Human Cytomegalovirus Manipulation of Latently Infected Cells


Abstract
:1. Introduction
2. Background—HCMV Latency and Reactivation
2.1. The Transcriptional Landscape of Latent HCMV

{kind=link}
{kind=link}
{kind=link}
| Gene Product | Latent Function | Lytic Function | References |
|---|---|---|---|
| CLTs | Unknown | Regulation of anti-viral 2’5’ OAS expression (ORF94) | [44,45,46,57] |
| UL138 | Regulation of TNFRI (up) and MRP1 (down), repression of the MIEP(?) | Regulation of TNFRI (up) and MRP1 (down), virus maturation (133-138 locus) | [53,58,59,60,61] |
| UL81-82ast | Promotes UL138 gene expression. | Unknown | [24,55,62] |
| LAvIL-10 | Down-regulation of MHC class II expression, immune evasion | Unknown—cmvIL-10 expressed during lytic infection | [54,63] |
| Lnc4.9 | Binds Polycomb repressor complex 2, Silencing of the MIEP | Unknown | [25] |
| UL84 | Genome maintenance | DNA replication, UTPase activity, transcriptional regulation | [25,64,65,66,67] |
| US28 | Unknown | GPCR, induces cell signalling and cell migration, agonist of the MIEP | [68,69,70,71,72,73,74] |
| UL144 | Unknown | TNF superfamily member, hijacks NF-kB signalling, immune evasion? | [75,76,77,78] |
3. Mechanisms Targeted during HCMV Latency
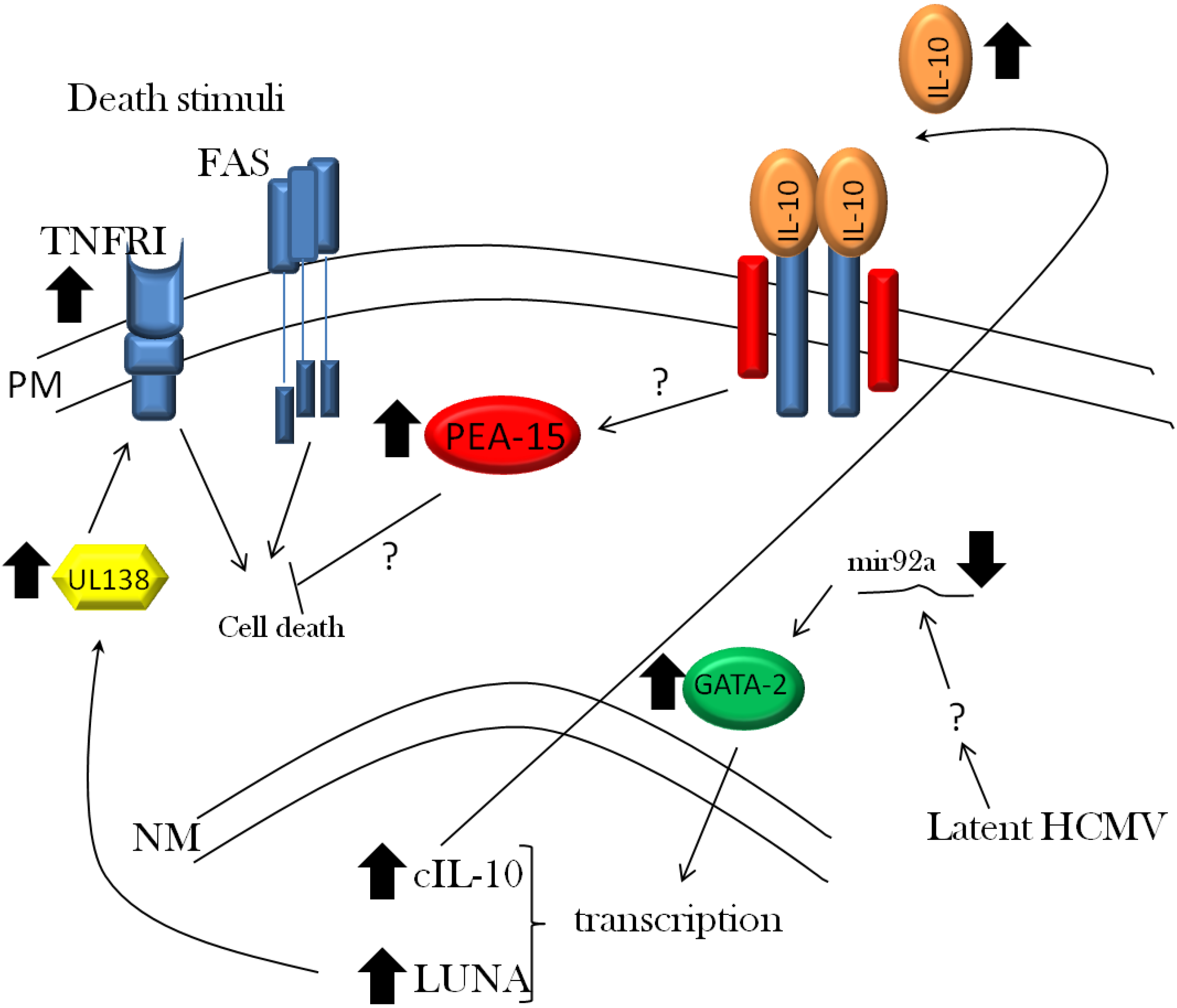
3.1. Viral Evasion of Cell Death
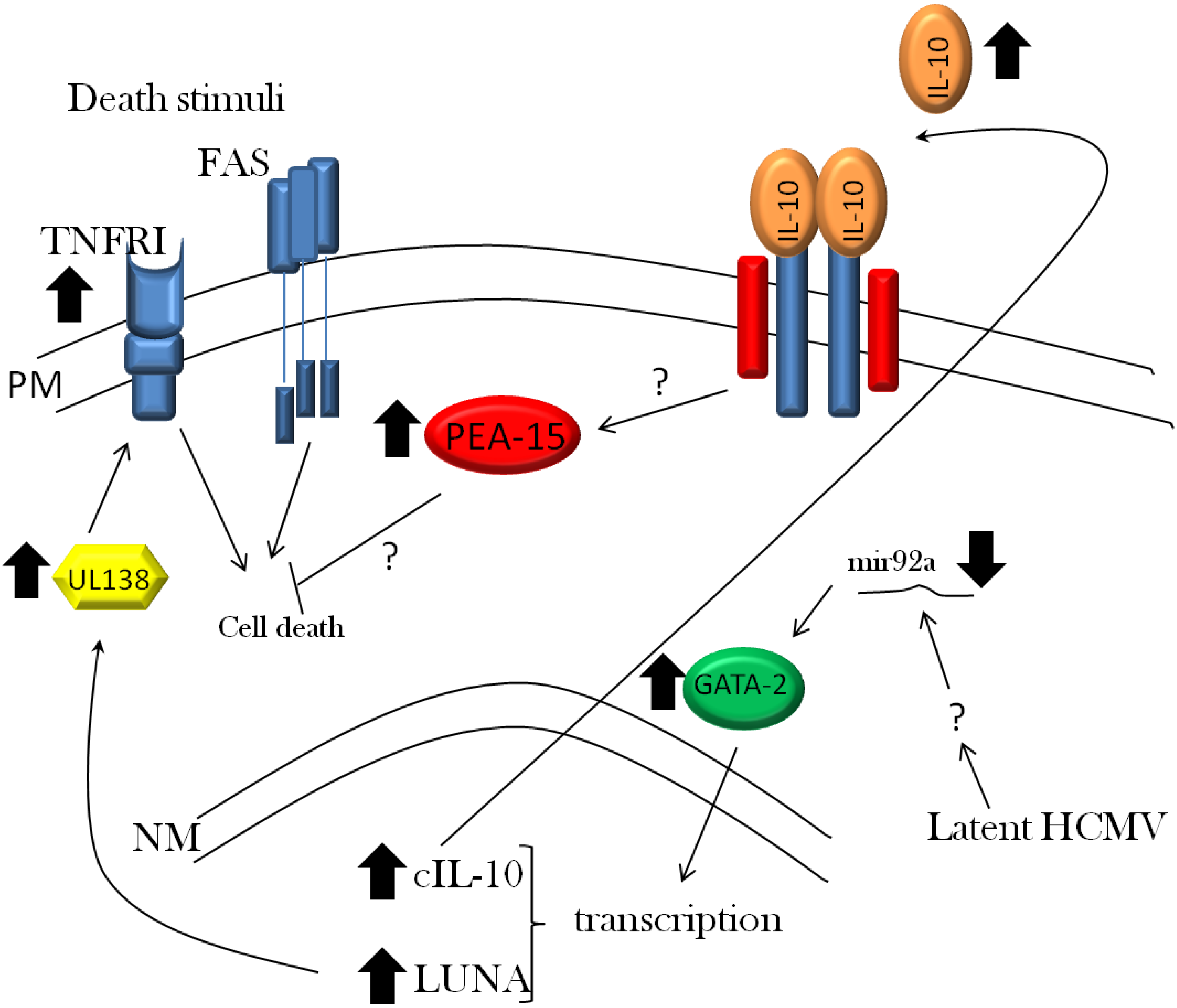
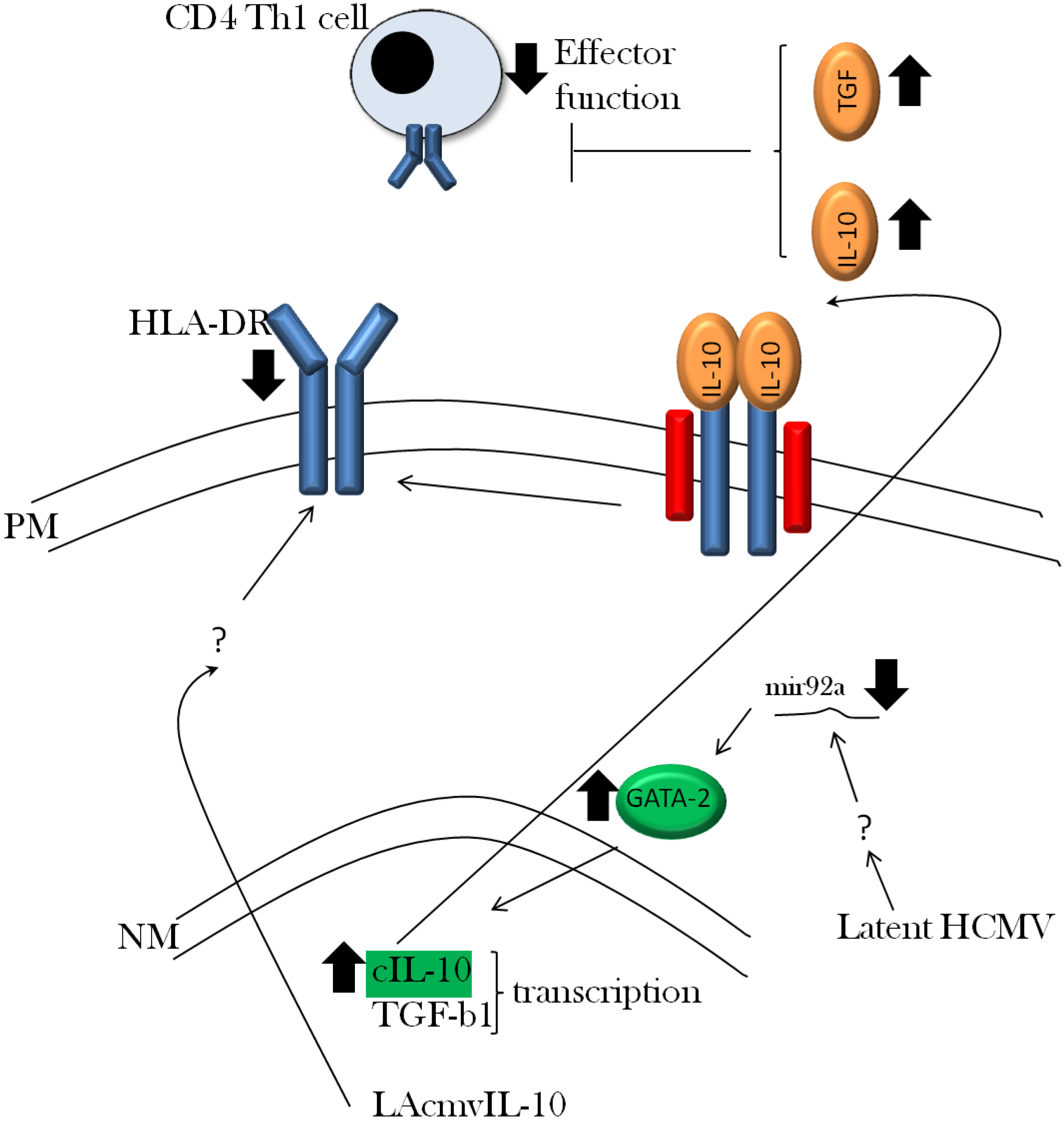
3.2. Viral Evasion of the Immune Response during Latent Infection
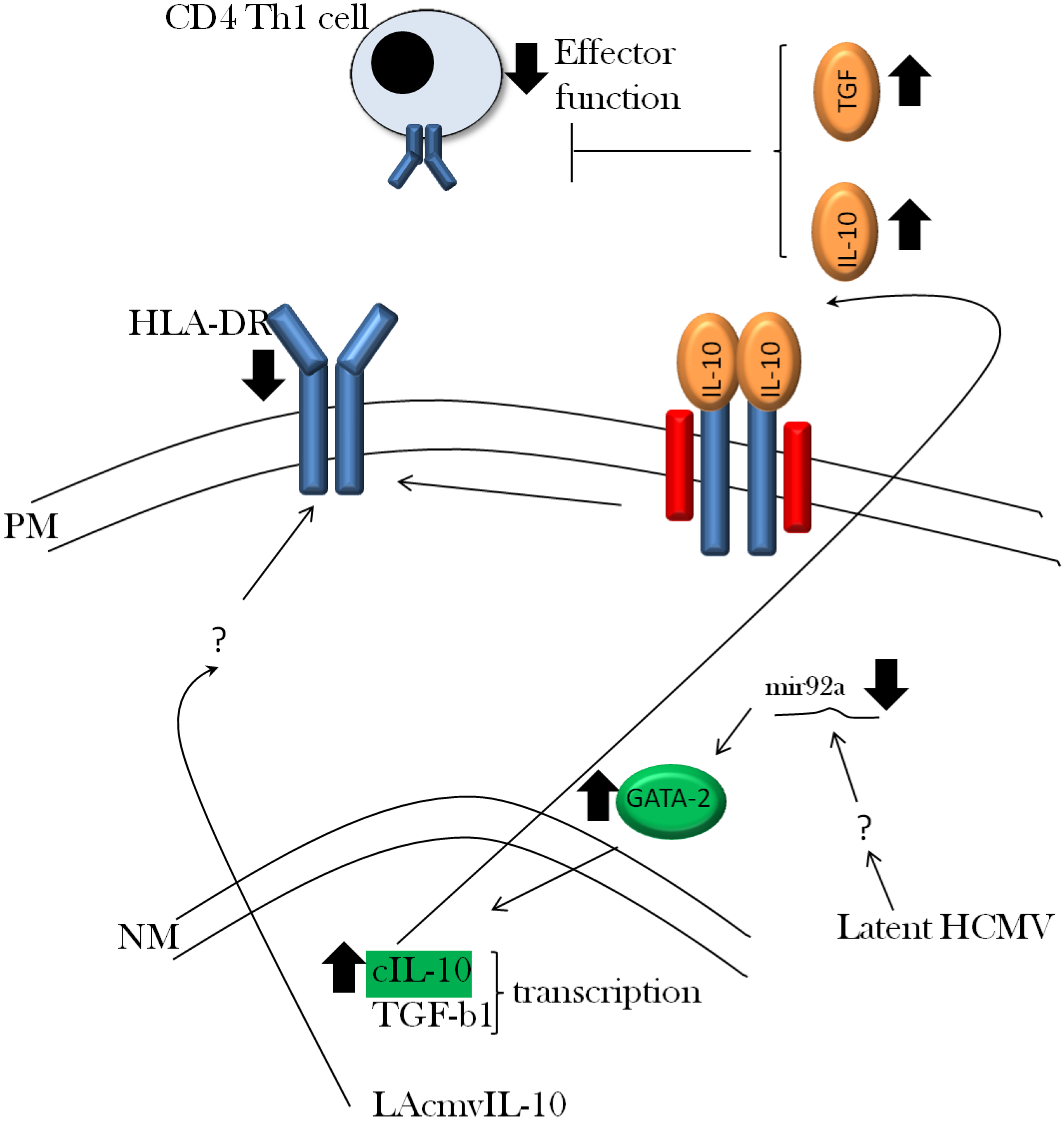
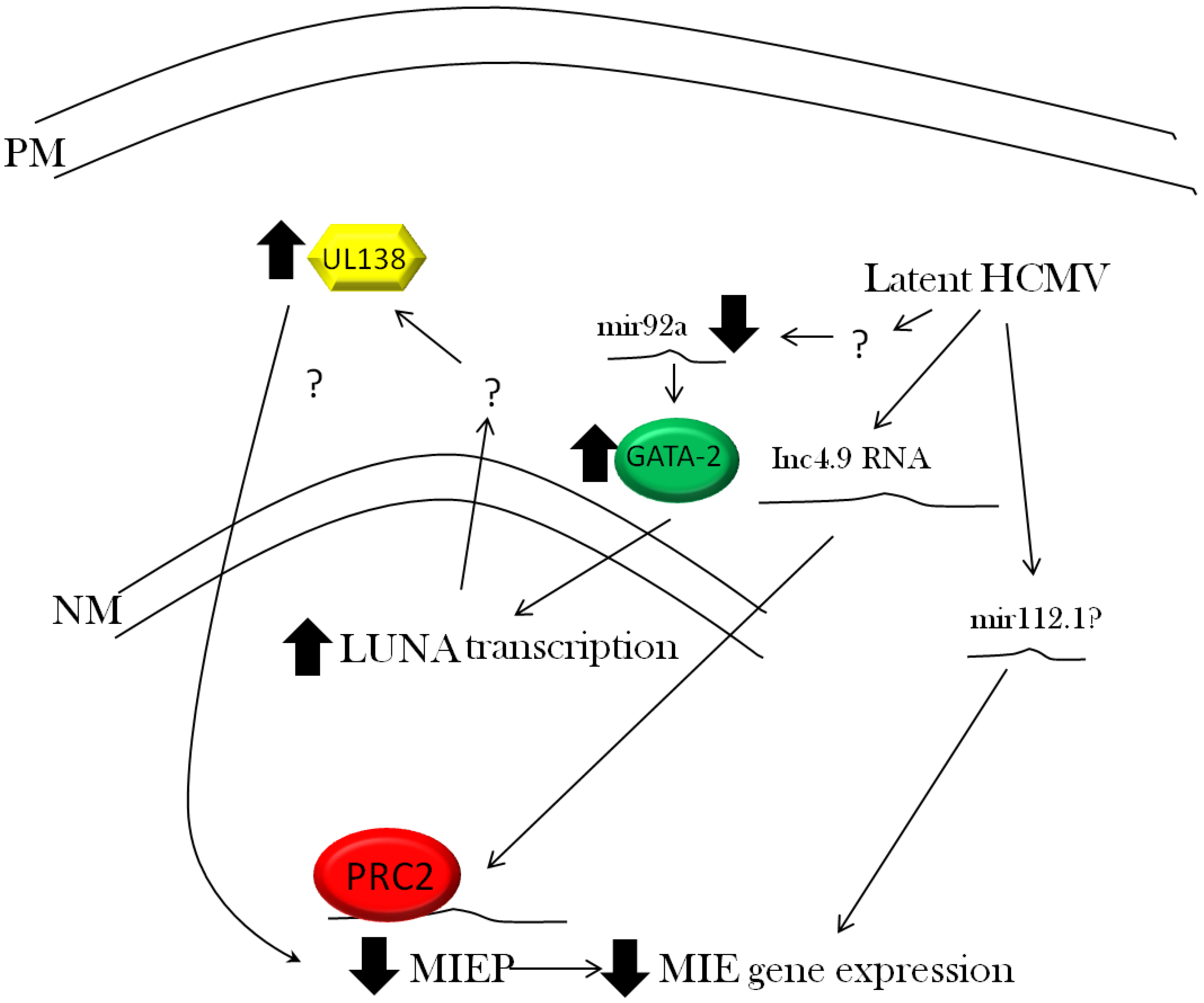
3.3. Viral Regulation of Immediate Early Gene Expression
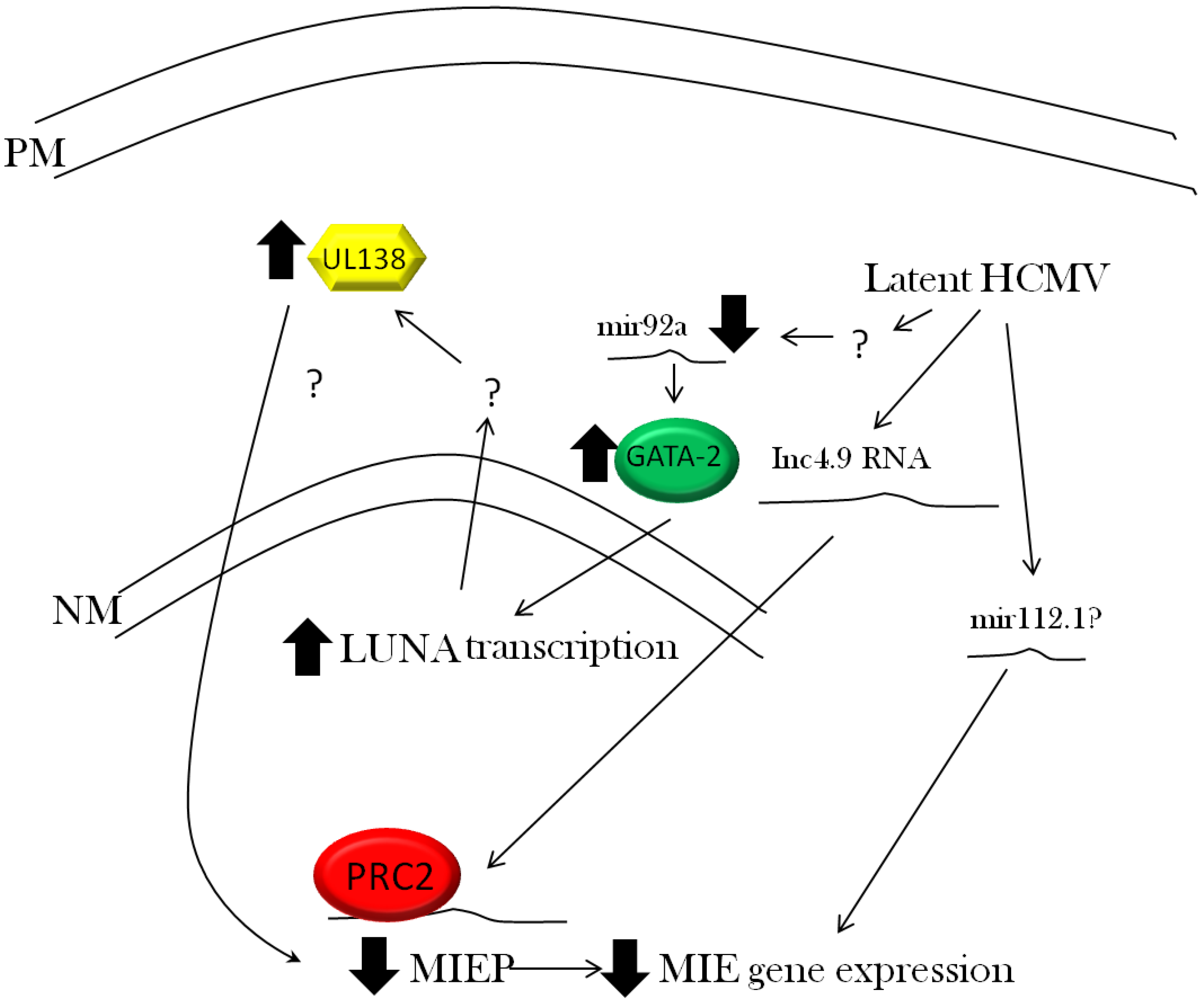
3.4. Viral Regulation of Latent Gene Expression
4. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Grilli, E.; Galati, V.; Bordi, L.; Taglietti, F.; Petrosillo, N. Cytomegalovirus pneumonia in immunocompetent host: Case report and literature review. J. Clin. Virol. 2012, 55, 356–359. [Google Scholar] [CrossRef]
- Limaye, A.P.; Kirby, K.A.; Rubenfeld, G.D.; Leisenring, W.M.; Bulger, E.M.; Neff, M.J.; Gibran, N.S.; Huang, M.L.; Santo Hayes, T.K.; Corey, L.; et al. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA 2008, 300, 413–422. [Google Scholar] [CrossRef]
- Legendre, C.; Pascual, M. Improving outcomes for solid-organ transplant recipients at risk from cytomegalovirus infection: Late-onset disease and indirect consequences. Clin. Infect. Dis. 2008, 46, 732–740. [Google Scholar] [CrossRef]
- Ljungman, P.; Hakki, M.; Boeckh, M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol. Oncol. Clin. N. Am. 2011, 25, 151–169. [Google Scholar] [CrossRef]
- Crough, T.; Khanna, R. Immunobiology of human cytomegalovirus: from bench to bedside. Clin. Microbiol. Rev. 2009, 22, 76–98. [Google Scholar] [CrossRef]
- Sinclair, J.; Sissons, P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef]
- Taylor-Wiedeman, J.; Sissons, P.; Sinclair, J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 1994, 68, 1597–1604. [Google Scholar]
- Soderberg-Naucler, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef]
- Zhuravskaya, T.; Maciejewski, J.P.; Netski, D.M.; Bruening, E.; Mackintosh, F.R.; St Jeor, S. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: Model of HCMV latency. Blood 1997, 90, 2482–2491. [Google Scholar]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef]
- Reeves, M.B.; Compton, T. Inhibition of inflammatory interleukin-6 activity via extracellular signal-regulated kinase-mitogen-activated protein kinase signaling antagonizes human cytomegalovirus reactivation from dendritic cells. J. Virol. 2011, 85, 12750–12758. [Google Scholar] [CrossRef]
- Huang, M.M.; Kew, V.G.; Jestice, K.; Wills, M.R.; Reeves, M.B. Efficient human cytomegalovirus reactivation is maturation dependent in the Langerhans dendritic cell lineage and can be studied using a CD14+ experimental latency model. J. Virol. 2012, 86, 8507–8515. [Google Scholar] [CrossRef]
- Minton, E.J.; Tysoe, C.; Sinclair, J.H.; Sissons, J.G. Human cytomegalovirus infection of the monocyte/macrophage lineage in bone marrow. J. Virol. 1994, 68, 4017–4021. [Google Scholar]
- Abraham, C.G.; Kulesza, C.A. Polycomb repressive complex 2 silences human cytomegalovirus transcription in quiescent infection models. J. Virol. 2013. [Google Scholar] [CrossRef]
- Murphy, J.C.; Fischle, W.; Verdin, E.; Sinclair, J.H. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002, 21, 1112–1120. [Google Scholar] [CrossRef]
- Reeves, M.B.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 2005, 86, 2949–2954. [Google Scholar] [CrossRef]
- Ioudinkova, E.; Arcangeletti, M.C.; Rynditch, A.; De Conto, F.; Motta, F.; Covan, S.; Pinardi, F.; Razin, S.V.; Chezzi, C. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 2006, 384, 120–128. [Google Scholar] [CrossRef]
- Meier, J.L. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: Role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 2001, 75, 1581–1593. [Google Scholar] [CrossRef]
- Keller, M.J.; Wu, A.W.; Andrews, J.I.; McGonagill, P.W.; Tibesar, E.E.; Meier, J.L. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J. Virol. 2007, 81, 6669–6681. [Google Scholar]
- Yuan, J.; Liu, X.; Wu, A.W.; McGonagill, P.W.; Keller, M.J.; Galle, C.S.; Meier, J.L. Breaking human cytomegalovirus major immediate-early gene silence by vasoactive intestinal peptide stimulation of the protein kinase A-CREB-TORC2 signaling cascade in human pluripotent embryonal NTera2 cells. J. Virol. 2009, 83, 6391–6403. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Murphy, E.A. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol. 2012, 86, 9854–9865. [Google Scholar] [CrossRef]
- Saffert, R.T.; Penkert, R.R.; Kalejta, R.F. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 2010, 84, 5594–5604. [Google Scholar] [CrossRef]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Simon, C.O.; Seckert, C.K.; Lemmermann, N.; Grzimek, N.K. Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 2008, 325, 315–331. [Google Scholar] [CrossRef]
- Schleiss, M.R. Nonprimate models of congenital cytomegalovirus (CMV) infection: gaining insight into pathogenesis and prevention of disease in newborns. ILAR J. 2006, 47, 65–72. [Google Scholar] [CrossRef]
- Yue, Y.; Barry, P.A. Rhesus cytomegalovirus a nonhuman primate model for the study of human cytomegalovirus. Adv. Virus Res. 2008, 72, 207–226. [Google Scholar] [CrossRef]
- Sindre, H.; Tjoonnfjord, G.E.; Rollag, H.; Ranneberg-Nilsen, T.; Veiby, O.P.; Beck, S.; Degre, M.; Hestdal, K. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 1996, 88, 4526–4533. [Google Scholar]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77, 3099–3102. [Google Scholar] [CrossRef]
- Reeves, M.B.; Sinclair, J.H. Circulating dendritic cells isolated from healthy seropositive donors are sites of human cytomegalovirus reactivation in vivo. J. Virol. 2013, 87, 10660–10667. [Google Scholar] [CrossRef]
- Hargett, D.; Shenk, T.E. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Tempera, I.; Lieberman, P.M. Chromatin organization of gammaherpesvirus latent genomes. Biochim. Biophys. Acta. 2010, 1799, 236–245. [Google Scholar]
- Reeves, M.B. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus. Res. 2011, 157, 134–143. [Google Scholar] [CrossRef]
- Sinclair, J. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta 2010, 1799, 286–295. [Google Scholar]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.; et al. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef]
- Murphy, E.; Yu, D.; Grimwood, J.; Schmutz, J.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2003, 100, 14976–14981. [Google Scholar] [CrossRef]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef]
- Grey, F.; Antoniewicz, A.; Allen, E.; Saugstad, J.; McShea, A.; Carrington, J.C.; Nelson, J. Identification and characterization of human cytomegalovirus-encoded microRNAs. J. Virol. 2005, 79, 12095–12099. [Google Scholar] [CrossRef]
- Dunn, W.; Trang, P.; Zhong, Q.; Yang, E.; van Belle, C.; Liu, F. Human cytomegalovirus expresses novel microRNAs during productive viral infection. Cell. Microbiol. 2005, 7, 1684–1695. [Google Scholar] [CrossRef]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar] [CrossRef]
- Kondo, K.; Xu, J.; Mocarski, E.S. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. USA 1996, 93, 11137–11142. [Google Scholar] [CrossRef]
- White, K.L.; Slobedman, B.; Mocarski, E.S. Human cytomegalovirus latency-associated protein pORF94 is dispensable for productive and latent infection. J. Virol. 2000, 74, 9333–9337. [Google Scholar] [CrossRef]
- Jackson, S.E.; Mason, G.M.; Wills, M.R. Human cytomegalovirus immunity and immune evasion. Virus. Res. 2011, 157, 151–160. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Hahn, G.; White, K.L.; Xu, J.; Slobedman, B.; Hertel, L.; Aguirre, S.A.; Noda, S. Myeloid Cell Recruitment and Function in Pathogenesis and Latency. In Cytomegaloviruses: Molecular Biology and Immunology; Reddehase, M.J., Ed.; Caister Academic Press: Poole, UK, 2006; Volume 1, pp. 463–482. [Google Scholar]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72, 2059–2064. [Google Scholar] [CrossRef]
- Taylor-Wiedeman, J.; Hayhurst, G.P.; Sissons, J.G.; Sinclair, J.H. Polymorphonuclear cells are not sites of persistence of human cytomegalovirus in healthy individuals. J. Gen. Virol. 1993, 74, 265–268. [Google Scholar] [CrossRef]
- Bevan, I.S.; Daw, R.A.; Day, P.J.; Ala, F.A.; Walker, M.R. Polymerase chain reaction for detection of human cytomegalovirus infection in a blood donor population. Br. J. Haematol. 1991, 78, 94–99. [Google Scholar] [CrossRef]
- Cheung, A.K.; Abendroth, A.; Cunningham, A.L.; Slobedman, B. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 2006, 108, 3691–3699. [Google Scholar] [CrossRef]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef]
- Jenkins, C.; Abendroth, A.; Slobedman, B. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 2004, 78, 1440–1447. [Google Scholar] [CrossRef]
- Bego, M.; Maciejewski, J.; Khaiboullina, S.; Pari, G.; St Jeor, S. Characterization of an antisense transcript spanning the UL81–82 locus of human cytomegalovirus. J. Virol. 2005, 79, 11022–11034. [Google Scholar] [CrossRef]
- Kalla, M.; Hammerschmidt, W. Human B cells on their route to latent infection—early but transient expression of lytic genes of Epstein-Barr virus. Eur. J. Cell. Biol. 2012, 91, 65–69. [Google Scholar] [CrossRef]
- Tan, J.C.; Avdic, S.; Cao, J.Z.; Mocarski, E.S.; White, K.L.; Abendroth, A.; Slobedman, B. Inhibition of 2',5'-oligoadenylate synthetase expression and function by the human cytomegalovirus ORF94 gene product. J. Virol. 2011, 85, 5696–5700. [Google Scholar] [CrossRef]
- Petrucelli, A.; Rak, M.; Grainger, L.; Goodrum, F. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J. Virol. 2009, 83, 5615–5629. [Google Scholar] [CrossRef]
- Le, V.T.; Trilling, M.; Hengel, H. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb'-encoded modulation of TNF-alpha signaling. J. Virol. 2011, 85, 13260–13270. [Google Scholar] [CrossRef]
- Montag, C.; Wagner, J.A.; Gruska, I.; Vetter, B.; Wiebusch, L.; Hagemeier, C. The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to tumor necrosis factor alpha (TNF-alpha) signaling by upregulating TNF-alpha receptor 1 cell surface expression. J. Virol. 2011, 85, 11409–11421. [Google Scholar] [CrossRef]
- Weekes, M.P.; Tan, S.Y.; Poole, E.; Talbot, S.; Antrobus, R.; Smith, D.L.; Montag, C.; Gygi, S.P.; Sinclair, J.H.; Lehner, P.J. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 2013, 340, 199–202. [Google Scholar] [CrossRef]
- Keyes, L.R.; Hargett, D.; Soland, M.; Bego, M.G.; Rossetto, C.C.; Almeida-Porada, G.; St Jeor, S. HCMV protein LUNA is required for viral reactivation from latently infected primary CD14(+) cells. PLoS One 2013, 7, e52827. [Google Scholar]
- Jenkins, C.; Garcia, W.; Godwin, M.J.; Spencer, J.V.; Stern, J.L.; Abendroth, A.; Slobedman, B. Immunomodulatory properties of a viral homolog of human interleukin-10 expressed by human cytomegalovirus during the latent phase of infection. J. Virol. 2008, 82, 3736–3750. [Google Scholar] [CrossRef]
- Pari, G.S.; Anders, D.G. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 1993, 67, 6979–6988. [Google Scholar]
- Sarisky, R.T.; Hayward, G.S. Evidence that the UL84 gene product of human cytomegalovirus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J. Virol. 1996, 70, 7398–7413. [Google Scholar]
- Spector, D.J.; Tevethia, M.J. Protein-protein interactions between human cytomegalovirus IE2–580aa and pUL84 in lytically infected cells. J. Virol. 1994, 68, 7549–7553. [Google Scholar]
- Colletti, K.S.; Xu, Y.; Yamboliev, I.; Pari, G.S. Human cytomegalovirus UL84 is a phosphoprotein that exhibits UTPase activity and is a putative member of the DExD/H box family of proteins. J. Biol. Chem. 2005, 280, 11955–11960. [Google Scholar] [CrossRef]
- Boomker, J.M.; The, T.H.; de Leij, L.F.; Harmsen, M.C. The human cytomegalovirus-encoded receptor US28 increases the activity of the major immediate-early promoter/enhancer. Virus. Res. 2006, 118, 196–200. [Google Scholar] [CrossRef]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef]
- Billstrom, M.A.; Johnson, G.L.; Avdi, N.J.; Worthen, G.S. Intracellular signaling by the chemokine receptor US28 during human cytomegalovirus infection. J. Virol. 1998, 72, 5535–5544. [Google Scholar]
- Casarosa, P.; Bakker, R.A.; Verzijl, D.; Navis, M.; Timmerman, H.; Leurs, R.; Smit, M.J. Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J. Biol. Chem. 2001, 276, 1133–1137. [Google Scholar]
- Gao, J.L.; Murphy, P.M. Human cytomegalovirus open reading frame US28 encodes a functional beta chemokine receptor. J. Biol. Chem. 1994, 269, 28539–28542. [Google Scholar]
- Miller, W.E.; Houtz, D.A.; Nelson, C.D.; Kolattukudy, P.E.; Lefkowitz, R.J. G-protein-coupled receptor (GPCR) kinase phosphorylation and beta-arrestin recruitment regulate the constitutive signaling activity of the human cytomegalovirus US28 GPCR. J. Biol. Chem. 2003, 278, 21663–21671. [Google Scholar]
- Streblow, D.N.; Soderberg-Naucler, C.; Vieira, J.; Smith, P.; Wakabayashi, E.; Ruchti, F.; Mattison, K.; Altschuler, Y.; Nelson, J.A. The human cytomegalovirus chemokine receptor US28 mediates vascular smooth muscle cell migration. Cell 1999, 99, 511–520. [Google Scholar] [CrossRef]
- Poole, E.; King, C.A.; Sinclair, J.H.; Alcami, A. The UL144 gene product of human cytomegalovirus activates NFkappaB via a TRAF6-dependent mechanism. EMBO J. 2006, 25, 4390–4399. [Google Scholar] [CrossRef]
- Poole, E.; Atkins, E.; Nakayama, T.; Yoshie, O.; Groves, I.; Alcami, A.; Sinclair, J. NF-kappaB-mediated activation of the chemokine CCL22 by the product of the human cytomegalovirus gene UL144 escapes regulation by viral IE86. J. Virol. 2008, 82, 4250–4256. [Google Scholar]
- Poole, E.; Walther, A.; Raven, K.; Benedict, C.A.; Mason, G.M.; Sinclair, J. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J. Virol. 2013, 87, 4261–4271. [Google Scholar] [CrossRef]
- Benedict, C.A.; Butrovich, K.D.; Lurain, N.S.; Corbeil, J.; Rooney, I.; Schneider, P.; Tschopp, J.; Ware, C.F. Cutting edge: A novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J. Immunol. 1999, 162, 6967–6970. [Google Scholar]
- Goldmacher, V.S.; Bartle, L.M.; Skaletskaya, A.; Dionne, C.A.; Kedersha, N.L.; Vater, C.A.; Han, J.W.; Lutz, R.J.; Watanabe, S.; Cahir McFarland, E.D.; et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA 1999, 96, 12536–12541. [Google Scholar] [CrossRef]
- McCormick, A.L. Control of apoptosis by human cytomegalovirus. Curr. Top. Microbiol. Immunol. 2008, 325, 281–295. [Google Scholar] [CrossRef]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell. Host. Microbe. 2008, 3, 253–262. [Google Scholar] [CrossRef]
- Terhune, S.; Torigoi, E.; Moorman, N.; Silva, M.; Qian, Z.; Shenk, T.; Yu, D. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 2007, 81, 3109–3123. [Google Scholar] [CrossRef]
- Everett, H.; McFadden, G. Apoptosis: An innate immune response to virus infection. Trends Microbiol. 1999, 7, 160–165. [Google Scholar] [CrossRef]
- Reeves, M.B.; Breidenstein, A.; Compton, T. Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc. Natl. Acad. Sci. USA 2012, 109, 588–593. [Google Scholar]
- Chan, G.; Nogalski, M.T.; Bentz, G.L.; Smith, M.S.; Parmater, A.; Yurochko, A.D. PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J. Immunol. 2010, 184, 3213–3222. [Google Scholar] [CrossRef]
- Perciavalle, R.M.; Opferman, J.T. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell. Biol. 2013, 23, 22–29. [Google Scholar] [CrossRef]
- Slobedman, B.; Stern, J.L.; Cunningham, A.L.; Abendroth, A.; Abate, D.A.; Mocarski, E.S. Impact of human cytomegalovirus latent infection on myeloid progenitor cell gene expression. J. Virol. 2004, 78, 4054–4062. [Google Scholar] [CrossRef]
- Condorelli, G.; Vigliotta, G.; Cafieri, A.; Trencia, A.; Andalo, P.; Oriente, F.; Miele, C.; Caruso, M.; Formisano, P.; Beguinot, F. PED/PEA-15: An anti-apoptotic molecule that regulates FAS/TNFR1-induced apoptosis. Oncogene 1999, 18, 4409–4415. [Google Scholar] [CrossRef]
- Poole, E.; McGregor Dallas, S.R.; Colston, J.; Joseph, R.S.; Sinclair, J. Virally induced changes in cellular microRNAs maintain latency of human cytomegalovirus in CD34(+) progenitors. J. Gen. Virol. 2011, 92, 1539–1549. [Google Scholar] [CrossRef]
- Mason, G.M.; Poole, E.; Sissons, J.G.; Wills, M.R.; Sinclair, J.H. Human cytomegalovirus latency alters the cellular secretome, inducing cluster of differentiation (CD)4+ T-cell migration and suppression of effector function. Proc. Natl. Acad. Sci. USA 2012, 109, 14538–14543. [Google Scholar] [CrossRef]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef]
- Tey, S.K.; Goodrum, F.; Khanna, R. CD8+ T-cell recognition of human cytomegalovirus latency-associated determinant pUL138. J. Gen. Virol. 2010, 91, 2040–2048. [Google Scholar] [CrossRef]
- Mason, G.; Jackson, S.E.; Okecha, G.; Poole, E.; Sissons, J.G.P.; Sinclair, J.; Wills, M.R. Human cytomegalovirus latency-associated proteins elicit immune-suppressive IL-10 producing CD4+ T cells. PLoS Pathog. 2013, 10, e1003635. [Google Scholar]
- Opal, S.M.; DePalo, V.A. Anti-inflammatory cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef]
- Slobedman, B.; Mocarski, E.S. Quantitative analysis of latent human cytomegalovirus. J. Virol. 1999, 73, 4806–4812. [Google Scholar]
- Kotenko, S.V.; Saccani, S.; Izotova, L.S.; Mirochnitchenko, O.V.; Pestka, S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc. Natl. Acad. Sci. USA 2000, 97, 1695–1700. [Google Scholar] [CrossRef]
- Lockridge, K.M.; Zhou, S.S.; Kravitz, R.H.; Johnson, J.L.; Sawai, E.T.; Blewett, E.L.; Barry, P.A. Primate cytomegaloviruses encode and express an IL-10-like protein. Virology 2000, 268, 272–280. [Google Scholar] [CrossRef]
- Nachtwey, J.; Spencer, J.V. HCMV IL-10 suppresses cytokine expression in monocytes through inhibition of nuclear factor-kappaB. Viral. Immunol. 2008, 21, 477–482. [Google Scholar] [CrossRef]
- Spencer, J.V. The cytomegalovirus homolog of interleukin-10 requires phosphatidylinositol 3-kinase activity for inhibition of cytokine synthesis in monocytes. J. Virol. 2007, 81, 2083–2086. [Google Scholar] [CrossRef]
- Spencer, J.V.; Lockridge, K.M.; Barry, P.A.; Lin, G.; Tsang, M.; Penfold, M.E.; Schall, T.J. Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J. Virol. 2002, 76, 1285–1292. [Google Scholar] [CrossRef]
- Jones, B.C.; Logsdon, N.J.; Josephson, K.; Cook, J.; Barry, P.A.; Walter, M.R. Crystal structure of human cytomegalovirus IL-10 bound to soluble human IL-10R1. Proc. Natl. Acad. Sci. USA 2002, 99, 9404–9409. [Google Scholar]
- Chang, W.L.; Barry, P.A. Attenuation of innate immunity by cytomegalovirus IL-10 establishes a long-term deficit of adaptive antiviral immunity. Proc. Natl. Acad. Sci. USA 2011, 107, 22647–22652. [Google Scholar] [CrossRef]
- Chang, W.L.; Baumgarth, N.; Yu, D.; Barry, P.A. Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J. Virol. 2004, 78, 8720–8731. [Google Scholar] [CrossRef]
- Raftery, M.J.; Wieland, D.; Gronewald, S.; Kraus, A.A.; Giese, T.; Schonrich, G. Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J. Immunol. 2004, 173, 3383–3391. [Google Scholar]
- Avdic, S.; Cao, J.Z.; McSharry, B.P.; Clancy, L.E.; Brown, R.; Steain, M.; Gottlieb, D.J.; Abendroth, A.; Slobedman, B. Human cytomegalovirus interleukin-10 polarizes monocytes toward a deactivated M2c phenotype to repress host immune responses. J. Virol. 2013, 87, 10273–10282. [Google Scholar] [CrossRef]
- Slobedman, B.; Mocarski, E.S.; Arvin, A.M.; Mellins, E.D.; Abendroth, A. Latent cytomegalovirus down-regulates major histocompatibility complex class II expression on myeloid progenitors. Blood 2002, 100, 2867–2873. [Google Scholar] [CrossRef]
- Cheung, A.K.; Gottlieb, D.J.; Plachter, B.; Pepperl-Klindworth, S.; Avdic, S.; Cunningham, A.L.; Abendroth, A.; Slobedman, B. The role of the human cytomegalovirus UL111A gene in down-regulating CD4+ T-cell recognition of latently infected cells: Implications for virus elimination during latency. Blood 2009, 114, 4128–4137. [Google Scholar] [CrossRef]
- Avdic, S.; Cao, J.Z.; Cheung, A.K.; Abendroth, A.; Slobedman, B. Viral interleukin-10 expressed by human cytomegalovirus during the latent phase of infection modulates latently infected myeloid cell differentiation. J. Virol. 2011, 85, 7465–7471. [Google Scholar] [CrossRef]
- Groves, I.J.; Reeves, M.B.; Sinclair, J.H. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic “pre-immediate-early” repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 2009, 90, 2364–2374. [Google Scholar] [CrossRef]
- Maul, G.G. Initiation of cytomegalovirus infection at ND10. Curr. Top. Microbiol. Immunol. 2008, 325, 117–132. [Google Scholar] [CrossRef]
- Kalejta, R.F. Functions of human cytomegalovirus tegument proteins prior to immediate early gene expression. Curr. Top. Microbiol. Immunol. 2008, 325, 101–115. [Google Scholar] [CrossRef]
- Stamminger, T. Interactions of human cytomegalovirus proteins with the nuclear transport machinery. Curr. Top. Microbiol. Immunol. 2008, 325, 167–185. [Google Scholar] [CrossRef]
- Sinclair, J.; Sissons, P. Latent and persistent infections of monocytes and macrophages. Intervirology 1996, 39, 293–301. [Google Scholar]
- Wright, E.; Bain, M.; Teague, L.; Murphy, J.; Sinclair, J. Ets-2 repressor factor recruits histone deacetylase to silence human cytomegalovirus immediate-early gene expression in non-permissive cells. J. Gen. Virol. 2005, 86, 535–544. [Google Scholar] [CrossRef]
- Liu, R.; Baillie, J.; Sissons, J.G.; Sinclair, J.H. The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic. Acids. Res. 1994, 22, 2453–2459. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef]
- Murphy, E.; Vanicek, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc. Natl. Acad. Sci. USA 2008, 105, 5453–5458. [Google Scholar] [CrossRef]
- Grey, F.; Meyers, H.; White, E.A.; Spector, D.H.; Nelson, J. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog. 2007, 3, e163. [Google Scholar] [CrossRef]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of viral and cellular gene expression by Kaposi's sarcoma-associated herpesvirus polyadenylated nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Coen, D.M.; Knipe, D.M. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. mBio 2013, 4. [Google Scholar] [CrossRef]
- Tsai, F.Y.; Keller, G.; Kuo, F.C.; Weiss, M.; Chen, J.; Rosenblatt, M.; Alt, F.W.; Orkin, S.H. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 1994, 371, 221–226. [Google Scholar] [CrossRef]
- Ling, K.W.; Ottersbach, K.; van Hamburg, J.P.; Oziemlak, A.; Tsai, F.Y.; Orkin, S.H.; Ploemacher, R.; Hendriks, R.W.; Dzierzak, E. GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J. Exp. Med. 2004, 200, 871–882. [Google Scholar] [CrossRef]
- Reeves, M.; Sinclair, J. Regulation of human cytomegalovirus transcription in latency: Beyond the major immediate-early promoter. Viruses 2013, 5, 1395–1413. [Google Scholar] [CrossRef]
- Reeves, M.; Woodhall, D.; Compton, T.; Sinclair, J. Human cytomegalovirus IE72 protein interacts with the transcriptional repressor hDaxx to regulate LUNA gene expression during lytic infection. J. Virol. 2010, 84, 7185–7194. [Google Scholar] [CrossRef]
- Smith, M.S.; Goldman, D.C.; Bailey, A.S.; Pfaffle, D.L.; Kreklywich, C.N.; Spencer, D.B.; Othieno, F.A.; Streblow, D.N.; Garcia, J.V.; Fleming, W.H.; et al. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 2010, 8, 284–291. [Google Scholar] [CrossRef]
- Jurak, I.; Brune, W. Induction of apoptosis limits cytomegalovirus cross-species infection. EMBO J. 2006, 25, 2634–2642. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sinclair, J.H.; Reeves, M.B. Human Cytomegalovirus Manipulation of Latently Infected Cells. Viruses 2013, 5, 2803-2824. https://doi.org/10.3390/v5112803
Sinclair JH, Reeves MB. Human Cytomegalovirus Manipulation of Latently Infected Cells. Viruses. 2013; 5(11):2803-2824. https://doi.org/10.3390/v5112803
Chicago/Turabian StyleSinclair, John H., and Matthew B. Reeves. 2013. "Human Cytomegalovirus Manipulation of Latently Infected Cells" Viruses 5, no. 11: 2803-2824. https://doi.org/10.3390/v5112803
APA StyleSinclair, J. H., & Reeves, M. B. (2013). Human Cytomegalovirus Manipulation of Latently Infected Cells. Viruses, 5(11), 2803-2824. https://doi.org/10.3390/v5112803