie, I.; Picard-Jean, F.; Despins, S.; Mayette, J.; Hobdey, S.E.; Bisaillon, M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 2009, 15, 2340–2350. [Google Scholar] [CrossRef]
- Bollati, M.; Milani, M.; Mastrangelo, E.; Ricagno, S.; Tedeschi, G.; Nonnis, S.; Decroly, E.; Selisko, B.; de Lamballerie, X.; Coutard, B.; et al. Recognition of RNA cap in the Wesselsbron virus NS5 methyltransferase domain: Implications for RNA-capping mechanisms in Flavivirus. J. Mol. Biol. 2009, 385, 40–52. [Google Scholar]
- Podvinc, M.; Lim, S.P.; Schmidt, T.; Scarsi, M.; Wen, D.; Sonntag, L.S.; Sanschagrin, P.; Shenkin, P.S.; Schwede, T. Novel inhibitors of dengue virus methyltransferase: Discovery by in vitro-driven virtual screening on a desktop computer grid. J. Med. Chem. 2010, 53, 1483–1495. [Google Scholar] [CrossRef]
- Dong, H.; Liu, L.; Zou, G.; Zhao, Y.; Li, Z.; Lim, S.P.; Shi, P-Y.; Li., H. Structural and Functional Analyses of a Conserved Hydrophobic Pocket of Flavivirus Methyltransferase. J. Biol. Chem. 2010, 285, 32586–32595. [Google Scholar]
- Lim., S.P.; Sonntag., L.S.; Noble, C.; Nilar, S.H.; Ng, R.H.; Zou, G.; Monaghan, P.; Chung, K.Y.; Dong, H.; Liu, B.; et al. Small molecule inhibitors that selectively block dengue virus methyltransferase. J. Biol. Chem. 2010, 286, 6233–6240. [Google Scholar]
- Geiss, B.J.; Stahla-Beek, H.J.; Hannah, A.M.; Gari, H.H.; Henderson, B.R.; Saeedi, B.J.; Keenan, S.M. A high-throughput screening assay for the identification of flavivirus NS5 capping enzyme GTP-binding inhibitors: Implications for antiviral drug development. J. Biomol. Screen. 2011, 16, 852–861. [Google Scholar] [CrossRef]
- Stahla-Beek, H.J.; April, D.G.; Saeedi, B.J.; Hannah, A.M.; Keenan, S.M.; Geiss, B.J. Identification of a novel antiviral inhibitor of the flavivirus guanylyltransferase enzyme. J. Virol. 2012, 86, 8730–8739. [Google Scholar] [CrossRef]
- Delang, L.; Vliegen, I.; Froeyen, M.; Neyts, J. Comparative study of the genetic barriers and pathways towards resistance of selective inhibitors of hepatitis C virus replication. Antimicrob. Agents Chemother. 2011, 55, 4103–4113. [Google Scholar]
- Ojwang, J.O.; Ali, S.; Smee, D.F.; Morrey, J.D.; Shimasaki, C.D.; Sidwell, R.W. Broad-spectrum inhibitor of viruses in the Flaviviridae family. Antivir. Res. 2005, 68, 49–55. [Google Scholar] [CrossRef]
- Yin, Z.; Chen, Y.L.; Schul, W.; Wang, Q.Y.; Gu, F.; Duraiswamy, J.; Kondreddi, R.R.; Niyomrattanakit, P.; Lakshminarayana, S.B.; Goh, A.; et al. An adenosine nucleoside inhibitor of dengue virus. Proc. Natl. Acad. Sci. USA 2009, 106, 20435–20439. [Google Scholar] [CrossRef]
- Chen, Y.L.; Yin, Z.; Lakshminarayana, S.B.; Qing, M.; Schul, W.; Duraiswamy, J.; Kondreddi, R.R.; Goh, A.; Xu, H.Y.; Yip, A.; et al. Inhibition of dengue virus by an ester prodrug of an adenosine analog. Antimicrob. Agents Chemother. 2010, 20154, 3255–3261. [Google Scholar]
- Migliaccio, G.; Tomassini, J.E.; Carroll, S.S.; Tomei, L.; Altamura, S.; Bhat, B.; Bartholomew, L.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 2003, 278, 49164–49170. [Google Scholar] [CrossRef]
- Nguyen, N.M.; Tran, C.N.; Phung, L.K.; Duong, K.T.; Huynh, H.L.; Farrar, J.; Nguyen, Q.T.; Tran, H.T.; Nguyen, C.V.; Merson, L.; et al. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J. Infect. Dis. 2013, 207, 1442–1450. [Google Scholar] [CrossRef]
- Ono, L.; Wollinger, W.; Rocco, I.M.; Coimbra, T.L.M.; Gorin, P.A.J.; Sierakowski, M.-R. In vitro and in vivo antiviral properties of sulfated galactomannans against yellow fever virus (BeH111 strain) and dengue 1 virus (Hawaii strain). Antivir. Res. 2003, 60, 201–208. [Google Scholar] [CrossRef]
- Malet, H.; Egloff, M.P.; Selisko, B.; Butcher, R.E.; Wright, P.J.; Roberts, M.; Gruez, A.; Sulzenbacher, G.; Vonrhein, C.; Bricogne, G.; et al. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J. Biol. Chem. 2007, 282, 10678–10689. [Google Scholar] [CrossRef]
- Selisko, B.; Dutartre, H.; Guillemot, J.C.; Debarnot, C.; Benarroch, D.; Khromykh, A.A.; Despres, P.; Egloff, M.P.; Canard, B. Comparative mechanistic studies of de novo RNA synthesis by flavivirus RNA-dependent RNA polymerases. Virology 2006, 351, 145–158. [Google Scholar] [CrossRef]
- Nomaguchi, M.; Teramoto, T.; Yu, L.; Markoff, L.; Padmanabhan, R. Requirements for West Nile virus (−)- and (+)-strand subgenomic RNA synthesis in vitro by the viral RNA-dependent RNA polymerase expressed in Escherichia coli. J. Biol. Chem. 2004, 279, 12141–12151. [Google Scholar]
- Niyomrattanakit, P.; Chen, Y.L.; Dong, H.; Yin, Z.; Qing, M.; Glickman, J.F.; Lin, K.; Mueller, D.; Voshol, H.; Lim, J.Y.; et al. Inhibition of dengue virus polymerase by blocking of the RNA tunnel. J. Virol. 2010, 84, 5678–5686. [Google Scholar] [CrossRef]
- Malet, H.; Massé, N.; Selisko, B.; Romette, J.L.; Alvarez, K.; Guillemot, J.C.; Tolou, H.; Yap, T.L.; Vasudevan, S.; Lescar, J.; et al. The flavivirus polymerase as a target for drug discovery. Antivir. Res. 2008, 80, 23–35. [Google Scholar] [CrossRef]
- Zou, G.; Chen, Y.L.; Dong, H.; Lim, C.C.; Yap, L.J.; Yau, Y.H.; Shochat, S.G.; Lescar, J.; Shi, P.Y. Functional analysis of two cavities in flavivirus NS5 polymerase. J. Biol. Chem. 2011, 286, 14362–14372. [Google Scholar] [CrossRef]
- Noble, C.G.; Lim, S.P.; Chen, Y.L.; Liew, C.W.; Yap, L.; Lescar, J.; Shi, P.Y. Conformational flexibility of the Dengue virus RNA-dependent RNA polymerase revealed by a complex with an inhibitor. J. Virol. 2013, 87, 5291–5295. [Google Scholar] [CrossRef]
- Schang, L.M. First demonstration of the effectiveness of inhibitors of cellular protein kinases in antiviral therapy. Expert Rev. Anti-Infect. Ther. 2006, 4, 953–956. [Google Scholar] [CrossRef]
- Hayes, E.B.; Sejvar, J.J.; Zaki, S.R.; Lanciotti, R.S.; Bode, A.V.; Campbell, G.L. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg. Infect. Dis. 2005, 11, 1174–1179. [Google Scholar] [CrossRef]
- Davis, L.E.; DeBiasi, R.; Goade, D.E.; Haaland, K.Y.; Harrington, J.A.; Harnar, J.B.; Pergam, S.A.; King, M.K.; DeMasters, B.K.; Tyler, K.L. West Nile virus neuroinvasive disease. Ann. Neurol. 2006, 60, 286–300. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Kondreddi, R.R.; Xie, X.; Rao, R.; Nilar, S.; Xu, H.Y.; Qing, M.; Chang, D.; Dong, H.; Yokokawa, F.; et al. A translation inhibitor that suppresses dengue virus in vitro and in vivo. Antimicrob. Agents Chemother. 2011, 55, 4072–4080. [Google Scholar] [CrossRef]
- Noueiry, A.O.; Olivo, P.D.; Slomczynska, U.; Zhou, Y.; Buscher, B.; Geiss, B.; Engle, M.; Roth, R.M.; Chung, K.M.; Samuel, M.; et al. Identification of novel small-molecule inhibitors of West Nile virus Infection. J. Virol. 2007, 81, 11992–12004. [Google Scholar] [CrossRef]
- Michaelis, M.; Kleinschmidt, M.C.; Doerr, H.W.; Cinatl, J., Jr. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. Antimicrob. Chemother. 2007, 60, 981–986. [Google Scholar] [CrossRef]
- Qing, M.; Yang, F.; Zhang, B.; Zou, G.; Robida, J.M.; Yuan, Z.; Tang, H.; Shi, P.Y. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob. Agents Chemother. 2009, 53, 3226–3235. [Google Scholar] [CrossRef]
- Diamond, M.S.; Zachariah, M.; Harris, E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA. Virology 2002, 304, 211–221. [Google Scholar] [CrossRef]
- Morrey, J.; Smee, D.; Sidwell, R.; Tseng, C. Identification of active antiviral compounds against a New York isolate of West Nile virus. Antivir. Res. 2002, 55, 107–116. [Google Scholar] [CrossRef]
- Leyssen, P.; Balzarini, J.; de Clercq, E.; Neyts, J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 2005, 79, 1943–1947. [Google Scholar] [CrossRef]
- Thomas, E.; Feld, J.J.; Li, Q.; Hu, Z.; Fried, M.W.; Liang, T.J. Ribavirin potentiates interferon action by augmenting interferon-stimulated gene induction in hepatitis C virus cell culture models. Hepatology 2011, 53, 32–41. [Google Scholar] [CrossRef]
- Pan, Q.; de Ruiter, P.E.; Metselaar, H.J.; Kwekkeboom, J.; de Jonge, J.; Tilanus, H.W.; Janssen, H.L.; van der Laan, L.J. Mycophenolic acid augments interferon-stimulated gene expression and inhibits hepatitis C Virus infection in vitro and in vivo. Hepatology 2012, 55, 1673–1683. [Google Scholar] [CrossRef]
- Crance, J.M.; Scaramozzino, N.; Jouan, A.; Garin, D. Interferon, ribavirin, 6-azauridine and glycyrrhizin: Antiviral compounds active against pathogenic flaviviruses. Antivir. Res. 2003, 58, 73–79. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Bushell, S.; Qing, M.; Xu, H.Y.; Bonavia, A.; Nunes, S.; Zhou, J.; Poh, M.K.; Florez de Sessions, P.; Niyomrattanakit, P.; et al. Inhibition of dengue virus through suppression of host pyrimidine biosynthesis. J. Virol. 2011, 85, 6548–6556. [Google Scholar] [CrossRef]
- Qing, M.; Zou, G.; Wang, Q.Y.; Xu, H.Y.; Dong, H.; Yuan, Z.; Shi, P.Y. Characterization of dengue virus resistance to brequinar in cell culture. Antimicrob. Agents Chemother. 2010, 54, 3686–3695. [Google Scholar] [CrossRef]
- Durantel, D. Celgosivir, an alpha-glucosidase I inhibitor for the potential treatment of HCV infection. Curr. Opin. Investig. Drugs. 2009, 10, 860–870. [Google Scholar]
- Whitby, K.; Pierson, T.C.; Geiss, B.; Lane, K.; Engle, M.; Zhou, Y.; Doms, R.W.; Diamond, M.S. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J. Virol. 2005, 79, 8698–8706. [Google Scholar]
- Rathore, A.P.; Paradkar, P.N.; Watanabe, S.; Tan, K.H.; Sung, C.; Connolly, J.E.; Low, J.; Ooi, E.E.; Vasudevan, S.G. Celgosivir treatment misfolds dengue virus NS1 protein, induces cellular pro-survival genes and protects against lethal challenge mouse model. Antivir. Res. 2011, 92, 453–460. [Google Scholar] [CrossRef]
- Watanabe, S.; Rathore, A.P.; Sung, C.; Lu, F.; Khoo, Y.M.; Connolly, J.; Low, J.; Ooi, E.E.; Lee, H.S.; Vasudevan, S.G. Dose- and schedule-dependent protective efficacy of celgosivir in a lethal mouse model for dengue virus infection informs dosing regimen for a proof of concept clinical trial. Antivir. Res. 2012, 96, 32–35. [Google Scholar]
- Gu, B.; Mason, P.; Wang, L.; Norton, P.; Bourne, N.; Moriarty, R.; Mehta, A.; Despande, M.; Shah, R.; Block, T. Antiviral profiles of novel iminocyclitol compounds against bovine viral diarrhea virus, West Nile virus, dengue virus and hepatitis B virus. Antivir. Chem. Chemother. 2007, 18, 49–59. [Google Scholar]
- Chang, J.; Wang, L.; Ma, D.; Qu, X.; Guo, H.; Xu, X.; Mason, P.M.; Bourne, N.; Moriarty, R.; Gu, B.; et al. Novel imino sugar derivatives demonstrate potent antiviral activity against flaviviruses. Antimicrob. Agents Chemother. 2009, 53, 1501–1508. [Google Scholar] [CrossRef]
- Xu, Z.; Anderson, R.; Hobman, T.C. The capsid-binding nucleolar helicase DDX56 is important for infectivity of West Nile virus. J. Virol. 2011, 85, 5571–5580. [Google Scholar] [CrossRef]
- Xu, Z.; Hobman, T.C. The helicase activity of DDX56 is required for its role in assembly of infectious West Nile virus particles. Virology 2012, 433, 226–235. [Google Scholar] [CrossRef]
- Hirsch, A.J.; Medigeshi, G.R.; Meyers, H.L.; DeFilippis, V.; Früh, K.; Briese, T.; Lipkin, W.I.; Nelson, J.A. The Src family kinase c-Yes is required for maturation of West Nile virus particles. J. Virol. 2005, 79, 11943–11951. [Google Scholar] [CrossRef]
- Becker, G.L.; Lu, Y.; Hardes, K.; Strehlow, B.; Levesque, C.; Lindberg, I.; Sandvig, K.; Bakowsky, U.; Day, R.; Garten, W.; et al. Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J. Biol. Chem. 2012, 287, 21992–22003. [Google Scholar] [CrossRef]
- Murray, K.O.; Walker, C.; Gould, E. The virology, epidemiology, and clinical impact of West Nile virus: A decade of advancements in research since its introduction into the Western Hemisphere. Epidemiol. Infect. 2011, 139, 807–817. [Google Scholar] [CrossRef]
- Najera, I. Resistance to HCV nucleoside analogue inhibitors of hepatitis C virus RNA-dependent RNA polymerase. Curr. Opin. Virol. 2013, 3, 508–513. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lim, S.P.; Shi, P.-Y. West Nile Virus Drug Discovery. Viruses 2013, 5, 2977-3006. https://doi.org/10.3390/v5122977
Lim SP, Shi P-Y. West Nile Virus Drug Discovery. Viruses. 2013; 5(12):2977-3006. https://doi.org/10.3390/v5122977
Chicago/Turabian StyleLim, Siew Pheng, and Pei-Yong Shi. 2013. "West Nile Virus Drug Discovery" Viruses 5, no. 12: 2977-3006. https://doi.org/10.3390/v5122977
APA StyleLim, S. P., & Shi, P.-Y. (2013). West Nile Virus Drug Discovery. Viruses, 5(12), 2977-3006. https://doi.org/10.3390/v5122977