Prevention of Cellular Suicide by Cytomegaloviruses



{kind=link}
{kind=link}
Abstract
:1. Introduction
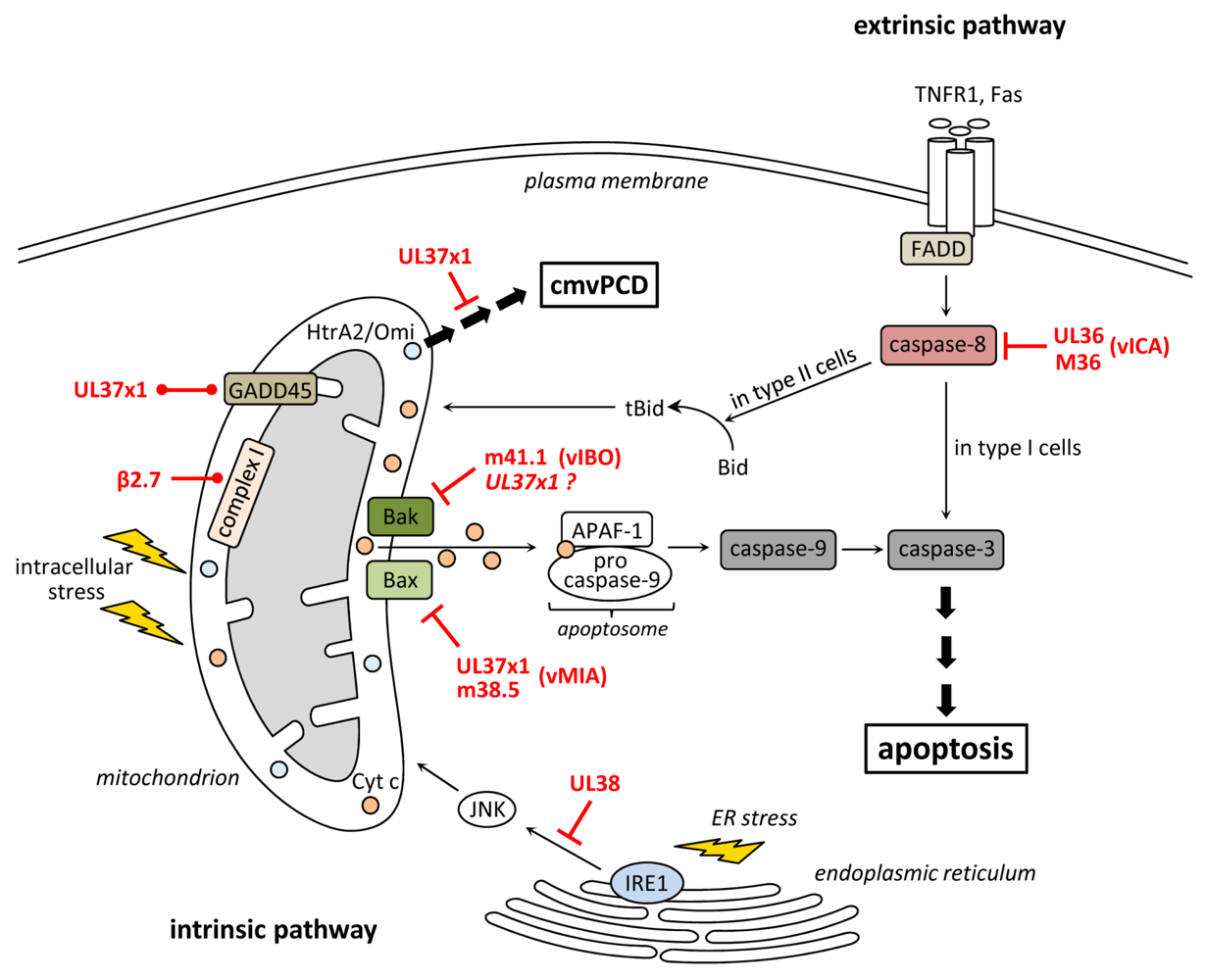
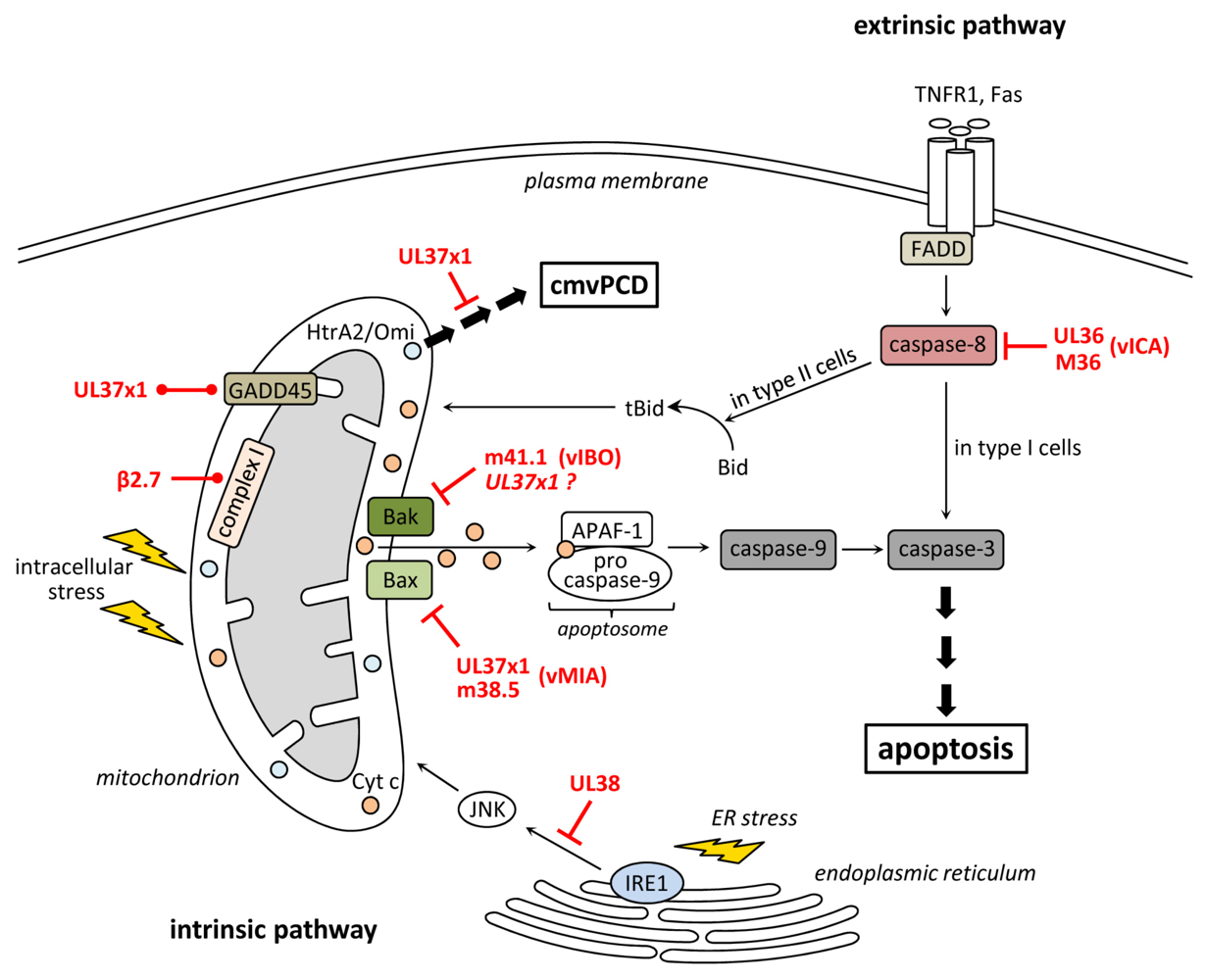
2. Inhibition of Apoptosis at the Level of Mitochondria
3. Inhibition of Death Receptor-Induced Apoptosis
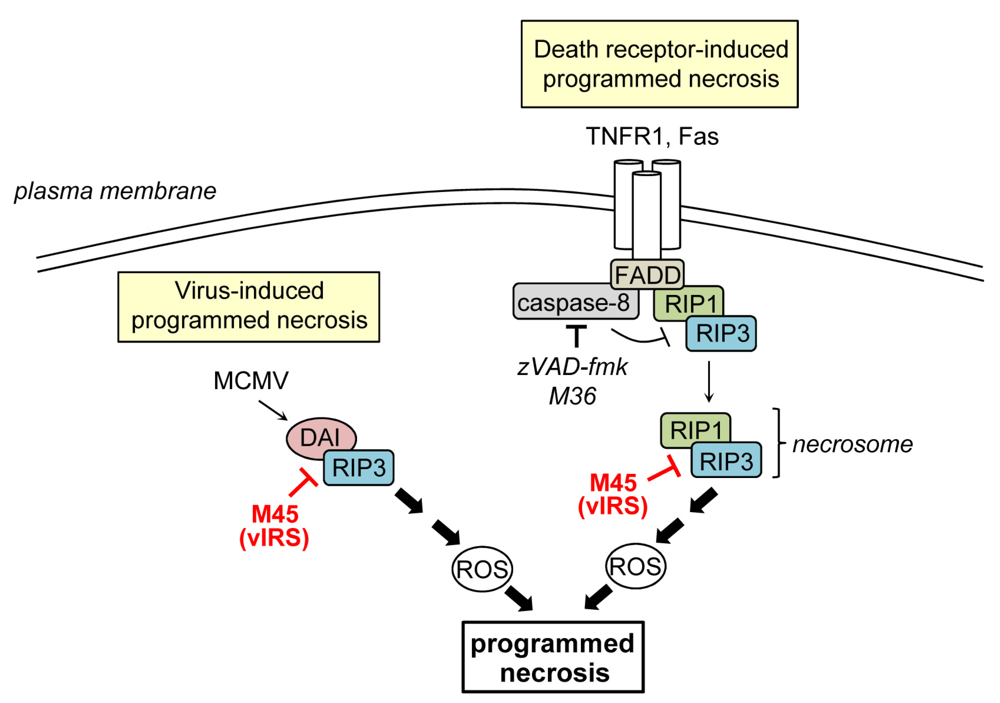
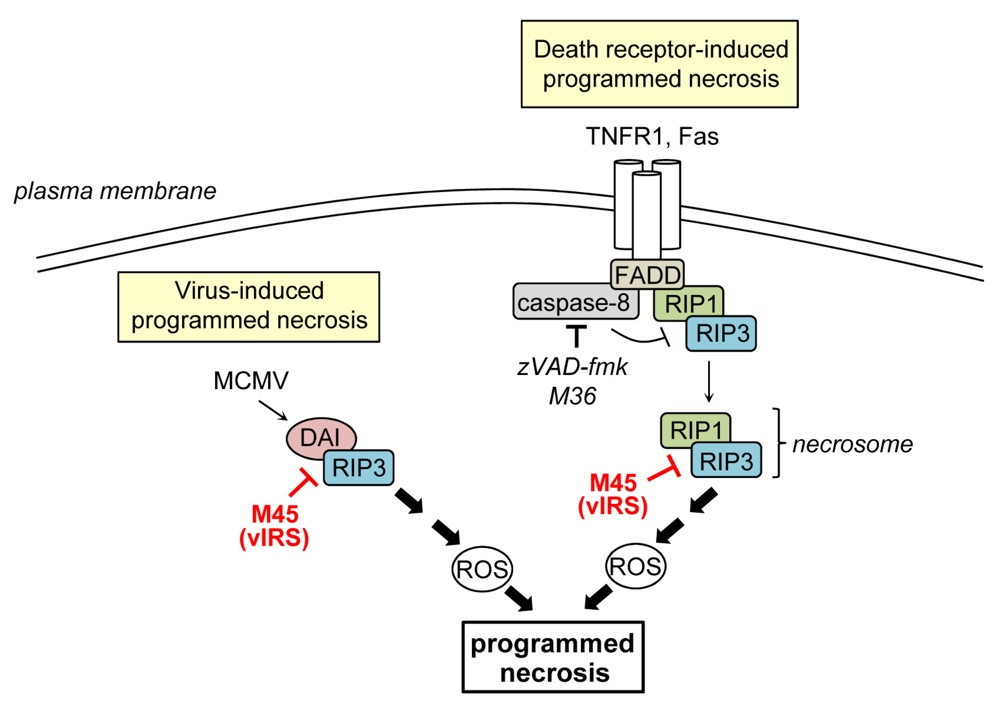
4. Inhibition of Programmed Necrosis
5. Inhibition of Stress-Induced Cell Death
6. Summary and Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Hacker, G. The morphology of apoptosis. Cell Tissue Res. 2000, 301, 5–17. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Kumar, S. Caspase function in programmed cell death. Cell Death Differ. 2007, 14, 32–43. [Google Scholar] [CrossRef]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 2006, 1757, 1371–1387. [Google Scholar]
- Zong, W.X.; Thompson, C.B. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Chan, F.K.; Shisler, J.; Bixby, J.G.; Felices, M.; Zheng, L.; Appel, M.; Orenstein, J.; Moss, B.; Lenardo, M.J. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 2003, 278, 51613–51621. [Google Scholar]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef]
- Han, J.; Zhong, C.Q.; Zhang, D.W. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat. Immunol. 2011, 12, 1143–1149. [Google Scholar] [CrossRef]
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634. [Google Scholar]
- Vanlangenakker, N.; Vanden Berghe, T.; Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012, 19, 75–86. [Google Scholar] [CrossRef]
- Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef]
- Steininger, C. Clinical relevance of cytomegalovirus infection in patients with disorders of the immune system. Clin. Microbiol. Infect. 2007, 13, 953–963. [Google Scholar] [CrossRef]
- Hudson, J.B. The murine cytomegalovirus as a model for the study of viral pathogenesis and persistent infections. Arch. Virol. 1979, 62, 1–29. [Google Scholar] [CrossRef]
- Krmpotic, A.; Bubic, I.; Polic, B.; Lucin, P.; Jonjic, S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 2003, 5, 1263–1277. [Google Scholar] [CrossRef]
- Voigt, S.; Ettinger, J.; Streblow, D.N. The rat model of cytomegalovirus infection and vascular disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intevention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013. [Google Scholar]
- Brune, W. Inhibition of programmed cell death by cytomegaloviruses. Virus Res. 2011, 157, 144–150. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar]
- Ow, Y.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Bouillet, P.; Strasser, A. BH3-only proteins—Evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J. Cell Sci. 2002, 115, 1567–1574. [Google Scholar]
- Uren, R.T.; Dewson, G.; Chen, L.; Coyne, S.C.; Huang, D.C.; Adams, J.M.; Kluck, R.M. Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J. Cell Biol. 2007, 177, 277–287. [Google Scholar] [CrossRef]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef]
- Gavathiotis, E.; Suzuki, M.; Davis, M.L.; Pitter, K.; Bird, G.H.; Katz, S.G.; Tu, H.C.; Kim, H.; Cheng, E.H.; Tjandra, N.; et al. BAX activation is initiated at a novel interaction site. Nature 2008, 455, 1076–1081. [Google Scholar] [CrossRef]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Nechushtan, A.; Smith, C.L.; Hsu, Y.T.; Youle, R.J. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 1999, 18, 2330–2341. [Google Scholar] [CrossRef]
- Antonsson, B.; Montessuit, S.; Sanchez, B.; Martinou, J.C. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 2001, 276, 11615–11623. [Google Scholar]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar]
- Dewson, G.; Kratina, T.; Sim, H.W.; Puthalakath, H.; Adams, J.M.; Colman, P.M.; Kluck, R.M. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell 2008, 30, 369–380. [Google Scholar] [CrossRef]
- Dewson, G.; Kratina, T.; Czabotar, P.; Day, C.L.; Adams, J.M.; Kluck, R.M. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol. Cell 2009, 36, 696–703. [Google Scholar] [CrossRef]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 2011, 1813, 521–531. [Google Scholar] [CrossRef]
- Belzacq, A.S.; El Hamel, C.; Vieira, H.L.; Cohen, I.; Haouzi, D.; Metivier, D.; Marchetti, P.; Brenner, C.; Kroemer, G. Adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene 2001, 20, 7579–7587. [Google Scholar] [CrossRef]
- Jan, G.; Belzacq, A.S.; Haouzi, D.; Rouault, A.; Metivier, D.; Kroemer, G.; Brenner, C. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ. 2002, 9, 179–188. [Google Scholar] [CrossRef]
- Arnoult, D.; Bartle, L.M.; Skaletskaya, A.; Poncet, D.; Zamzami, N.; Park, P.U.; Sharpe, J.; Youle, R.J.; Goldmacher, V.S. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7988–7993. [Google Scholar]
- Goldmacher, V.S.; Bartle, L.M.; Skaletskaya, A.; Dionne, C.A.; Kedersha, N.L.; Vater, C.A.; Han, J.W.; Lutz, R.J.; Watanabe, S.; Cahir McFarland, E.D.; et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 12536–12541. [Google Scholar]
- Boya, P.; Cohen, I.; Zamzami, N.; Vieira, H.L.; Kroemer, G. Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 2002, 9, 465–467. [Google Scholar] [CrossRef]
- McCormick, A.L.; Meiering, C.D.; Smith, G.B.; Mocarski, E.S. Mitochondrial cell death suppressors carried by human and murine cytomegalovirus confer resistance to proteasome inhibitor-induced apoptosis. J. Virol. 2005, 79, 12205–12217. [Google Scholar] [CrossRef]
- Pauleau, A.L.; Larochette, N.; Giordanetto, F.; Scholz, S.R.; Poncet, D.; Zamzami, N.; Goldmacher, V.S.; Kroemer, G. Structure-function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene 2007, 26, 7067–7080. [Google Scholar] [CrossRef]
- Poncet, D.; Larochette, N.; Pauleau, A.L.; Boya, P.; Jalil, A.A.; Cartron, P.F.; Vallette, F.; Schnebelen, C.; Bartle, L.M.; Skaletskaya, A.; et al. An anti-apoptotic viral protein that recruits Bax to mitochondria. J. Biol. Chem. 2004, 279, 22605–22614. [Google Scholar]
- Hayajneh, W.A.; Colberg-Poley, A.M.; Skaletskaya, A.; Bartle, L.M.; Lesperance, M.M.; Contopoulos-Ioannidis, D.G.; Kedersha, N.L.; Goldmacher, V.S. The sequence and antiapoptotic functional domains of the human cytomegalovirus UL37 exon 1 immediate early protein are conserved in multiple primary strains. Virology 2001, 279, 233–240. [Google Scholar] [CrossRef]
- Smith, G.B.; Mocarski, E.S. Contribution of GADD45 family members to cell death suppression by cellular Bcl-xL and cytomegalovirus vMIA. J. Virol. 2005, 79, 14923–14932. [Google Scholar] [CrossRef]
- McCormick, A.L.; Roback, L.; Mocarski, E.S. HtrA2/Omi terminates cytomegalovirus infection and is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA). PLoS Pathog. 2008, 4, e1000063. [Google Scholar] [CrossRef]
- McCormick, A.L.; Skaletskaya, A.; Barry, P.A.; Mocarski, E.S.; Goldmacher, V.S. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 2003, 316, 221–233. [Google Scholar] [CrossRef]
- Norris, K.L.; Youle, R.J. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J. Virol. 2008, 82, 6232–6243. [Google Scholar] [CrossRef]
- Andoniou, C.E.; Andrews, D.M.; Manzur, M.; Ricciardi-Castagnoli, P.; Degli-Esposti, M.A. A novel checkpoint in the Bcl-2-regulated apoptotic pathway revealed by murine cytomegalovirus infection of dendritic cells. J. Cell Biol. 2004, 166, 827–837. [Google Scholar] [CrossRef]
- Jurak, I.; Schumacher, U.; Simic, H.; Voigt, S.; Brune, W. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J. Virol. 2008, 82, 4812–4822. [Google Scholar] [CrossRef]
- Degenhardt, K.; Sundararajan, R.; Lindsten, T.; Thompson, C.; White, E. Bax and Bak independently promote cytochrome C release from mitochondria. J. Biol. Chem. 2002, 277, 14127–14134. [Google Scholar]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Manzur, M.; Fleming, P.; Huang, D.C.; Degli-Esposti, M.A.; Andoniou, C.E. Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ. 2009, 16, 312–320. [Google Scholar] [CrossRef]
- Cam, M.; Handke, W.; Picard-Maureau, M.; Brune, W. Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ. 2010, 17, 655–665. [Google Scholar] [CrossRef]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.Y.; Youle, R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006, 443, 658–662. [Google Scholar] [CrossRef]
- Hanson, L.K.; Slater, J.S.; Karabekian, Z.; Virgin, H.W.t.; Biron, C.A.; Ruzek, M.C.; van Rooijen, N.; Ciavarra, R.P.; Stenberg, R.M.; Campbell, A.E. Replication of murine cytomegalovirus in differentiated macrophages as a determinant of viral pathogenesis. J. Virol. 1999, 73, 5970–5980. [Google Scholar]
- Reboredo, M.; Greaves, R.F.; Hahn, G. Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. J. Gen. Virol. 2004, 85, 3555–3567. [Google Scholar] [CrossRef]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12396–12401. [Google Scholar]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef]
- McDonough, S.H.; Staprans, S.I.; Spector, D.H. Analysis of the major transcripts encoded by the long repeat of human cytomegalovirus strain AD169. J. Virol. 1985, 53, 711–718. [Google Scholar]
- Greenaway, P.J.; Wilkinson, G.W. Nucleotide sequence of the most abundantly transcribed early gene of human cytomegalovirus strain AD169. Virus Res. 1987, 7, 17–31. [Google Scholar] [CrossRef]
- McSharry, B.P.; Tomasec, P.; Neale, M.L.; Wilkinson, G.W. The most abundantly transcribed human cytomegalovirus gene (beta 2.7) is non-essential for growth in vitro. J. Gen. Virol. 2003, 84, 2511–2516. [Google Scholar] [CrossRef]
- Zhao, J.; Sinclair, J.; Houghton, J.; Bolton, E.; Bradley, A.; Lever, A. Cytomegalovirus beta2.7 RNA transcript protects endothelial cells against apoptosis during ischemia/reperfusion injury. J. Heart Lung Transplant. 2010, 29, 342–345. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar]
- Hoglinger, G.U.; Carrard, G.; Michel, P.P.; Medja, F.; Lombes, A.; Ruberg, M.; Friguet, B.; Hirsch, E.C. Dysfunction of mitochondrial complex I and the proteasome: Interactions between two biochemical deficits in a cellular model of Parkinson's disease. J. Neurochem. 2003, 86, 1297–1307. [Google Scholar] [CrossRef]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000, 12, 599–609. [Google Scholar] [CrossRef]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef]
- Schutze, S.; Tchikov, V.; Schneider-Brachert, W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 655–662. [Google Scholar]
- Morgan, M.J.; Kim, Y.S.; Liu, Z.G. Membrane-bound Fas ligand requires RIP1 for efficient activation of caspase-8 within the death-inducing signaling complex. J. Immunol. 2009, 183, 3278–3284. [Google Scholar] [CrossRef]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O'Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death—Inducing signaling complex. Cell 1996, 85, 817–827. [Google Scholar] [CrossRef]
- Medema, J.P.; Scaffidi, C.; Kischkel, F.C.; Shevchenko, A.; Mann, M.; Krammer, P.H.; Peter, M.E. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 1997, 16, 2794–2804. [Google Scholar] [CrossRef]
- Yin, X.M.; Wang, K.; Gross, A.; Zhao, Y.; Zinkel, S.; Klocke, B.; Roth, K.A.; Korsmeyer, S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999, 400, 886–891. [Google Scholar] [CrossRef]
- Barnhart, B.C.; Alappat, E.C.; Peter, M.E. The CD95 type I/type II model. Semin. Immunol. 2003, 15, 185–193. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptor. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 7829–7834. [Google Scholar]
- McCormick, A.L.; Roback, L.; Livingston-Rosanoff, D.; St Clair, C. The human cytomegalovirus UL36 gene controls caspase-dependent and -independent cell death programs activated by infection of monocytes differentiating to macrophages. J. Virol. 2010, 84, 5108–5123. [Google Scholar] [CrossRef]
- Carrington, P.E.; Sandu, C.; Wei, Y.; Hill, J.M.; Morisawa, G.; Huang, T.; Gavathiotis, E.; Werner, M.H. The structure of FADD and its mode of interaction with procaspase-8. Mol. Cell 2006, 22, 599–610. [Google Scholar] [CrossRef]
- McCormick, A.L.; Skaletskaya, A.; Barry, P.A.; Mocarski, E.S.; Goldmacher, V.S. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 2003, 316, 221–233. [Google Scholar] [CrossRef]
- Menard, C.; Wagner, M.; Ruzsics, Z.; Holak, K.; Brune, W.; Campbell, A.E.; Koszinowski, U.H. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 2003, 77, 5557–5570. [Google Scholar]
- Patterson, C.E.; Shenk, T. Human cytomegalovirus UL36 protein is dispensable for viral replication in cultured cells. J. Virol. 1999, 73, 7126–7131. [Google Scholar]
- Chang, W.L.; Tarantal, A.F.; Zhou, S.S.; Borowsky, A.D.; Barry, P.A. A recombinant rhesus cytomegalovirus expressing enhanced green fluorescent protein retains the wild-type phenotype and pathogenicity in fetal macaques. J. Virol. 2002, 76, 9493–9504. [Google Scholar] [CrossRef]
- Cicin-Sain, L.; Ruzsics, Z.; Podlech, J.; Bubic, I.; Menard, C.; Jonjic, S.; Reddehase, M.J.; Koszinowski, U.H. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J. Virol. 2008, 82, 2056–2064. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Kalai, M.; Van Loo, G.; Vanden Berghe, T.; Meeus, A.; Burm, W.; Saelens, X.; Vandenabeele, P. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ. 2002, 9, 981–994. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, V.; Liu, H.; Pope, R.M. NF-kappaB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J. Biol. Chem. 2005, 280, 41827–41834. [Google Scholar] [CrossRef]
- Lalanne, A.I.; Moraga, I.; Hao, Y.; Pereira, J.P.; Alves, N.L.; Huntington, N.D.; Freitas, A.A.; Cumano, A.; Vieira, P. CpG inhibits pro-B cell expansion through a cathepsin B-dependent mechanism. J. Immunol. 2010, 184, 5678–5685. [Google Scholar] [CrossRef]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 20054–20059. [Google Scholar]
- Lenardo, M.J.; Angleman, S.B.; Bounkeua, V.; Dimas, J.; Duvall, M.G.; Graubard, M.B.; Hornung, F.; Selkirk, M.C.; Speirs, C.K.; Trageser, C.; et al. Cytopathic killing of peripheral blood CD4(+) T lymphocytes by human immunodeficiency virus type 1 appears necrotic rather than apoptotic and does not require env. J. Virol. 2002, 76, 5082–5093. [Google Scholar]
- Petit, F.; Arnoult, D.; Lelievre, J.D.; Moutouh-de Parseval, L.; Hance, A.J.; Schneider, P.; Corbeil, J.; Ameisen, J.C.; Estaquier, J. Productive HIV-1 infection of primary CD4+ T cells induces mitochondrial membrane permeabilization leading to a caspase-independent cell death. J. Biol. Chem. 2002, 277, 1477–1487. [Google Scholar]
- Peri, P.; Nuutila, K.; Vuorinen, T.; Saukko, P.; Hukkanen, V. Cathepsins are involved in virus-induced cell death in ICP4 and Us3 deletion mutant herpes simplex virus type 1-infected monocytic cells. J. Gen. Virol. 2011, 92, 173–180. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Vercammen, D.; Brouckaert, G.; Denecker, G.; Van de Craen, M.; Declercq, W.; Fiers, W.; Vandenabeele, P. Dual signaling of the Fas receptor: Initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 1998, 188, 919–930. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar]
- Vandenabeele, P.; Declercq, W.; Van Herreweghe, F.; Vanden Berghe, T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 2010, 3, re4. [Google Scholar] [CrossRef]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 2007, 19, 2056–2067. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef]
- Gunther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011, 477, 335–339. [Google Scholar] [CrossRef]
- Sun, X.; Yin, J.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 2002, 277, 9505–9511. [Google Scholar]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Vanden Berghe, T.; Krysko, D.V.; Festjens, N.; Vandenabeele, P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 2008, 8, 207–220. [Google Scholar] [CrossRef]
- Brune, W.; Menard, C.; Heesemann, J.; Koszinowski, U.H. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 2001, 291, 303–305. [Google Scholar]
- Mack, C.; Sickmann, A.; Lembo, D.; Brune, W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3094–3099. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 2008, 283, 16966–16970. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Rebsamen, M.; Heinz, L.X.; Meylan, E.; Michallet, M.C.; Schroder, K.; Hofmann, K.; Vazquez, J.; Benedict, C.A.; Tschopp, J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009, 10, 916–922. [Google Scholar] [CrossRef]
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral mediated redirection of NEMO/IKKgamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef]
- Irrinki, K.M.; Mallilankaraman, K.; Thapa, R.J.; Chandramoorthy, H.C.; Smith, F.J.; Jog, N.R.; Gandhirajan, R.K.; Kelsen, S.G.; Houser, S.R.; May, M.J.; et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol. Cell Biol. 2011, 31, 3745–3758. [Google Scholar] [CrossRef]
- Lembo, D.; Donalisio, M.; Hofer, A.; Cornaglia, M.; Brune, W.; Koszinowski, U.; Thelander, L.; Landolfo, S. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J. Virol. 2004, 78, 4278–4288. [Google Scholar] [CrossRef]
- Rawlinson, W.D.; Farrell, H.E.; Barrell, B.G. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar]
- Lembo, D.; Brune, W. Tinkering with a viral ribonucleotide reductase. Trends Biochem. Sci. 2009, 34, 25–32. [Google Scholar] [CrossRef]
- Hahn, G.; Khan, H.; Baldanti, F.; Koszinowski, U.H.; Revello, M.G.; Gerna, G. The human cytomegalovirus ribonucleotide reductase homolog UL45 is dispensable for growth in endothelial cells, as determined by a BAC-cloned clinical isolate of human cytomegalovirus with preserved wild-type characteristics. J. Virol. 2002, 76, 9551–9555. [Google Scholar] [CrossRef]
- Patrone, M.; Percivalle, E.; Secchi, M.; Fiorina, L.; Pedrali-Noy, G.; Zoppe, M.; Baldanti, F.; Hahn, G.; Koszinowski, U.H.; Milanesi, G.; et al. The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection. J. Gen. Virol. 2003, 84, 3359–3370. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 2432–2437. [Google Scholar] [CrossRef]
- Putcha, G.V.; Le, S.; Frank, S.; Besirli, C.G.; Clark, K.; Chu, B.; Alix, S.; Youle, R.J.; LaMarche, A.; Maroney, A.C.; et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 2003, 38, 899–914. [Google Scholar] [CrossRef]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef]
- Terhune, S.; Torigoi, E.; Moorman, N.; Silva, M.; Qian, Z.; Shenk, T.; Yu, D. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 2007, 81, 3109–3123. [Google Scholar]
- Xuan, B.; Qian, Z.; Torigoi, E.; Yu, D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 2009, 83, 3463–3474. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Gualberto, N.; Yu, D. The human cytomegalovirus protein pUL38 suppresses endoplasmic reticulum stress-mediated cell death independently of its ability to induce mTORC1 activation. J. Virol. 2011, 85, 9103–9113. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Chapa, T.J.; Gualberto, N.; Yu, D. Murine cytomegalovirus targets transcription factor ATF4 to exploit the unfolded-protein response. J. Virol. 2012, 86, 6712–6723. [Google Scholar] [CrossRef]
- Ruzek, M.C.; Miller, A.H.; Opal, S.M.; Pearce, B.D.; Biron, C.A. Characterization of early cytokine responses and an interleukin (IL)-6-dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J. Exp. Med. 1997, 185, 1185–1192. [Google Scholar] [CrossRef]
- Orange, J.S.; Biron, C.A. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J. Immunol. 1996, 156, 4746–4756. [Google Scholar]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar]
- Kijpittayarit, S.; Eid, A.J.; Brown, R.A.; Paya, C.V.; Razonable, R.R. Relationship between Toll-like receptor 2 polymorphism and cytomegalovirus disease after liver transplantation. Clin. Infect. Dis. 2007, 44, 1315–1320. [Google Scholar] [CrossRef]
- Tabeta, K.; Georgel, P.; Janssen, E.; Du, X.; Hoebe, K.; Crozat, K.; Mudd, S.; Shamel, L.; Sovath, S.; Goode, J.; et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 3516–3521. [Google Scholar]
- Delale, T.; Paquin, A.; Asselin-Paturel, C.; Dalod, M.; Brizard, G.; Bates, E.E.; Kastner, P.; Chan, S.; Akira, S.; Vicari, A.; et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J. Immunol. 2005, 175, 6723–6732. [Google Scholar]
- Gaspar, M.; Shenk, T. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2821–2826. [Google Scholar] [CrossRef]
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008, 4, e1000018. [Google Scholar] [CrossRef]
- Brune, W.; Nevels, M.; Shenk, T. Murine cytomegalovirus m41 open reading frame encodes a Golgi-localized antiapoptotic protein. J. Virol. 2003, 77, 11633–11643. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fliss, P.M.; Brune, W. Prevention of Cellular Suicide by Cytomegaloviruses. Viruses 2012, 4, 1928-1949. https://doi.org/10.3390/v4101928
Fliss PM, Brune W. Prevention of Cellular Suicide by Cytomegaloviruses. Viruses. 2012; 4(10):1928-1949. https://doi.org/10.3390/v4101928
Chicago/Turabian StyleFliss, Patricia M., and Wolfram Brune. 2012. "Prevention of Cellular Suicide by Cytomegaloviruses" Viruses 4, no. 10: 1928-1949. https://doi.org/10.3390/v4101928
APA StyleFliss, P. M., & Brune, W. (2012). Prevention of Cellular Suicide by Cytomegaloviruses. Viruses, 4(10), 1928-1949. https://doi.org/10.3390/v4101928