Contributions of Epstein–Barr Nuclear Antigen 1 (EBNA1) to Cell Immortalization and Survival

{kind=link}
Abstract
:1. Introduction
2. Evidence for Cellular Effects of EBNA1
3. Molecular Mechanisms of EBNA1 Cellular Effects
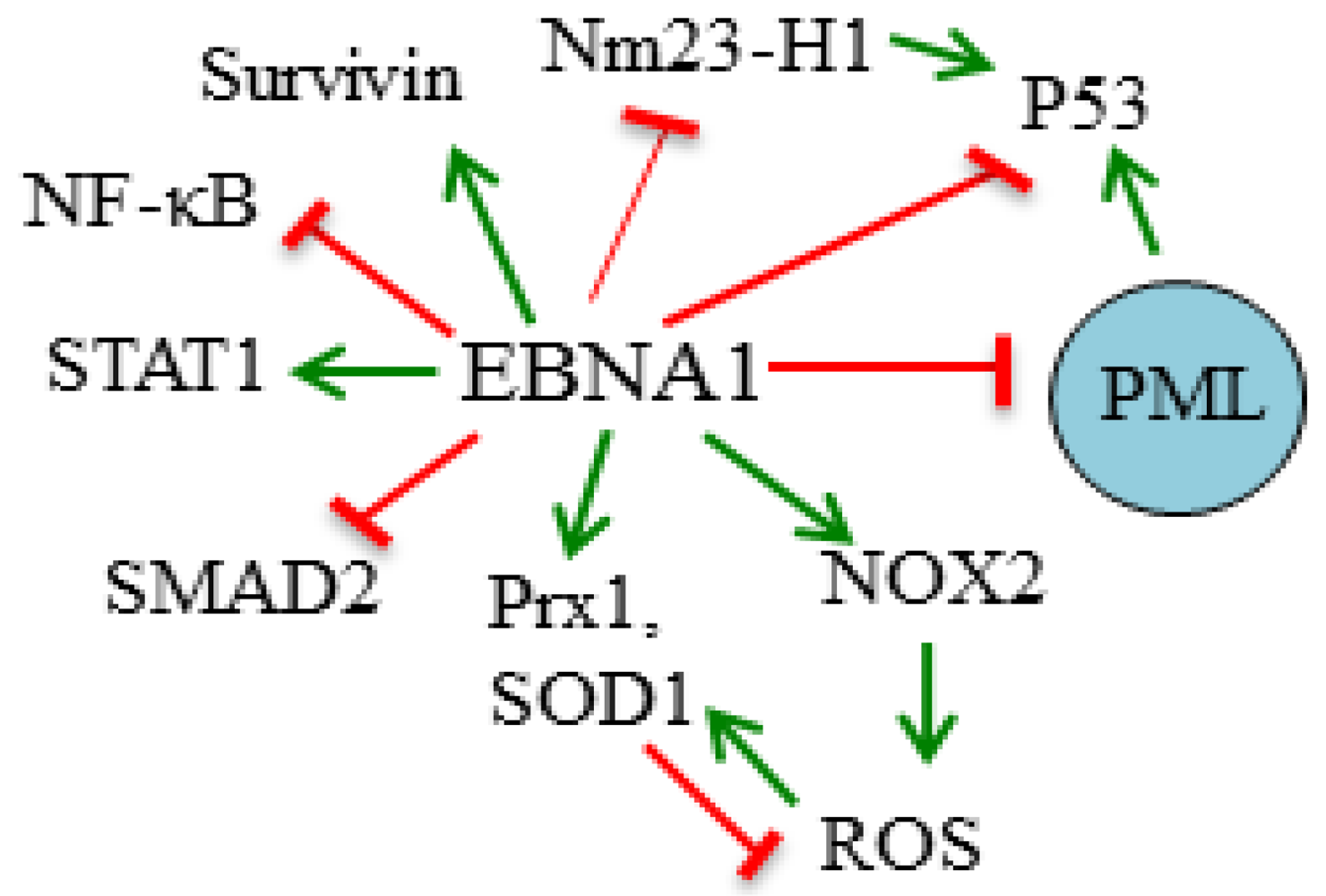
3.1. Destabilization of p53
3.2. Disruption of PML Nuclear Bodies
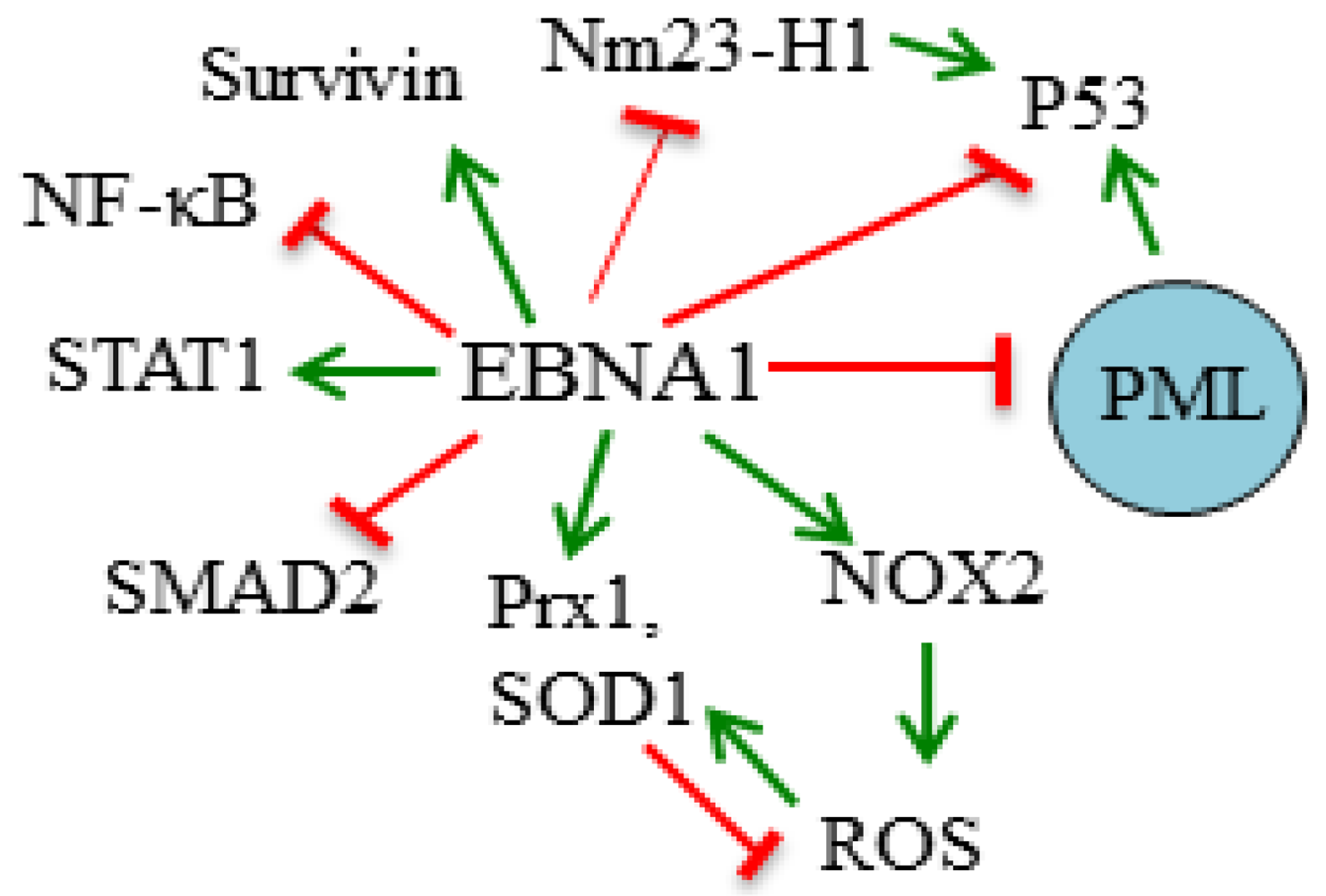
3.3. Modulation of Signaling Pathways
3.4. Induction of Oxidative Stress
3.5. Inhibition of Nm23-H1
3.6. Induction of Survivin
4. Summary
Acknowledgements
Conflict of Interest
References and Notes
- Klein, G. Viral latency and transformation: The strategy of Epstein-Barr virus. Cell 1989, 58, 5–8. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 1985, 313, 812–815. [Google Scholar] [CrossRef]
- Lupton, S.; Levine, A.J. Mapping of genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells. Mol. Cell. Biol. 1985, 5, 2533–2542. [Google Scholar]
- Reisman, D.; Yates, J.; Sugden, B. A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol. Cell. Biol. 1985, 5, 1822–1832. [Google Scholar]
- Rawlins, D.R.; Milman, G.; Hayward, S.D.; Hayward, G.S. Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA1) to clustered sites in the plasmid maintenance region. Cell 1985, 42, 859–868. [Google Scholar] [CrossRef]
- Frappier, L. EBNA1 in viral DNA replication and persistence. In Epstein-Barr Virus: Latency and Transformation; Robertson, E.S., Ed.; Caister Academic Press: Norwich, UK, 2010; pp. 37–59. [Google Scholar]
- Gahn, T.; Sugden, B. An EBNA1 dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein-Barr virus lmp gene. J. Virol. 1995, 69, 2633–2636. [Google Scholar]
- Reisman, D.; Sugden, B. Trans activation of an Epstein-Barr viral transcripitonal enhancer by the Epstein-Barr viral nuclear antigen 1. Mol. Cell. Biol. 1986, 6, 3838–3846. [Google Scholar]
- Klein, G. Viral latency and transformation: The strategy of Epstein-Barr virus. Cell 1989, 58, 5–8. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Hume, S.; Reisbach, G.; Feederle, R.; Delecluse, H.-J.; Bousset, K.; Hammerschmidt, W.; Schepers, A. The EBV nuclear antigen 1 (EBNA 1) enhances B cell immortalization several thousand fold. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 10989–10994. [Google Scholar]
- Altmann, M.; Pich, D.; Ruiss, R.; Wang, J.; Sugden, B.; Hammerschmidt, W. Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 14188–14193. [Google Scholar]
- Kennedy, G.; Komano, J.; Sugden, B. Epstein-Barr virus provide a survival factor to Burkitt’s lymphomas. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 14269–14274. [Google Scholar]
- Hong, M.; Murai, Y.; Kutsuna, T.; Takahashi, H.; Nomoto, K.; Cheng, C.M.; Ishizawa, S.; Zhao, Q.L.; Ogawa, R.; Harmon, B.V.; et al. Suppression of Epstein-Barr nuclear antigen 1 (EBNA 1) by rna interference inhibits proliferation of EBV-positive Burkitt’s lymphoma cells. J. Cancer Res. Clin. Oncol. 2006, 132, 1–8. [Google Scholar]
- Yin, Q.; Flemington, E.K. Sirnas against the Epstein-Barr virus latency replication factor, EBNA 1, inhibit its function and growth of EBV-dependent tumor cells. Virology 2006, 346, 385–393. [Google Scholar] [CrossRef]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBO J. 1996, 15, 3117–3126. [Google Scholar]
- Tsimbouri, P.; Drotar, M.E.; Coy, J.L.; Wilson, J.B. Bcl-xL and rag genes are induced and the response to IL-2 enhanced in EmuEBNA-1 transgenic mouse lymphocytes. Oncogene 2002, 21, 5182–5187. [Google Scholar] [CrossRef]
- Kang, M.S.; Lu, H.; Yasui, T.; Sharpe, A.; Warren, H.; Cahir-McFarland, E.; Bronson, R.; Hung, S.C.; Kieff, E. Epstein-Barr virus nuclear antigen 1 does not induce lymphoma in transgenic fvb mice. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 820–825. [Google Scholar]
- Kang, M.S.; Soni, V.; Bronson, R.; Kieff, E. Epstein-Barr virus nuclear antigen 1 does not cause lymphoma in c57bl/6j mice. J. Virol. 2008, 82, 4180–4183. [Google Scholar] [CrossRef]
- Kang, M.S.; Hung, S.C.; Kieff, E. Epstein-Barr virus nuclear antigen 1 activates transcription from episomal but not integrated DNA and does not alter lymphocyte growth. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 15233–15238. [Google Scholar]
- Sheu, L.F.; Chen, A.; Meng, C.L.; Ho, K.C.; Lee, W.H.; Leu, F.J.; Chao, C.F. Enhanced malignant progression of nasopharyngeal carcinoma cells mediated by the expression of Epstein-Barr nuclear antigen 1 in vivo. J. Pathol. 1996, 180, 243–248. [Google Scholar] [CrossRef]
- Cheng, T.C.; Hsieh, S.S.; Hsu, W.L.; Chen, Y.F.; Ho, H.H.; Sheu, L.F. Expression of Epstein-Barr nuclear antigen 1 in gastric carcinoma cells is associated with enhanced tumorigenicity and reduced cisplatin sensitivity. Int. J. Oncol. 2010, 36, 151–160. [Google Scholar]
- Kube, D.; Vockerodt, M.; Weber, O.; Hell, K.; Wolf, J.; Haier, B.; Grasser, F.A.; Muller-Lantzsch, N.; Kieff, E.; Diehl, V.; et al. Expression of Epstein-Barr virus nuclear antigen 1 is associated with enhanced expression of cd25 in the hodgkin cell line l428. J. Virol. 1999, 73, 1630–1636. [Google Scholar]
- Kaul, R.; Murakami, M.; Choudhuri, T.; Robertson, E.S. Epstein-Barr virus latent nuclear antigens can induce metastasis in a nude mouse model. J. Virol. 2007, 81, 10352–10361. [Google Scholar] [CrossRef]
- Holowaty, M.N.; Zeghouf, M.; Wu, H.; Tellam, J.; Athanasopoulos, V.; Greenblatt, J.; Frappier, L. Protein profiling with Epstein-Barr nuclear antigen 1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 2003, 278, 29987–29994. [Google Scholar]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar]
- Cummins, J.M.; Rago, C.; Kohli, M.; Kinzler, K.W.; Lengauer, C.; Vogelstein, B. Tumour suppression: Disruption of HAUSP gene stabilizes p53. Nature 2004, 428, 486–487. [Google Scholar]
- Holowaty, M.N.; Sheng, Y.; Nguyen, T.; Arrowsmith, C.; Frappier, L. Protein interaction domains of the ubiqutin specific protease, USP7/HAUSP. J. Biol. Chem. 2003, 278, 47753–47761. [Google Scholar]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 2005, 18, 25–36. [Google Scholar]
- Sheng, Y.; Saridakis, V.; Sarkari, F.; Duan, S.; Wu, T.; Arrowsmith, C.H.; Frappier, L. Molecular recognition of p53 and mdm2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 2006, 13, 285–291. [Google Scholar] [CrossRef]
- Hu, M.; Gu, L.; Li, M.; Jeffrey, P.D.; Gu, W.; Shi, Y. Structural basis of competitive recognition of p53 and mdm2 by HAUSP/USP7: Implications for the regulation of the p53-mdm2 pathway. PLoS Biol. 2006, 4, e27. [Google Scholar]
- Sivachandran, N.; Sarkari, F.; Frappier, L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 2008, 4, e1000170. [Google Scholar] [CrossRef]
- Sivachandran, N.; Dawson, C.W.; Young, L.S.; Liu, F.F.; Middeldorp, J.; Frappier, L. Contributions of the Epstein-Barr virus EBNA 1 protein to gastric carcinoma. J. Virol. 2012, 86, 60–68. [Google Scholar] [CrossRef]
- Salomoni, P.; Ferguson, B.J.; Wyllie, A.H.; Rich, T. New insights into the role of PML in tumour suppression. Cell Res. 2008, 18, 622–640. [Google Scholar] [CrossRef]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef]
- Takahashi, Y.; Lallemand-Breitenbach, V.; Zhu, J.; de The, H. PML nuclear bodies and apoptosis. Oncogene 2004, 23, 2819–2824. [Google Scholar] [CrossRef]
- Guo, A.; Salomoni, P.; Luo, J.; Shih, A.; Zhong, S.; Gu, W.; Pandolfi, P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000, 2, 730–736. [Google Scholar] [CrossRef]
- Wang, Z.G.; Ruggero, D.; Ronchetti, S.; Zhong, S.; Gaboli, M.; Rivi, R.; Pandolfi, P.P. PML is essential for multiple apoptotic pathways. Nat. Genet. 1998, 20, 266–272. [Google Scholar] [CrossRef]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef]
- Gurrieri, C.; Capodieci, P.; Bernardi, R.; Scaglioni, P.P.; Nafa, K.; Rush, L.J.; Verbel, D.A.; Cordon-Cardo, C.; Pandolfi, P.P. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J. Natl. Cancer Inst. 2004, 96, 269–279. [Google Scholar] [CrossRef]
- Geoffroy, M.C.; Chelbi-Alix, M.K. Role of promyelocytic leukemia protein in host antiviral defense. J. Interf. Cytokine Res. 2011, 31, 145–158. [Google Scholar] [CrossRef]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef]
- Reichelt, M.; Wang, L.; Sommer, M.; Perrino, J.; Nour, A.M.; Sen, N.; Baiker, A.; Zerboni, L.; Arvin, A.M. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS Pathog. 2011, 7, e1001266. [Google Scholar]
- Everett, R.D. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 2001, 20, 7266–7273. [Google Scholar] [CrossRef]
- Sivachandran, N.; Cao, J.Y.; Frappier, L. Epstein-Barr virus nuclear antigen 1 hijacks the host kinase CK2 to disrupt PML nuclear bodies. J. Virol. 2010, 84, 11113–11123. [Google Scholar] [CrossRef]
- Sivachandran, N.; Wang, X.; Frappier, L. Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. J. Virol. 2012, 86, 6146–6158. [Google Scholar] [CrossRef]
- Scaglioni, P.P.; Yung, T.M.; Cai, L.F.; Erdjument-Bromage, H.; Kaufman, A.J.; Singh, B.; Teruya-Feldstein, J.; Tempst, P.; Pandolfi, P.P. A CK2-dependent mechanism for degradation of thePML tumor suppressor. Cell 2006, 126, 269–283. [Google Scholar] [CrossRef]
- Scaglioni, P.P.; Yung, T.M.; Choi, S.C.; Baldini, C.; Konstantinidou, G.; Pandolfi, P.P. CK2 mediates phosphorylation and ubiquitin-mediated degradation of the PML tumor suppressor. Mol. Cell Biochem. 2008, 316, 149–154. [Google Scholar] [CrossRef]
- Sarkari, F.; Wang, X.; Nguyen, T.; Frappier, L. The herpesvirus associated ubiquitin specific protease, USP7, is a negative regulator of PML proteins and PML nuclear bodies. PLoS One 2011, 6, e16598. [Google Scholar]
- Sides, M.D.; Block, G.J.; Shan, B.; Esteves, K.C.; Lin, Z.; Flemington, E.K.; Lasky, J.A. Arsenic mediated disruption of promyelocytic leukemia protein nuclear bodies induces ganciclovir susceptibility in Epstein-Barr positive epithelial cells. Virology 2011, 416, 86–97. [Google Scholar] [CrossRef]
- Wood, V.H.; O’Neil, J.D.; Wei, W.; Stewart, S.E.; Dawson, C.W.; Young, L.S. Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFβ signaling pathways. Oncogene 2007, 26, 4135–4147. [Google Scholar]
- Kim, H.S.; Lee, M.S. STAT1 as a key modulator of cell death. Cell. Signal. 2007, 19, 454–465. [Google Scholar] [CrossRef]
- Flavell, J.R.; Baumforth, K.R.; Wood, V.H.; Davies, G.L.; Wei, W.; Reynolds, G.M.; Morgan, S.; Boyce, A.; Kelly, G.L.; Young, L.S.; et al. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA 1 contributes to the growth and survival of hodgkin lymphoma cells. Blood 2008, 111, 292–301. [Google Scholar] [CrossRef]
- Valentine, R.; Dawson, C.W.; Hu, C.; Shah, K.M.; Owen, T.J.; Date, K.L.; Maia, S.P.; Shao, J.; Arrand, J.R.; Young, L.S.; et al. Epstein-Barr virus-encoded EBNA 1 inhibits the canonical NF-κB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef]
- Avery, S.V. Molecular targets of oxidative stress. Biochem. J. 2011, 434, 201–210. [Google Scholar] [CrossRef]
- Lassoued, S.; Ben Ameur, R.; Ayadi, W.; Gargouri, B.; Ben Mansour, R.; Attia, H. Epstein-Barr virus induces an oxidative stress during the early stages of infection in B lymphocytes, epithelial, and lymphoblastoid cell lines. Mol. Cell Biochem. 2008, 313, 179–186. [Google Scholar] [CrossRef]
- Cerimele, F.; Battle, T.; Lynch, R.; Frank, D.A.; Murad, E.; Cohen, C.; Macaron, N.; Sixbey, J.; Smith, K.; Watnick, R.S.; et al. Reactive oxygen signaling and MAPK activation distinguish Epstein-Barr virus (EBV)-positive versus EBV-negative Burkitt's lymphoma. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 175–179. [Google Scholar]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2313–2318. [Google Scholar]
- Kamranvar, S.A.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 2011, 25, 1017–1025. [Google Scholar] [CrossRef]
- Cao, J.Y.; Mansouri, S.; Frappier, L. Changes in the nasopharyngeal carcinoma nuclear proteome induced by the EBNA 1 protein of Epstein-Barr virus reveal potential roles for EBNA1 in metastasis and oxidative stress responses. J. Virol. 2012, 86, 382–394. [Google Scholar] [CrossRef]
- Murakami, M.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr virus nuclear antigen 1 interacts with Nm23-H1 in lymphoblastoid cell lines and inhibits its ability to suppress cell migration. J. Virol. 2005, 79, 1559–1568. [Google Scholar] [CrossRef]
- Choudhuri, T.; Murakami, M.; Kaul, R.; Sahu, S.K.; Mohanty, S.; Verma, S.C.; Kumar, P.; Robertson, E.S. Nm23-H1 can induce cell cycle arrest and apoptosis in B cells. Cancer Biol. Ther. 2010, 9, 1065–1078. [Google Scholar] [CrossRef]
- Lu, J.; Murakami, M.; Verma, S.C.; Cai, Q.; Haldar, S.; Kaul, R.; Wasik, M.A.; Middeldorp, J.; Robertson, E.S. Epstein-Barr virus nuclear antigen 1 (EBNA 1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 2011, 410, 64–75. [Google Scholar] [CrossRef]
- Wu, H.; Kapoor, P.; Frappier, L. Separation of the DNA replication, segregation, and transcriptional activation functions of Epstein-Barr nuclear antigen 1. J. Virol. 2002, 76, 2480–2490. [Google Scholar] [CrossRef]
- Kennedy, G.; Sugden, B. EBNA 1, a bifunctional transcriptional activator. Mol. Cell Biol. 2003, 23, 6901–6908. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Frappier, L. Contributions of Epstein–Barr Nuclear Antigen 1 (EBNA1) to Cell Immortalization and Survival. Viruses 2012, 4, 1537-1547. https://doi.org/10.3390/v4091537
Frappier L. Contributions of Epstein–Barr Nuclear Antigen 1 (EBNA1) to Cell Immortalization and Survival. Viruses. 2012; 4(9):1537-1547. https://doi.org/10.3390/v4091537
Chicago/Turabian StyleFrappier, Lori. 2012. "Contributions of Epstein–Barr Nuclear Antigen 1 (EBNA1) to Cell Immortalization and Survival" Viruses 4, no. 9: 1537-1547. https://doi.org/10.3390/v4091537
APA StyleFrappier, L. (2012). Contributions of Epstein–Barr Nuclear Antigen 1 (EBNA1) to Cell Immortalization and Survival. Viruses, 4(9), 1537-1547. https://doi.org/10.3390/v4091537