Pharmacological Inhibition of Feline Immunodeficiency Virus (FIV)

Abstract
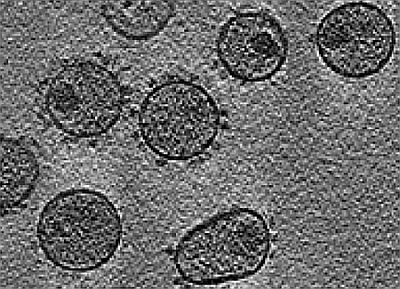
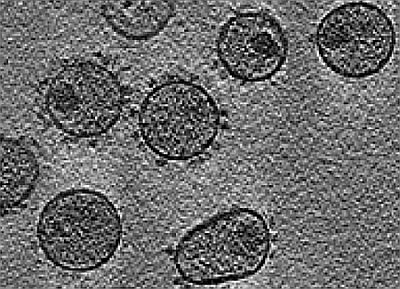
:
1. Introduction
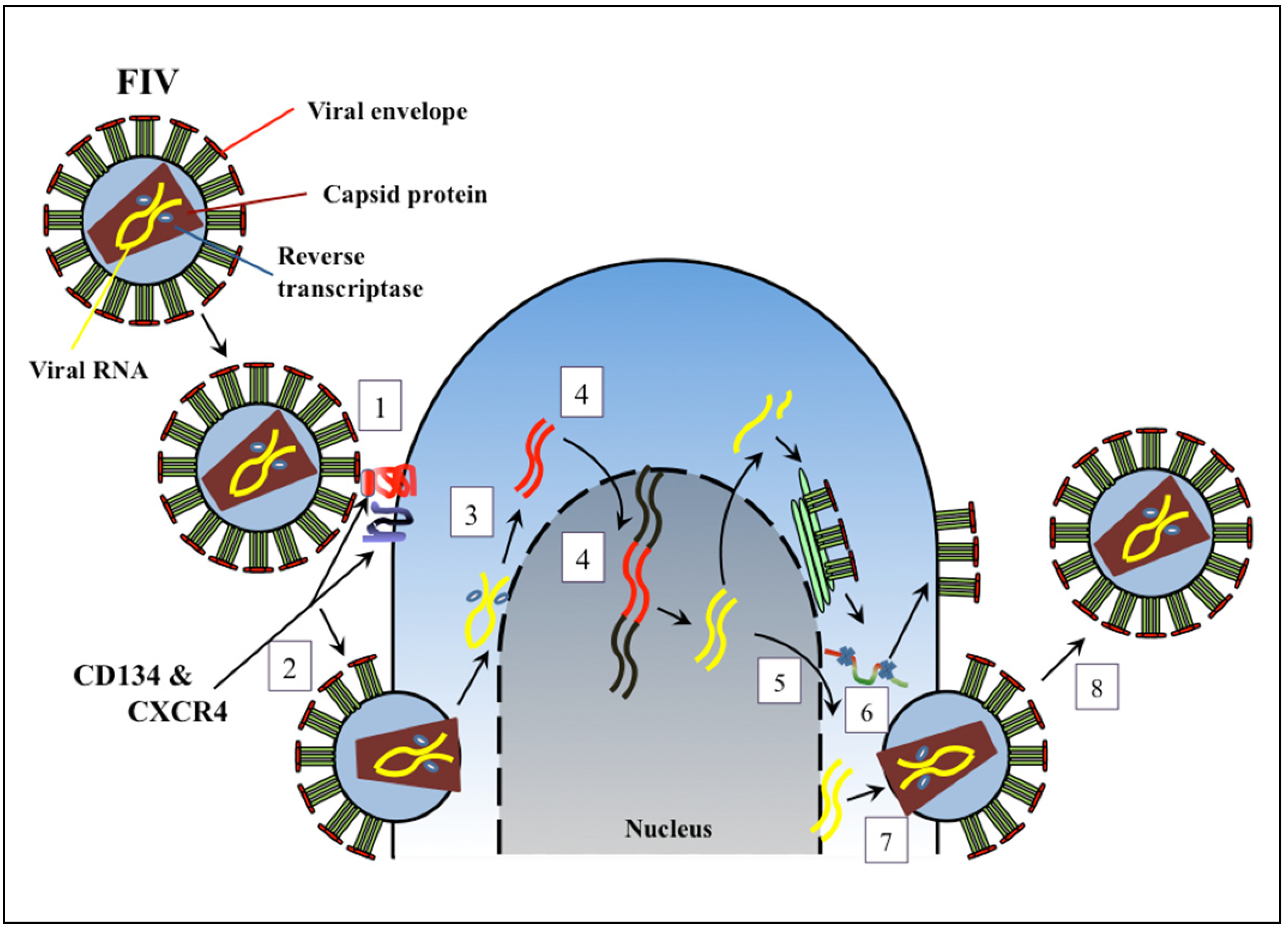

{kind=link}
{kind=link}
{kind=link}
| Nature of interference | Agent |
|---|---|
| Virus entry | Maraviroc (CCR5 antagonist), Enfuvirtide (fusion inhibitor) |
| Nucleoside reverse transcriptase inhibition | Zidovudine, Stavudine, Lamivudine, Didanosine, Abacavir, Emtricitabine |
| Nucleotide reverse transcriptase inhibition | Tenofovir disoproxil fumarate |
| Non-nucleoside reverse transcriptase inhibition | Efavirenz, Nevirapine, Delavirdine, Etravirine |
| Protease inhibition | Indinavir, Ritonavir, Nelfinavir, Saquinavir, Atazanavir, Darunavir, Fosamprenavir, Tipranavir, Lopinavir |
| Integrase inhibition | Raltegravir |
| Approach | Agent |
|---|---|
| CD4 attachment inhibition | PRO-542, BMS-806 |
| Chemokine receptor inhibition | Vicriviroc, PRO 140 |
| Fusion inhibition | ADS-J1 |
| Nucleoside reverse transcriptase inhibition | Emtricitabine, Amdoxovir |
| Non-nucleoside reverse transcriptase inhibition | DPC-083, Etravirine, Calanolide A |
| Protease inhibition | Darunavir |
| Integrase inhibition | Elvitegravir, MK-2048 |
2. Inhibitors of Virus Entry
2.1. Attachment Inhibitors
2.2. Inhibitors of Co-receptor Interaction
2.3. Fusion Inhibitors
3. Inhibitors of Reverse Transcription of Viral Genomic RNA
3.1. NRTIs and NtRTIs
3.2. Non-nucleoside Reverse Transcriptase Inhibitors (NNRTIs)
4. Inhibitors of Nuclear Translocation and Integration of Viral DNA into Host Genome
5. Inhibitors of Viral Transcription and Nuclear Export
6. Inhibitors of Viral Protease and Virion Assembly
7. Miscellaneous
8. Conclusions
| Approach | Compounds | Outcome |
|---|---|---|
| CXCR4 entry inhibitor | AMD3100 | Reduced FIV replication in vitro and tolerated by cats [38,39]. |
| Nucleoside reverse transcriptase inhibitor | Zidovudine, Stavudine, PMEA, Dideoxycytidine, Fozivudine, WHI-07 (derivative of zidovudine), Stampidine, Lamivudine | Prevented virus replication in vitro and in vivo, decreased FIV load in chronically infected cats, tolerated [48,49,50,51,52,53,54,55,56,57,58,59,60,61]. |
| Protease inhibitor | Atazanavir, Tipranavir, Lopinavir, TL-3 | Inhibited FIV replication, reduced neurodegeneration and in vivo tolerated [86,87,88]. |
| Integrase inhibitor | L-870810 | Reduced FIV replication in vitro [75]. |
Acknowledgments
References
- Pedersen, N.C.; Ho, E.W.; Brown, M.L.; Yamamoto, J.K. Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science 1987, 235, 790–793. [Google Scholar]
- Bendinelli, M.; Pistello, M.; Lombardi, S.; Poli, A.; Garzelli, C.; Matteucci, D.; Ceccherini-Nelli, L.; Malvaldi, G.; Tozzini, F. Feline immunodeficiency virus: An interesting model for aids studies and an important cat pathogen. Clin. Microbiol. Rev. 1995, 8, 87–112. [Google Scholar]
- Pedersen, N.C.; Yamamoto, J.K.; Ishida, T.; Hansen, H. Feline immunodeficiency virus infection. Vet. Immunol. Immunopathol. 1989, 21, 111–129. [Google Scholar]
- Miyazawa, T.; Tomonaga, K.; Kawaguchi, Y.; Mikami, T. The genome of feline immunodeficiency virus. Arch. Virol. 1994, 134, 221–234. [Google Scholar]
- Shimojima, M.; Miyazawa, T.; Ikeda, Y.; McMonagle, E.L.; Haining, H.; Akashi, H.; Takeuchi, Y.; Hosie, M.J.; Willett, B.J. Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science 2004, 303, 1192–1195. [Google Scholar]
- De Parseval, A.; Chatterji, U.; Morris, G.; Sun, P.; Olson, A.J.; Elder, J.H. Structural mapping of CD134 residues critical for interaction with feline immunodeficiency virus. Nat. Struct. Mol. Biol. 2005, 12, 60–66. [Google Scholar]
- De Parseval, A.; Grant, C.K.; Sastry, K.J.; Elder, J.H. Sequential CD134-CXCR4 interactions in feline immunodeficiency virus (FIV): Soluble CD134 activates FIV Env for CXCR4-dependent entry and reveals a cryptic neutralization epitope. J. Virol. 2006, 80, 3088–3091. [Google Scholar]
- Phillips, T.R.; Lamont, C.; Konings, D.A.; Shacklett, B.L.; Hamson, C.A.; Luciw, P.A.; Elder, J.H. Identification of the rev transactivation and rev-responsive elements of feline immunodeficiency virus. J. Virol. 1992, 66, 5464–5471. [Google Scholar]
- De Clercq, E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 2002, 1, 13–25. [Google Scholar]
- Reeves, J.D.; Piefer, A.J. Emerging drug targets for antiretroviral therapy. Drugs 2005, 65, 1747–1766. [Google Scholar]
- Fischl, M.A.; Richman, D.D.; Grieco, M.H.; Gottlieb, M.S.; Volberding, P.A.; Laskin, O.L.; Leedom, J.M.; Groopman, J.E.; Mildvan, D.; Schooley, R.T. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N. Engl. J. Med. 1987, 317, 185–191. [Google Scholar]
- Dau, B.; Holodniy, M. Novel targets for antiretroviral therapy: Clinical progress to date. Drugs 2009, 69, 31–50. [Google Scholar]
- Ghosh, R.K.; Ghosh, S.M.; Chawla, S. Recent advances in antiretroviral drugs. Expert Opin. Pharmacother. 2011, 12, 31–46. [Google Scholar]
- Pereira, C.F.; Paridaen, J.T. Anti-HIV drug development—An overview. Curr. Pharm. Des. 2004, 10, 4005–4037. [Google Scholar]
- Gulick, R.M. New antiretroviral drugs. Clin. Microbiol. Infect. 2003, 9, 186–193. [Google Scholar]
- Gulick, R.M. Antiretroviral treatment 2010: Progress and controversies. J. Acquir. Immune Defic. Syndr. 2010, 55, S43–S48. [Google Scholar]
- Troia-Cancio, P.; Asmuth, D.M. Lessons from maraviroc clinical trials. Expert Rev. Anti Infect. Ther. 2011, 9, 649–651. [Google Scholar]
- Peters, B.S.; Conway, K. Therapy for HIV: Past, present, and future. Adv. Dent. Res. 2011, 23, 23–27. [Google Scholar] [CrossRef]
- Dias, A.S.; Bester, M.J.; Britz, R.F.; Apostolides, Z. Animal models used for the evaluation of antiretroviral therapies. Curr. HIV Res. 2006, 4, 431–446. [Google Scholar]
- Elder, J.H.; Lin, Y.C.; Fink, E.; Grant, C.K. Feline Immunodeficiency Virus (FIV) as a Model for Study of Lentivirus Infections: Parallels with HIV. Curr. HIV Res. 2010, 8, 73–80. [Google Scholar]
- North, T.W.; LaCasse, R.A. Testing anti-HIV drugs in the FIV model. Nat. Med. 1995, 1, 410–411. [Google Scholar]
- Singh, I.P.; Chauthe, S.K. Small molecule HIV entry inhibitors: Part II. Attachment and fusion inhibitors: 2004–2010. Expert Opin. Ther. Pat. 2011, 21, 399–416. [Google Scholar] [CrossRef]
- Tanabe-Tochikura, A.; Tochikura, T.S.; Blakeslee, J.R. Anti-human immunodeficiency virus (HIV) agents are also potent and selective inhibitors of feline immunodeficiency virus (FIV)-induced cytopathic effect: Development of a new method for screening of anti-FIV substances in vitro. Antivir. Res. 1992, 19, 161–172. [Google Scholar] [CrossRef]
- Hu, Q.Y.; Fink, E.; Happer, M.; Elder, J.H. Identification of amino acid residues important for heparan sulfate proteoglycan interaction within variable region 3 of the feline immunodeficiency virus surface glycoprotein. J. Virol. 2011, 85, 7108–7117. [Google Scholar]
- Marradi, M.; di Gianvincenzo, P.; Enriquez-Navas, P.M.; Martinez-Avila, O.M.; Chiodo, F.; Yuste, E.; Angulo, J.; Penades, S. Gold nanoparticles coated with oligomannosides of HIV-1 glycoprotein gp120 mimic the carbohydrate epitope of antibody 2G12. J. Mol. Biol. 2011, 410, 798–810. [Google Scholar]
- Becer, C.R.; Gibson, M.I.; Geng, J.; Ilyas, R.; Wallis, R.; Mitchell, D.A.; Haddleton, D.M. High-affinity glycopolymer binding to human DC-SIGN and disruption of DC-SIGN interactions with HIV envelope glycoprotein. J. Am. Chem. Soc. 2010, 132, 15130–15132. [Google Scholar]
- Balzarini, J. Inhibition of HIV entry by carbohydrate-binding proteins. Antivir. Res. 2006, 71, 237–247. [Google Scholar]
- Van der Meer, F.J.; Schuurman, N.M.; Balzarini, J.; Egberink, H.F. Comparative evaluation of the activity of antivirals towards feline immunodeficiency virus in different cell culture systems. Antivir. Res. 2007, 76, 198–201. [Google Scholar]
- Toma, J.; Weinheimer, S.P.; Stawiski, E.; Whitcomb, J.M.; Lewis, S.T.; Petropoulos, C.J.; Huang, W. Loss of asparagine-linked glycosylation sites in variable region 5 of human immunodeficiency virus type 1 envelope is associated with resistance to CD4 antibody ibalizumab. J. Virol. 2011, 85, 3872–3880. [Google Scholar]
- Alexander, L.; Zhang, S.; McAuliffe, B.; Connors, D.; Zhou, N.; Wang, T.; Agler, M.; Kadow, J.; Lin, P.F. Inhibition of envelope-mediated CD4+-T-cell depletion by human immunodeficiency virus attachment inhibitors. Antimicrob. Agents Chemother. 2009, 53, 4726–4732. [Google Scholar]
- Willett, B.J.; McMonagle, E.L.; Bonci, F.; Pistello, M.; Hosie, M.J. Mapping the domains of CD134 as a functional receptor for feline immunodeficiency virus. J. Virol. 2006, 80, 7744–7747. [Google Scholar]
- Singh, I.P.; Chauthe, S.K. Small molecule HIV entry inhibitors: Part I. Chemokine receptor antagonists: 2004–2010. Expert Opin. Ther. Pat. 2011, 21, 227–269. [Google Scholar] [CrossRef]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar]
- Ayouba, A.; Cannou, C.; Nugeyre, M.T.; Barre-Sinoussi, F.; Menu, E. Distinct efficacy of HIV-1 entry inhibitors to prevent cell-to-cell transfer of R5 and X4 viruses across a human placental trophoblast barrier in a reconstitution model in vitro. Retrovirology 2008, 5. [Google Scholar] [CrossRef]
- Duda, D.G.; Kozin, S.V.; Kirkpatrick, N.D.; Xu, L.; Fukumura, D.; Jain, R.K. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: An emerging sensitizer for anticancer therapies? Clin. Cancer Res. 2011, 17, 2074–2080. [Google Scholar] [CrossRef]
- Pettersson, S.; Perez-Nueno, V.I.; Mena, M.P.; Clotet, B.; Este, J.A.; Borrell, J.I.; Teixido, J. Novel monocyclam derivatives as HIV entry inhibitors: Design, synthesis, anti-HIV evaluation, and their interaction with the CXCR4 co-receptor. ChemMedChem 2010, 5, 1272–1281. [Google Scholar] [CrossRef]
- Willett, B.J.; Picard, L.; Hosie, M.J.; Turner, J.D.; Adema, K.; Clapham, P.R. Shared usage of the chemokine receptor CXCR4 by the feline and human immunodeficiency viruses. J. Virol. 1997, 71, 6407–6415. [Google Scholar]
- Egberink, H.F.; de Clercq, E.; van Vliet, A.L.; Balzarini, J.; Bridger, G.J.; Henson, G.; Horzinek, M.C.; Schols, D. Bicyclams, selective antagonists of the human chemokine receptor CXCR4, potently inhibit feline immunodeficiency virus replication. J. Virol. 1999, 73, 6346–6352. [Google Scholar]
- Hartmann, E.; Stengel, S.; Klein, D.; Egberink, H.; Balzarini, J. Efficacy of the Chemokine Receptor Inhibitor 1,1’-Bis-1,4,8,11-Tetraazacyclotetradekan Against Feline Immunodeficiency Virus Infection. In Proceedings of the 6th International Feline Retrovirus Research Symposium, Amelia Island, FL, USA, 2–5 December 2002; p. 26.
- Ray, S.; Fatima, Z.; Saxena, A. Drugs for AIDS. Mini Rev. Med. Chem. 2010, 10, 147–161. [Google Scholar]
- Fletcher, C.V. Enfuvirtide, a new drug for HIV infection. Lancet 2003, 361, 1577–1578. [Google Scholar]
- Medinas, R.J.; Lambert, D.M.; Tompkins, W.A. C-terminal gp40 peptide analogs inhibit feline immunodeficiency virus: Cell fusion and virus spread. J. Virol. 2002, 76, 9079–9086. [Google Scholar]
- Mizukoshi, F.; Baba, K.; Goto, Y.; Setoguchi, A.; Fujino, Y.; Ohno, K.; Oishi, S.; Kodera, Y.; Fujii, N.; Tsujimoto, H. Antiviral activity of membrane fusion inhibitors that target gp40 of the feline immunodeficiency virus envelope protein. Vet. Microbiol. 2009, 136, 155–159. [Google Scholar]
- Oishi, S.; Kodera, Y.; Nishikawa, H.; Kamitani, H.; Watabe, T.; Ohno, H.; Tochikura, T.; Shimane, K.; Kodama, E.; Matsuoka, M.; et al. Design and synthesis of membrane fusion inhibitors against the feline immunodeficiency virus. Bioorg. Med. Chem. 2009, 17, 4916–4920. [Google Scholar]
- Giannecchini, S.; di Fenza, A.; D’Ursi, A.M.; Matteucci, D.; Rovero, P.; Bendinelli, M. Antiviral activity and conformational features of an octapeptide derived from the membrane-proximal ectodomain of the feline immunodeficiency virus transmembrane glycoprotein. J. Virol. 2003, 77, 3724–3733. [Google Scholar]
- Giannecchini, S.; Alcaro, M.C.; Isola, P.; Sichi, O.; Pistello, M.; Papini, A.M.; Rovero, P.; Bendinelli, M. Feline immunodeficiency virus plasma load reduction by a retroinverso octapeptide reproducing the Trp-rich motif of the transmembrane glycoprotein. Antivir Ther. 2005, 10, 671–680. [Google Scholar]
- Jiang, S.; Debnath, A.K. A salt bridge between an N-terminal coiled coil of gp41gp41 and an antiviral agent targeted to the gp41gp41 core is important for anti-HIV-1 activity. Biochem. Biophys. Res. Commun. 2000, 270, 153–157. [Google Scholar]
- North, T.W.; North, G.L.; Pedersen, N.C. Feline immunodeficiency virus, a model for reverse transcriptase-targeted chemotherapy for acquired immune deficiency syndrome. Antimicrob. Agents Chemother. 1989, 33, 915–919. [Google Scholar]
- Connell, S. Manufacturer addresses concerns about FIV vaccine. J. Am. Vet. Med. Assoc. 2003, 222, 149. [Google Scholar]
- Cronn, R.C.; Remington, K.M.; Preston, B.D.; North, T.W. Inhibition of reverse transcriptase from feline immunodeficiency virus by analogs of 2'-deoxyadenosine-5'-triphosphate. Biochem. Pharmacol. 1992, 44, 1375–1381. [Google Scholar]
- Egberink, H.; Borst, M.; Niphuis, H.; Balzarini, J.; Neu, H.; Schellekens, H.; de Clercq, E.; Horzinek, M.; Koolen, M. Suppression of feline immunodeficiency virus infection in vivo by 9-(2-phosphonomethoxyethyl)adenine. Proc. Natl. Acad. Sci. USA 1990, 87, 3087–3091. [Google Scholar]
- Hosie, M.J.; Addie, D.; Belak, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Lloret, A.; Lutz, H.; et al. Feline immunodeficiency. ABCD guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 575–584. [Google Scholar] [CrossRef]
- Hartmann, K.; Donath, A.; Beer, B.; Egberink, H.F.; Horzinek, M.C.; Lutz, H.; Hoffmann-Fezer, G.; Thum, I.; Thefeld, S. Use of two virustatica (AZT, PMEA) in the treatment of FIV and of FeLV seropositive cats with clinical symptoms. Vet. Immunol. Immunopathol. 1992, 35, 167–175. [Google Scholar]
- Philpott, M.S.; Ebner, J.P.; Hoover, E.A. Evaluation of 9-(2-phosphonylmethoxyethyl) adenine therapy for feline immunodeficiency virus using a quantitative polymerase chain reaction. Vet. Immunol. Immunopathol. 1992, 35, 155–166. [Google Scholar]
- Meers, J.; del Fierro, G.M.; Cope, R.B.; Park, H.S.; Greene, W.K.; Robinson, W.F. Feline immunodeficiency virus infection: Plasma, but not peripheral blood mononuclear cell virus titer is influenced by zidovudine and cyclosporine. Arch. Virol. 1993, 132, 67–81. [Google Scholar]
- Magnani, M.; Rossi, L.; Fraternale, A.; Silvotti, L.; Quintavalla, F.; Piedimonte, G.; Matteucci, D.; Baldinotti, F.; Bendinelli, M. Feline immunodeficiency virus infection of macrophages: In vitro and in vivo inhibition by dideoxycytidine-5'-triphosphate-loaded erythrocytes. AIDS Res. Hum. Retrovir. 1994, 10, 1179–1186. [Google Scholar] [CrossRef]
- Fogle, J.E.; Tompkins, W.A.; Campbell, B.; Sumner, D.; Tompkins, M.B. Fozivudine tidoxil as single-agent therapy decreases plasma and cell-associated viremia during acute feline immunodeficiency virus infection. J. Vet. Intern. Med. 2011, 25, 413–418. [Google Scholar]
- Uckun, F.M.; Chen, C.L.; Samuel, P.; Pendergrass, S.; Venkatachalam, T.K.; Waurzyniak, B.; Qazi, S. In vivo antiretroviral activity of stampidine in chronically feline immunodeficiency virus-infected cats. Antimicrob. Agents Chemother. 2003, 47, 1233–1240. [Google Scholar] [CrossRef]
- D’Cruz, O.J.; Waurzyniak, B.; Uckun, F.M. Antiretroviral spermicide WHI-07 prevents vaginal and rectal transmission of feline immunodeficiency virus in domestic cats. Antimicrob. Agents Chemother. 2004, 48, 1082–1088. [Google Scholar]
- Bisset, L.R.; Lutz, H.; Boni, J.; Hofmann-Lehmann, R.; Luthy, R.; Schupbach, J. Combined effect of zidovudine (ZDV), lamivudine (3TC) and abacavir (ABC) antiretroviral therapy in suppressing in vitro FIV replication. Antivir. Res. 2002, 53, 35–45. [Google Scholar]
- Arai, M.; Earl, D.D.; Yamamoto, J.K. Is AZT/3TC therapy effective against FIV infection or immunopathogenesis? Vet. Immunol. Immunopathol. 2002, 85, 189–204. [Google Scholar] [CrossRef]
- Auwerx, J.; Esnouf, R.; de Clercq, E.; Balzarini, J. Susceptibility of feline immunodeficiency virus/human immunodeficiency virus type 1 reverse transcriptase chimeras to non-nucleoside RT inhibitors. Mol. Pharmacol. 2004, 65, 244–251. [Google Scholar]
- Smyth, N.R.; McCracken, C.; Gaskell, R.M.; Cameron, J.M.; Coates, J.A.; Gaskell, C.J.; Hart, C.A.; Bennett, M. Susceptibility in cell culture of feline immunodeficiency virus to eighteen antiviral agents. J. Antimicrob. Chemother. 1994, 34, 589–594. [Google Scholar]
- Piller, S.C.; Caly, L.; Jans, D.A. Nuclear import of the pre-integration complex (PIC): The achilles heel of HIV? Curr. Drug Targets 2003, 4, 409–429. [Google Scholar] [CrossRef]
- Zhan, P.; Liu, X.; de Clercq, E. Blocking nuclear import of pre-integration complex: An emerging anti-HIV-1 drug discovery paradigm. Curr. Med. Chem. 2010, 17, 495–503. [Google Scholar]
- Hoelz, A.; Debler, E.W.; Blobel, G. The structure of the nuclear pore complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar]
- Gorlich, D.; Kutay, U. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 1999, 15, 607–660. [Google Scholar]
- Gorlich, D. Nuclear protein import. Curr. Opin. Cell Biol. 1997, 9, 412–419. [Google Scholar]
- Sherman, M.P.; Greene, W.C. Slipping through the door: HIV entry into the nucleus. Microbes Infect. 2002, 4, 67–73. [Google Scholar]
- Al-Abed, Y.; Dubrovsky, L.; Ruzsicska, B.; Seepersaud, M.; Bukrinsky, M. Inhibition of HIV-1 nuclear import via schiff base formation with arylene bis(methylketone) compounds. Bioorg. Med. Chem. Lett. 2002, 12, 3117–3119. [Google Scholar]
- Suzuki, T.; Yamamoto, N.; Nonaka, M.; Hashimoto, Y.; Matsuda, G.; Takeshima, S.N.; Matsuyama, M.; Igarashi, T.; Miura, T.; Tanaka, R.; et al. Inhibition of human immunodeficiency virus type 1 (HIV-1) nuclear import via vpr-importin alpha interactions as a novel HIV-1 therapy. Biochem. Biophys. Res. Commun. 2009, 380, 838–843. [Google Scholar]
- Zeinalipour-Loizidou, E.; Nicolaou, C.; Nicolaides, A.; Kostrikis, L.G. HIV-1 integrase: From biology to chemotherapeutics. Curr. HIV Res. 2007, 5, 365–388. [Google Scholar]
- Ciuffi, A.; Bushman, F.D. Retroviral DNA integration: HIV and the role of LEDGF/p75. Trends Genet. 2006, 22, 388–395. [Google Scholar]
- Du, L.; Zhao, Y.; Chen, J.; Yang, L.; Zheng, Y.; Tang, Y.; Shen, X.; Jiang, H. D77, one benzoic acid derivative, functions as a novel anti-HIV-1 inhibitor targeting the interaction between integrase and cellular LEDGF/p7. Biochem. Biophys. Res. Commun. 2008, 375, 139–144. [Google Scholar]
- Savarino, A.; Pistello, M.; D’Ostilio, D.; Zabogli, E.; Taglia, F.; Mancini, F.; Ferro, S.; Matteucci, D.; de Luca, L.; Barreca, M.L.; et al. Human immunodeficiency virus integrase inhibitors efficiently suppress feline immunodeficiency virus replication in vitro and provide a rationale to redesign antiretroviral treatment for feline AIDS. Retrovirology 2007, 4. [Google Scholar] [CrossRef]
- Matteucci, D.; Mazzetti, P.; Baldinotti, F.; Zaccaro, L.; Bendinelli, M. The feline lymphoid cell line MBM and its use for feline immunodeficiency virus isolation and quantitation. Vet. Immunol. Immunopathol. 1995, 46, 71–82. [Google Scholar]
- Zapp, M.L.; Young, D.W.; Kumar, A.; Singh, R.; Boykin, D.W.; Wilson, W.D.; Green, M.R. Modulation of the rev-RRE interaction by aromatic heterocyclic compounds. Bioorg. Med. Chem. 1997, 5, 1149–1155. [Google Scholar]
- Daelemans, D.; Afonina, E.; Nilsson, J.; Werner, G.; Kjems, J.; de Clercq, E.; Pavlakis, G.N.; Vandamme, A.M. A synthetic HIV-1 rev inhibitor interfering with the CRM1-mediated nuclear export. Proc. Natl. Acad. Sci. USA 2002, 99, 14440–14445. [Google Scholar]
- Kudo, N.; Wolff, B.; Sekimoto, T.; Schreiner, E.P.; Yoneda, Y.; Yanagida, M.; Horinouchi, S.; Yoshida, M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 1998, 242, 540–547. [Google Scholar]
- Tamura, S.; Shimizu, N.; Fujiwara, K.; Kaneko, M.; Kimura, T.; Murakami, N. Bioisostere of valtrate, anti-HIV principle by inhibition for nuclear export of rev. Bioorg. Med. Chem. Lett. 2010, 20, 2159–2162. [Google Scholar]
- Von der Helm, K. Retroviral proteases: Structure, function and inhibition from a non-anticipated viral enzyme to the target of a most promising HIV therapy. Biol. Chem. 1996, 377, 765–774. [Google Scholar]
- Anderson, J.; Schiffer, C.; Lee, S.K.; Swanstrom, R. Viral protease inhibitors. Handb. Exp. Pharmacol. 2009, 189, 85–110. [Google Scholar]
- Perryman, A.L.; Zhang, Q.; Soutter, H.H.; Rosenfeld, R.; McRee, D.E.; Olson, A.J.; Elder, J.E.; Stout, C.D. Fragment-based screen against HIV protease. Chem. Biol. Drug Des. 2010, 75, 257–268. [Google Scholar]
- Chang, M.W.; Giffin, M.J.; Muller, R.; Savage, J.; Lin, Y.C.; Hong, S.; Jin, W.; Whitby, L.R.; Elder, J.H.; Boger, D.L.; et al. Identification of broad-based HIV-1 protease inhibitors from combinatorial libraries. Biochem. J. 2010, 429, 527–532. [Google Scholar] [CrossRef]
- Elder, J.H.; Schnolzer, M.; Hasselkus-Light, C.S.; Henson, M.; Lerner, D.A.; Phillips, T.R.; Wagaman, P.C.; Kent, S.B. Identification of proteolytic processing sites within the gag and pol polyproteins of feline immunodeficiency virus. J. Virol. 1993, 67, 1869–1876. [Google Scholar]
- Lee, T.; Laco, G.S.; Torbett, B.E.; Fox, H.S.; Lerner, D.L.; Elder, J.H.; Wong, C.H. Analysis of the S3 and S3’ Subsite specificities of feline immunodeficiency virus (FIV) protease: Development of a broad-based protease inhibitor efficacious against FIV, SIV, and HIV in vitro and ex vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 939–944. [Google Scholar]
- Huitron-Resendiz, S.; de Rozieres, S.; Sanchez-Alavez, M.; Buhler, B.; Lin, Y.C.; Lerner, D.L.; Henriksen, N.W.; Burudi, M.; Fox, H.S.; Torbett, B.E.; et al. Resolution and prevention of feline immunodeficiency virus-induced neurological deficits by treatment with the protease inhibitor TL-3. J. Virol. 2004, 78, 4525–4532. [Google Scholar]
- Norelli, S.; El Daker, S.; D’Ostilio, D.; Mele, F.; Mancini, F.; Taglia, F.; Ruggieri, A.; Ciccozzi, M.; Cauda, R.; Ciervo, A.; et al. Response of feline immunodeficiency virus (FIV) to tipranavir may provide new clues for development of broad-based inhibitors of retroviral proteases acting on drug-resistant HIV-1. Curr. HIV Res. 2008, 6, 306–317. [Google Scholar]
- Neira, J.L. The capsid protein of human immunodeficiency virus: Designing inhibitors of capsid assembly. FEBS J. 2009, 276, 6110–6117. [Google Scholar]
- Prevelige, P.E., Jr. New approaches for antiviral targeting of HIV assembly. J. Mol. Biol. 2011, 410, 634–640. [Google Scholar]
- Dietrich, I.; Macintyre, A.; McMonagle, E.; Price, A.J.; James, L.C.; McEwan, W.A.; Hosie, M.J.; Willett, B.J. Potent lentiviral restriction by a synthetic feline TRIM5 cyclophilin a fusion. J. Virol. 2010, 84, 8980–8985. [Google Scholar]
- Dietrich, I.; McEwan, W.A.; Hosie, M.J.; Willett, B.J. Restriction of the felid lentiviruses by a synthetic feline TRIM5-CypA fusion. Vet. Immunol. Immunopathol. 2011, 143, 235–242. [Google Scholar]
- Towers, G.J. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology 2007, 4. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mohammadi, H.; Bienzle, D. Pharmacological Inhibition of Feline Immunodeficiency Virus (FIV). Viruses 2012, 4, 708-724. https://doi.org/10.3390/v4050708
Mohammadi H, Bienzle D. Pharmacological Inhibition of Feline Immunodeficiency Virus (FIV). Viruses. 2012; 4(5):708-724. https://doi.org/10.3390/v4050708
Chicago/Turabian StyleMohammadi, Hakimeh, and Dorothee Bienzle. 2012. "Pharmacological Inhibition of Feline Immunodeficiency Virus (FIV)" Viruses 4, no. 5: 708-724. https://doi.org/10.3390/v4050708
APA StyleMohammadi, H., & Bienzle, D. (2012). Pharmacological Inhibition of Feline Immunodeficiency Virus (FIV). Viruses, 4(5), 708-724. https://doi.org/10.3390/v4050708