West Nile Virus: Immunity and Pathogenesis


{kind=link}
{kind=link}
Abstract
:1. Introduction
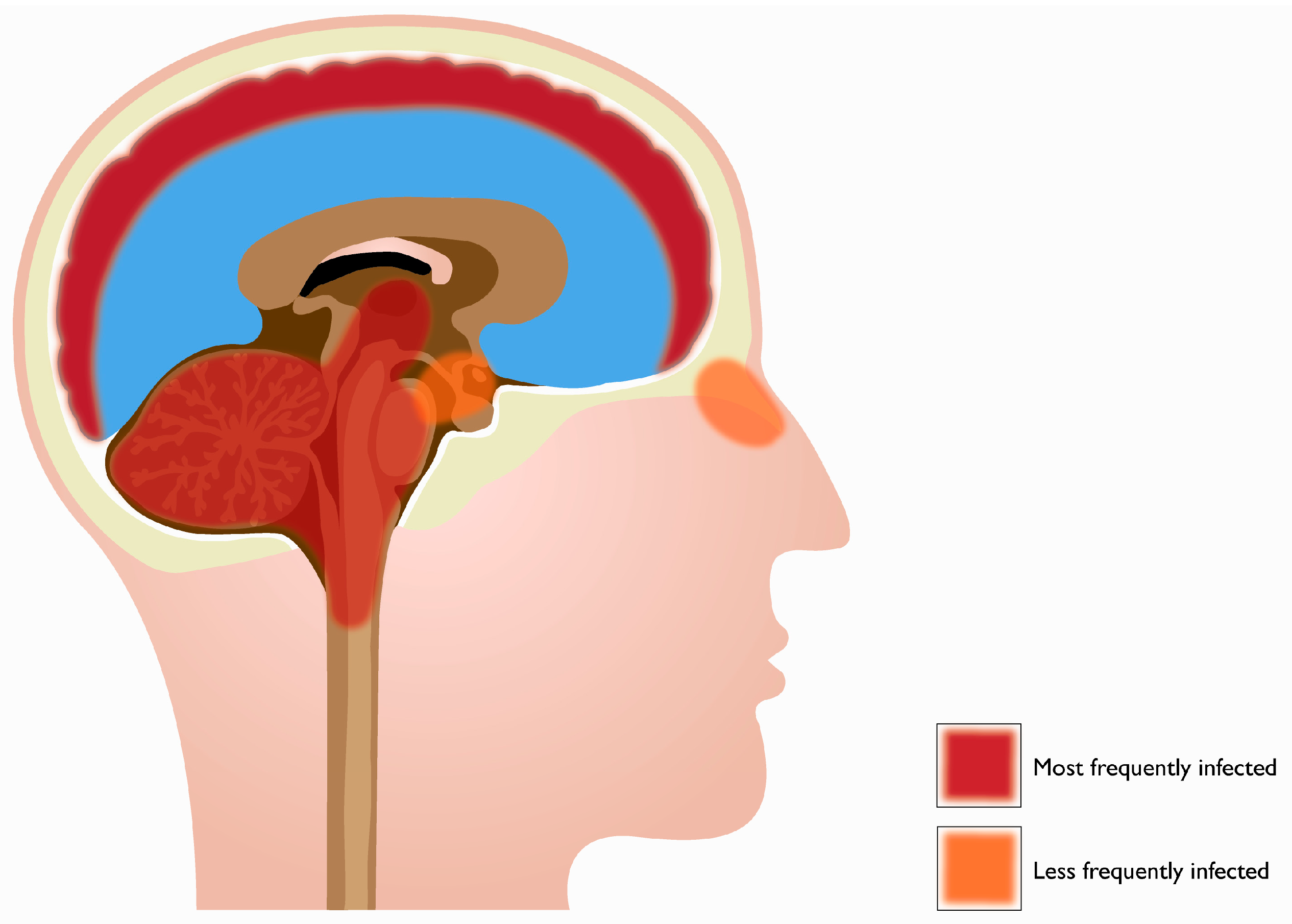
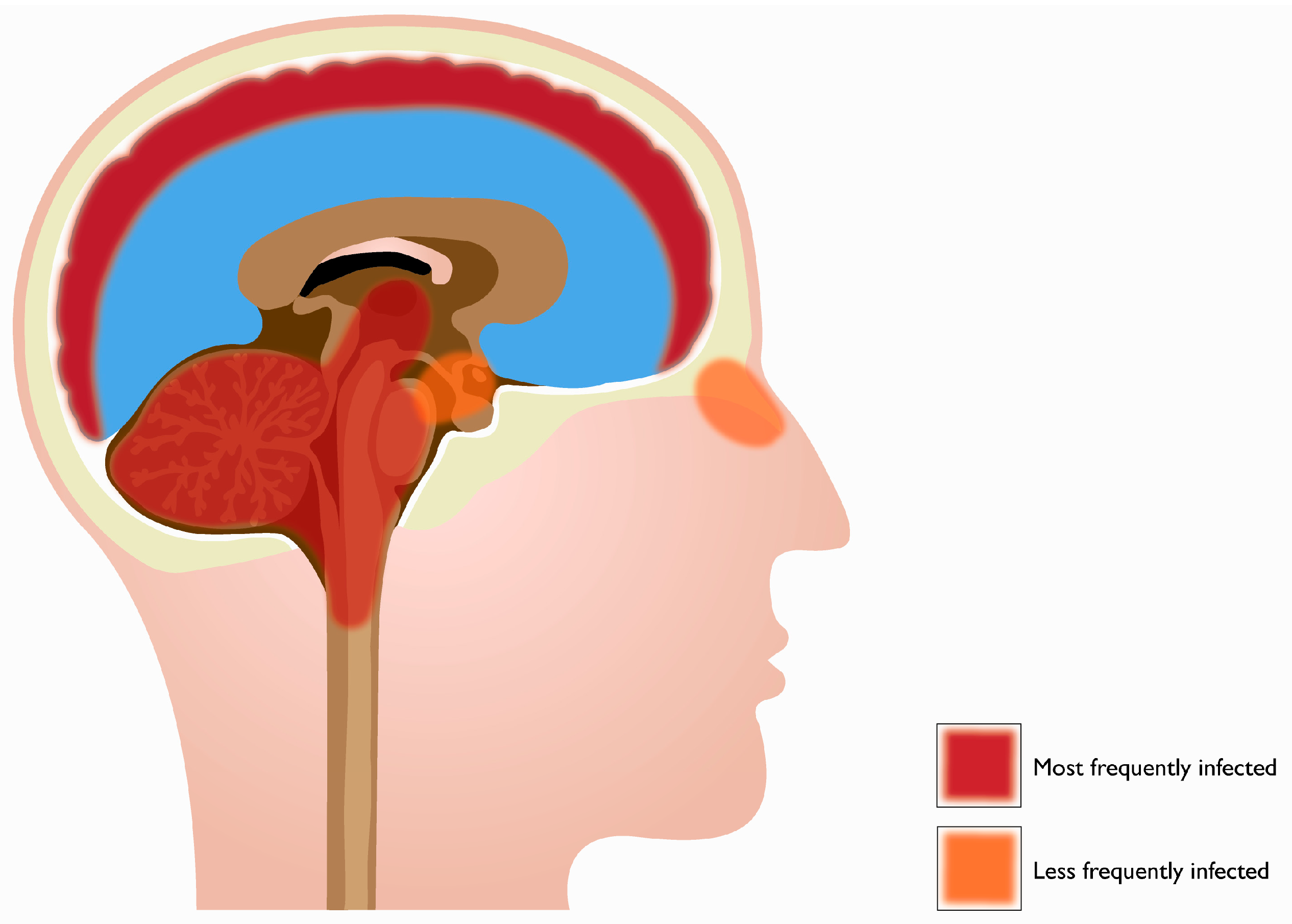
2. Tropism
3. Adaptive Immune Response Protects against Severe WNV Infection
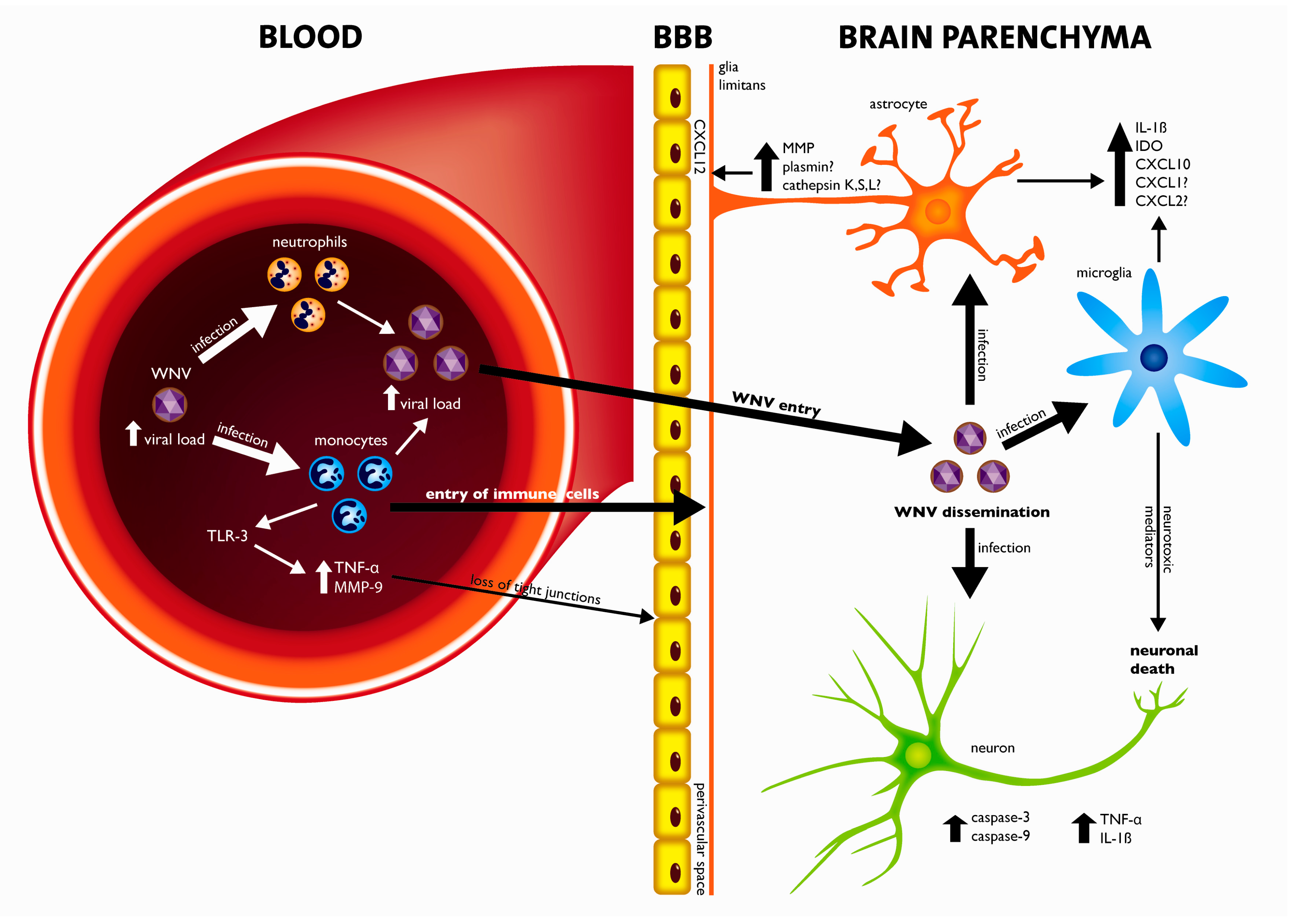
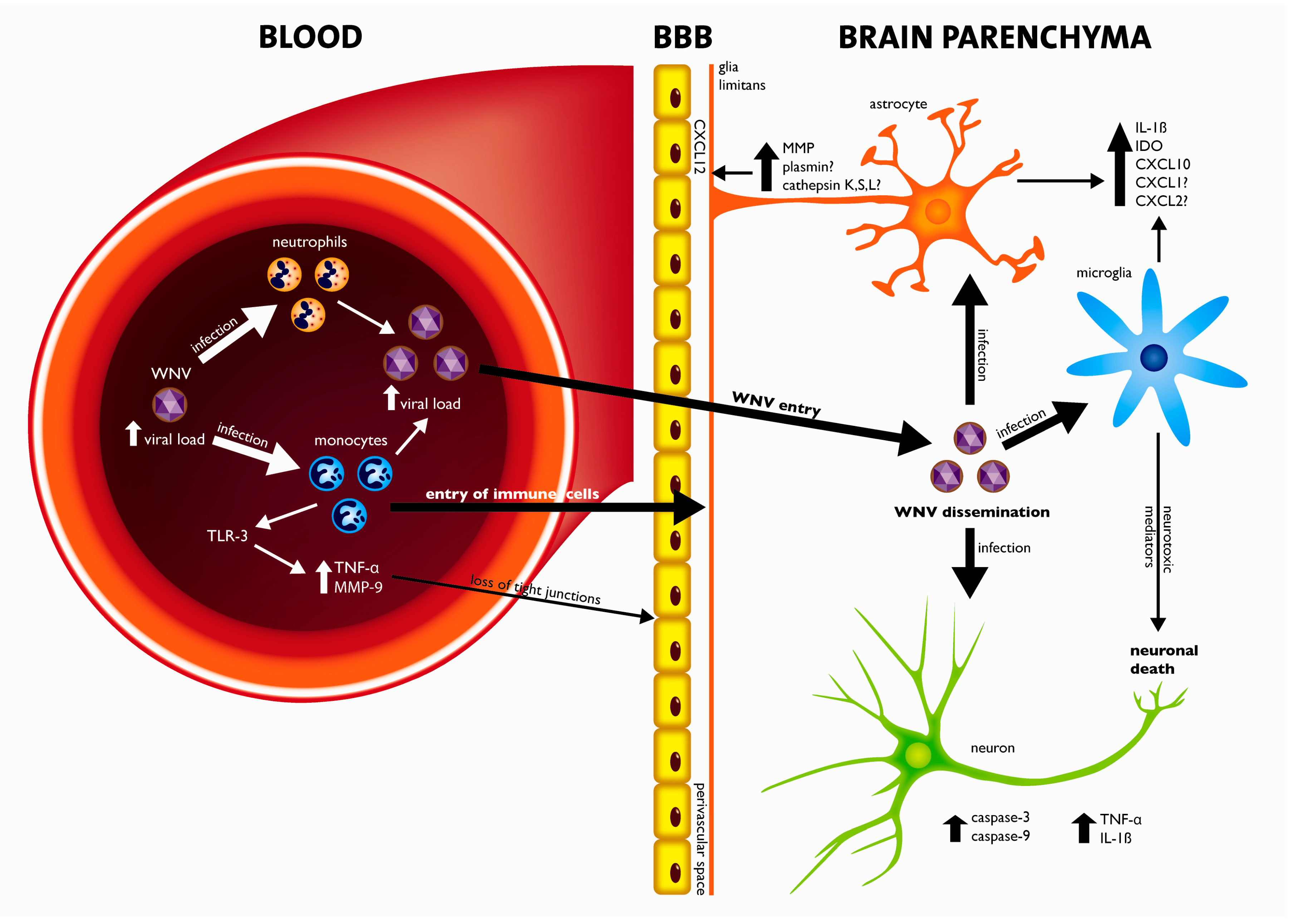
4. Crossing the Blood-Brain-Barrier (BBB)
5. Pathogenesis of WNV Neuroinvasive Disease: A Balance between Viral Cytopathology and the Immune System
5.1. Mechanisms of Cell Death
5.2. Immunopathology
6. Concluding Remarks
Conflict of Interest
Acknowledgements
References and Notes
- Dauphin, G.; Zientara, S.; Zeller, H.; Murgue, B. West Nile: Worldwide current situation in animals and humans. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 343–355. [Google Scholar] [CrossRef]
- Deardorff, E.; Estrada-Franco, J.; Brault, A.C.; Navarro-Lopez, R.; Campomanes-Cortes, A.; Paz-Ramirez, P.; Solis-Hernandez, M.; Ramey, W.N.; Davis, C.T.; Beasley, D.W.; et al. Introductions of West Nile virus strains to Mexico. Emerg. Infect. Dis. 2006, 12, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Komar, N.; Clark, G.G. West Nile virus activity in Latin America and the Caribbean. Rev. Panam. Salud Public. 2006, 19, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Ebel, G.D.; Deubel, V.; Kerst, A.J.; Murri, S.; Meyer, R.; Bowen, M.; McKinney, N.; Morrill, W.E.; Crabtree, M.B.; et al. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology 2002, 298, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Nash, D.; Mostashari, F.; Fine, A.; Miller, J.; O'Leary, D.; Murray, K.; Huang, A.; Rosenberg, A.; Greenberg, A.; Sherman, M.; et al. The outbreak of West Nile virus infection in the New York City area in 1999. New Engl. J. Med. 2001, 344, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.F.; Popovici, F.; Cernescu, C.; Campbell, G.L.; Nedelcu, N.I. West Nile encephalitis epidemic in southeastern Romania. Lancet 1998, 352, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Mostashari, F.; Bunning, M.L.; Kitsutani, P.T.; Singer, D.A.; Nash, D.; Cooper, M.J.; Katz, N.; Liljebjelke, K.A.; Biggerstaff, B.J.; Fine, A.D.; et al. Epidemic West Nile encephalitis, New York, 1999: Results of a household-based seroepidemiological survey. Lancet 2001, 358, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Marfin, A.A. West Nile virus: A primer for the clinician. Ann. Intern. Med. 2002, 137, 173–179. [Google Scholar] [CrossRef]
- Watson, J.T.; Pertel, P.E.; Jones, R.C.; Siston, A.M.; Paul, W.S.; Austin, C.C.; Gerber, S.I. Clinical characteristics and functional outcomes of West Nile Fever. Ann. Intern. Med. 2004, 141, 360–365. [Google Scholar] [CrossRef]
- Campbell, G.L.; Marfin, A.A.; Lanciotti, R.S.; Gubler, D.J. West Nile virus. Lancet Infect. Dis. 2002, 2, 519–529. [Google Scholar] [CrossRef]
- Goldblum, N.; Jasinska-Klingberg, W.; Klingberg, M.A.; Marberg, K.; Sterk, V.V. The natural history of West Nile Fever. I. Clinical observations during an epidemic in Israel. Am. J. Hyg. 1956, 64, 259–269. [Google Scholar] [PubMed]
- Hubalek, Z. Comparative symptomatology of West Nile fever. Lancet 2001, 358, 254–255. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Possible West Nile Virus Transmission to an Infant through Breast Feeding; Centers for Disease Control and Prevention: Michigan, MI, USA, 2002; pp. 1133–1135. [Google Scholar]
- Southam, C.M.; Moore, A.E. Induced virus infections in man by the Egypt isolates of West Nile virus. Am. J. Trop. Med. Hyg. 1954, 3, 19–50. [Google Scholar] [CrossRef]
- Ceausu, E.; Erscoiu, S.; Calistru, P.; Ispas, D.; Dorobat, O.; Homos, M.; Barbulescu, C.; Cojocaru, I.; Simion, C.V.; Cristea, C.; et al. Clinical manifestations in the West Nile virus outbreak. Rom. J. Virol. 1997, 48, 3–11. [Google Scholar] [PubMed]
- Pepperell, C.; Rau, N.; Krajden, S.; Kern, R.; Humar, A.; Mederski, B.; Simor, A.; Low, D.E.; McGeer, A.; Mazzulli, T.; et al. West Nile virus infection in 2002: Morbidity and mortality among patients admitted to hospital in southcentral Ontario. Can. Med. Assoc. J. 2003, 168, 1399–1405. [Google Scholar]
- Sejvar, J.J.; Leis, A.A.; Stokic, D.S.; Van Gerpen, J.A.; Marfin, A.A.; Webb, R.; Haddad, M.B.; Tierney, B.C.; Slavinski, S.A.; Polk, J.L.; et al. Acute flaccid paralysis and West Nile virus infection. Emerg. Infect. Dis. 2003, 9, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Klee, A.L.; Maidin, B.; Edwin, B.; Poshni, I.; Mostashari, F.; Fine, A.; Layton, M.; Nash, D. Long-term prognosis for clinical West Nile virus infection. Emerg. Infect. Dis. 2004, 10, 1405–1411. [Google Scholar] [CrossRef]
- Murray, K.; Baraniuk, S.; Resnick, M.; Arafat, R.; Kilborn, C.; Cain, K.; Shallenberger, R.; York, T.L.; Martinez, D.; Hellums, J.S.; et al. Risk factors for encephalitis and death from West Nile virus infection. Epidemiol. Infect. 2006, 134, 1325–1332. [Google Scholar] [CrossRef]
- Wang, Y.; Lobigs, M.; Lee, E.; Mullbacher, A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J. Virol. 2003, 77, 13323–13334. [Google Scholar] [CrossRef]
- Beasley, D.W.; Li, L.; Suderman, M.T.; Barrett, A.D. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology 2002, 296, 17–23. [Google Scholar] [CrossRef]
- Martina, B.E.; Koraka, P.; Osterhaus, A.D. Dengue virus pathogenesis: An integrated view. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef] [PubMed]
- Wasay, M.; Channa, R.; Jumani, M.; Shabbir, G.; Azeemuddin, M.; Zafar, A. Encephalitis and myelitis associated with dengue viral infection clinical and neuroimaging features. Clin. Neurol. Neurosurg. 2008, 110, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Tripathi, S.; Tambe, J.J.; Arora, V.; Srivastava, A.; Nag, V.L. Dengue encephalopathy in children in Northern India: Clinical features and comparison with non dengue. J. Neurol. Sci. 2008, 269, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Barker, W.C.; Mazumder, R.; Vasudevan, S.; Sagripanti, J.L.; Wu, C.H. Sequence signatures in envelope protein may determine whether flaviviruses produce hemorrhagic or encephalitic syndromes. Virus Genes 2009, 39, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.Y.; Behr, M.J.; Chadwick, C.M.; Shi, P.Y.; Bernard, K.A. Keratinocytes are cell targets of west nile virus in vivo. J. Virol. 85, 5197–5201. [CrossRef]
- Ho, L.J.; Wang, J.J.; Shaio, M.F.; Kao, C.L.; Chang, D.M.; Han, S.W.; Lai, J.H. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J. Immunol. 2001, 166, 1499–1506. [Google Scholar] [CrossRef]
- Johnston, L.J.; Halliday, G.M.; King, N.J. Phenotypic changes in Langerhans' cells after infection with arboviruses: A role in the immune response to epidermally acquired viral infection? J. Virol. 1996, 70, 4761–4766. [Google Scholar] [CrossRef]
- Libraty, D.H.; Pichyangkul, S.; Ajariyakhajorn, C.; Endy, T.P.; Ennis, F.A. Human dendritic cells are activated by dengue virus infection: Enhancement by gamma interferon and implications for disease pathogenesis. J. Virol. 2001, 75, 3501–3508. [Google Scholar] [CrossRef]
- Marovich, M.; Grouard-Vogel, G.; Louder, M.; Eller, M.; Sun, W.; Wu, S.J.; Putvatana, R.; Murphy, G.; Tassaneetrithep, B.; Burgess, T.; et al. Human dendritic cells as targets of dengue virus infection. J. Investig. Dermatol. Symp. Proc. 2001, 6, 219–224. [Google Scholar] [CrossRef]
- Wu, S.J.; Grouard-Vogel, G.; Sun, W.; Mascola, J.R.; Brachtel, E.; Putvatana, R.; Louder, M.K.; Filgueira, L.; Marovich, M.A.; Wong, H.K.; et al. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 2000, 6, 816–820. [Google Scholar] [CrossRef]
- Rios, M.; Zhang, M.J.; Grinev, A.; Srinivasan, K.; Daniel, S.; Wood, O.; Hewlett, I.K.; Dayton, A.I. Monocytes-macrophages are a potential target in human infection with West Nile virus through blood transfusion. Transfusion 2006, 46, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, N. Slow viruses and chronic disease: The contribution of epidemiology. Publ. Health Rep. 1980, 95, 436–443. [Google Scholar]
- Beasley, D.W.; Barrett, A.D. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J. Virol. 2002, 76, 13097–13100. [Google Scholar] [CrossRef] [PubMed]
- Deubel, V.; Fiette, L.; Gounon, P.; Drouet, M.T.; Khun, H.; Huerre, M.; Banet, C.; Malkinson, M.; Despres, P. Variations in biological features of West Nile viruses. Ann. New York Acad. Sci. 2001, 951, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Halevy, M.; Nestorowicz, A.; Rice, C.M.; Lustig, S. West Nile virus envelope proteins: Nucleotide sequence analysis of strains differing in mouse neuroinvasiveness. J. Gen. Virol. 1998, 79, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Hammerle, S.; Rothenaigner, I.; Wolff, H.; Bell, J.E.; Brack-Werner, R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005, 111, 194–213. [Google Scholar] [CrossRef]
- Monath, T.P.; Cropp, C.B.; Harrison, A.K. Mode of entry of a neurotropic arbovirus into the central nervous system. Reinvestigation of an old controversy. Lab. Invest. 1983, 48, 399–410. [Google Scholar]
- Garcia-Tapia, D.; Loiacono, C.M.; Kleiboeker, S.B. Replication of West Nile virus in equine peripheral blood mononuclear cells. Vet. Immunol. Immunopathol. 2006, 110, 229–244. [Google Scholar] [CrossRef]
- Hunsperger, E.A.; Roehrig, J.T. Temporal analyses of the neuropathogenesis of a West Nile virus infection in mice. J. Neurovirol. 2006, 12, 129–139. [Google Scholar] [CrossRef]
- Johnson, R.T. Viral Infections of the Nervous System, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1998. [Google Scholar]
- Samuel, M.A.; Wang, H.; Siddharthan, V.; Morrey, J.D.; Diamond, M.S. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17140–17145. [Google Scholar] [CrossRef]
- Armah, H.B.; Wang, G.; Omalu, B.I.; Tesh, R.B.; Gyure, K.A.; Chute, D.J.; Smith, R.D.; Dulai, P.; Vinters, H.V.; Kleinschmidt-DeMasters, B.K.; et al. Systemic distribution of West Nile virus infection: postmortem immunohistochemical study of six cases. Brain Pathol. 2007, 17, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, M.C.; Hu, S.; Sheng, W.S.; Rashid, A.; Peterson, P.K.; Lokensgard, J.R. Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 2005, 11, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, M.; Gurjav, U.; Lum, S.; Nerurkar, V.R. Reversal of West Nile virus-induced blood-brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 2009, 397, 130–138. [Google Scholar] [CrossRef]
- Jordan, I.; Briese, T.; Fischer, N.; Lau, J.Y.; Lipkin, W.I. Ribavirin inhibits West Nile virus replication and cytopathic effect in neural cells. J. Infect. Dis. 2000, 182, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; King, N.; Kesson, A.; Blanden, R.V.; Mullbacher, A. West Nile virus infection modulates the expression of class I and class II MHC antigens on astrocytes in vitro. Ann. New York Acad. Sci. 1988, 540, 483–485. [Google Scholar] [CrossRef]
- Parquet, M.C.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 2001, 500, 17–24. [Google Scholar] [CrossRef]
- Shrestha, B.; Gottlieb, D.; Diamond, M.S. Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 2003, 77, 13203–13213. [Google Scholar] [CrossRef]
- Xiao, S.Y.; Guzman, H.; Zhang, H.; Travassos da Rosa, A.P.; Tesh, R.B. West Nile virus infection in the golden hamster (Mesocricetus auratus): A model for West Nile encephalitis. Emerg. Infect. Dis. 2001, 7, 714–721. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef]
- Sampson, B.A.; Ambrosi, C.; Charlot, A.; Reiber, K.; Veress, J.F.; Armbrustmacher, V. The pathology of human West Nile Virus infection. Hum. Pathol. 2000, 31, 527–531. [Google Scholar] [CrossRef]
- Shieh, W.J.; Guarner, J.; Layton, M.; Fine, A.; Miller, J.; Nash, D.; Campbell, G.L.; Roehrig, J.T.; Gubler, D.J.; Zaki, S.R. The role of pathology in an investigation of an outbreak of West Nile encephalitis in New York, 1999. Emerg. Infect. Dis. 2000, 6, 370–372. [Google Scholar] [CrossRef] [PubMed]
- McElhaney, J.E.; Effros, R.B. Immunosenescence: what does it mean to health outcomes in older adults? Curr. Opin. Immunol. 2009, 21, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Sitati, E.M.; Diamond, M.S. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J. Virol. 2006, 80, 12060–12069. [Google Scholar] [CrossRef]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef]
- Bear, M.F. , Connors, B., Paradiso, M. Neuroscience—Exploring the Brain, 3rd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Kandel, E.R. , Schwartz, J.H., Jessell, T.M. Principles of Neural Science, Nerve Cells and Behavior, 4th ed.; McGraw-Hill: New York, NY, USA, 2000. [Google Scholar]
- Hilgetag, C.C.; Barbas, H. Are there ten times more glia than neurons in the brain? Brain Struct. Funct. 2009, 213, 365–366. [Google Scholar] [CrossRef]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Halevy, M.; Akov, Y.; Ben-Nathan, D.; Kobiler, D.; Lachmi, B.; Lustig, S. Loss of active neuroinvasiveness in attenuated strains of West Nile virus: Pathogenicity in immunocompetent and SCID mice. Arch. Virol. 1994, 137, 355–370. [Google Scholar] [CrossRef]
- Lustig, S.; Danenberg, H.D.; Kafri, Y.; Kobiler, D.; Ben-Nathan, D. Viral neuroinvasion and encephalitis induced by lipopolysaccharide and its mediators. J. Exp. Med. 1992, 176, 707–712. [Google Scholar] [CrossRef]
- McCandless, E.E.; Wang, Q.; Woerner, B.M.; Harper, J.M.; Klein, R.S. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J. Immunol. 2006, 177, 8053–8064. [Google Scholar] [CrossRef]
- Cruz-Orengo, L.; Holman, D.W.; Dorsey, D.; Zhou, L.; Zhang, P.; Wright, M.; McCandless, E.E.; Patel, J.R.; Luker, G.D.; Littman, D.R.; et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J. Exp. Med. 2011, 208, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Savarin, C.; Stohlman, S.A.; Atkinson, R.; Ransohoff, R.M.; Bergmann, C.C. Monocytes regulate T cell migration through the glia limitans during acute viral encephalitis. J. Virol. 2010, 84, 4878–4888. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Touchard, A.; Henry, T.D.; Sangiorgi, G.; Spagnoli, L.G.; Mauriello, A.; Conover, C.; Schwartz, R.S. Extracellular proteases in atherosclerosis and restenosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1119–1127. [Google Scholar] [CrossRef]
- Reddy, V.Y.; Zhang, Q.Y.; Weiss, S.J. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 3849–3853. [Google Scholar] [CrossRef]
- Reijerkerk, A.; Kooij, G.; van der Pol, S.M.; Leyen, T.; van Het Hof, B.; Couraud, P.O.; Vivien, D.; Dijkstra, C.D.; de Vries, H.E. Tissue-type plasminogen activator is a regulator of monocyte diapedesis through the brain endothelial barrier. J. Immunol. 2008, 181, 3567–3574. [Google Scholar] [CrossRef] [PubMed]
- Jaattela, M. Programmed cell death: many ways for cells to die decently. Ann. Med. 2002, 34, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Lockshin, R.A.; Zakeri, Z. Programmed cell death and apoptosis: origins of the theory. Nat. Rev. 2001, 2, 545–550. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Brennan, M.A.; Cookson, B.T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef]
- Chen, Y.; Smith, M.R.; Thirumalai, K.; Zychlinsky, A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. 1996, 15, 3853–3860. [Google Scholar] [CrossRef]
- Hersh, D.; Monack, D.M.; Smith, M.R.; Ghori, N.; Falkow, S.; Zychlinsky, A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 2396–2401. [Google Scholar] [CrossRef] [PubMed]
- Hilbi, H.; Chen, Y.; Thirumalai, K.; Zychlinsky, A. The interleukin 1beta-converting enzyme, caspase 1, is activated during Shigella flexneri-induced apoptosis in human monocyte-derived macrophages. Infect. Immun. 1997, 65, 5165–5170. [Google Scholar] [CrossRef] [PubMed]
- Hilbi, H.; Moss, J.E.; Hersh, D.; Chen, Y.; Arondel, J.; Banerjee, S.; Flavell, R.A.; Yuan, J.; Sansonetti, P.J.; Zychlinsky, A. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 1998, 273, 32895–32900. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G.; Dinarello, C.A. Interleukin-18 and interleukin-1 beta: Two cytokine substrates for ICE (caspase-1). J. Clin. Immunol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, G.; MacDonald, G.; Bergeron, L.; Li, H.; Miura, M.; Rotello, R.J.; Miller, D.K.; Li, P.; Seshadri, T.; et al. Activation of an interleukin 1 converting enzyme-dependent apoptosis pathway by granzyme B. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 11002–11007. [Google Scholar] [CrossRef]
- Liu, X.H.; Kwon, D.; Schielke, G.P.; Yang, G.Y.; Silverstein, F.S.; Barks, J.D. Mice deficient in interleukin-1 converting enzyme are resistant to neonatal hypoxic-ischemic brain damage. J. Cereb. Blood Flow Metab. 1999, 19, 1099–1108. [Google Scholar] [CrossRef]
- Zhang, W.H.; Wang, X.; Narayanan, M.; Zhang, Y.; Huo, C.; Reed, J.C.; Friedlander, R.M. Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 16012–16017. [Google Scholar] [CrossRef]
- Frantz, S.; Ducharme, A.; Sawyer, D.; Rohde, L.E.; Kobzik, L.; Fukazawa, R.; Tracey, D.; Allen, H.; Lee, R.T.; Kelly, R.A. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J. Mol. Cell Cardiol. 2003, 35, 685–694. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Narula, J.; Burke, A.P.; Haider, N.; Farb, A.; Hui-Liang, Y.; Smialek, J.; Virmani, R. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Amer. J. Pathol. 2000, 157, 1259–1268. [Google Scholar] [CrossRef]
- Raung, S.L.; Kuo, M.D.; Wang, Y.M.; Chen, C.J. Role of reactive oxygen intermediates in Japanese encephalitis virus infection in murine neuroblastoma cells. Neurosci. Lett. 2001, 315, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Weissenbock, H.; Bakonyi, T.; Chvala, S.; Nowotny, N. Experimental Usutu virus infection of suckling mice causes neuronal and glial cell apoptosis and demyelination. Acta Neuropathol. 2004, 108, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Ramanathan, M.P.; Muthumani, K.; Choo, A.Y.; Jin, S.H.; Yu, Q.C.; Hwang, D.S.; Choo, D.K.; Lee, M.D.; Dang, K.; et al. Induction of inflammation by West Nile virus capsid through the caspase-9 apoptotic pathway. Emerg. Infect. Dis. 2002, 8, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Morrey, J.D.; Diamond, M.S. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J. Virol. 2007, 81, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Hail, N., Jr.; Carter, B.Z.; Konopleva, M.; Andreeff, M. Apoptosis effector mechanisms: a requiem performed in different keys. Apoptosis 2006, 11, 889–904. [Google Scholar] [CrossRef]
- Kroemer, G.; Martin, S.J. Caspase-independent cell death. Nat. Med. 2005, 11, 725–730. [Google Scholar] [CrossRef]
- Chu, J.J.; Ng, M.L. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J. Gen. Virol. 2003, 84, 3305–3314. [Google Scholar] [CrossRef]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef]
- Wang, P.; Arjona, A.; Zhang, Y.; Sultana, H.; Dai, J.; Yang, L.; LeBlanc, P.M.; Doiron, K.; Saleh, M.; Fikrig, E. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat. Immunol. 2010, 11, 912–919. [Google Scholar] [CrossRef]
- Yu, C.Y.; Hsu, Y.W.; Liao, C.L.; Lin, Y.L. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 2006, 80, 11868–11880. [Google Scholar] [CrossRef]
- van Marle, G.; Antony, J.; Ostermann, H.; Dunham, C.; Hunt, T.; Halliday, W.; Maingat, F.; Urbanowski, M.D.; Hobman, T.; Peeling, J.; et al. West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J. Virol. 2007, 81, 10933–10949. [Google Scholar] [CrossRef]
- Klein, R.S.; Lin, E.; Zhang, B.; Luster, A.D.; Tollett, J.; Samuel, M.A.; Engle, M.; Diamond, M.S. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005, 79, 11457–11466. [Google Scholar] [CrossRef]
- Kumar, M.; Verma, S.; Nerurkar, V.R. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflammation 2010, 7, 73. [Google Scholar] [CrossRef]
- Swarup, V.; Das, S.; Ghosh, S.; Basu, A. Tumor necrosis factor receptor-1-induced neuronal death by TRADD contributes to the pathogenesis of Japanese encephalitis. J. Neurochemistry 2007, 103, 771–783. [Google Scholar] [CrossRef]
- Ghoshal, A.; Das, S.; Ghosh, S.; Mishra, M.K.; Sharma, V.; Koli, P.; Sen, E.; Basu, A. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 2007, 55, 483–496. [Google Scholar] [CrossRef]
- Johnson, R.T.; Irani, D.N. West Nile virus encephalitis in the United States. Curr. Neurol. Neurosci. Rep. 2002, 2, 496–500. [Google Scholar] [CrossRef]
- Kelley, T.W.; Prayson, R.A.; Isada, C.M. Spinal cord disease in West Nile virus infection. New Engl. J. Med. 2003, 348, 564–566; author reply 564–566. [Google Scholar]
- Kelley, T.W.; Prayson, R.A.; Ruiz, A.I.; Isada, C.M.; Gordon, S.M. The neuropathology of West Nile virus meningoencephalitis. A report of two cases and review of the literature. Am. J. Clin. Pathol. 2003, 119, 749–753. [Google Scholar] [CrossRef]
- Kong, K.F.; Delroux, K.; Wang, X.; Qian, F.; Arjona, A.; Malawista, S.E.; Fikrig, E.; Montgomery, R.R. Dysregulation of TLR3 impairs the innate immune response to West Nile virus in the elderly. J. Virol. 2008, 82, 7613–7623. [Google Scholar] [CrossRef]
- Cinque, P.; Bestetti, A.; Marenzi, R.; Sala, S.; Gisslen, M.; Hagberg, L.; Price, R.W. Cerebrospinal fluid interferon-gamma-inducible protein 10 (IP-10, CXCL10) in HIV-1 infection. J. Neuroimmunol. 2005, 168, 154–163. [Google Scholar] [CrossRef]
- Sui, Y.; Potula, R.; Dhillon, N.; Pinson, D.; Li, S.; Nath, A.; Anderson, C.; Turchan, J.; Kolson, D.; Narayan, O.; et al. Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Amer. J. Pathol. 2004, 164, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Stehno-Bittel, L.; Li, S.; Loganathan, R.; Dhillon, N.K.; Pinson, D.; Nath, A.; Kolson, D.; Narayan, O.; Buch, S. CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur. J. Neurosci. 2006, 23, 957–964. [Google Scholar] [CrossRef] [PubMed]
- van Marle, G.; Henry, S.; Todoruk, T.; Sullivan, A.; Silva, C.; Rourke, S.B.; Holden, J.; McArthur, J.C.; Gill, M.J.; Power, C. Human immunodeficiency virus type 1 Nef protein mediates neural cell death: a neurotoxic role for IP-10. Virology 2004, 329, 302–318. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Kong, K.F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J. Infect. Dis. 2010, 202, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Reisen, W.K.; Hahn, D.C. Comparison of immune responses of brown-headed cowbird and related blackbirds to west Nile and other mosquito-borne encephalitis viruses. J. Wildl. Dis. 2007, 43, 439–449. [Google Scholar] [CrossRef] [PubMed]
- For details go to http://www.erasmusmc.nl/ (accessed on 26 May 2011).
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lim, S.M.; Koraka, P.; Osterhaus, A.D.M.E.; Martina, B.E.E. West Nile Virus: Immunity and Pathogenesis. Viruses 2011, 3, 811-828. https://doi.org/10.3390/v3060811
Lim SM, Koraka P, Osterhaus ADME, Martina BEE. West Nile Virus: Immunity and Pathogenesis. Viruses. 2011; 3(6):811-828. https://doi.org/10.3390/v3060811
Chicago/Turabian StyleLim, Stephanie M., Penelope Koraka, Albert D.M.E. Osterhaus, and Byron E.E. Martina. 2011. "West Nile Virus: Immunity and Pathogenesis" Viruses 3, no. 6: 811-828. https://doi.org/10.3390/v3060811
APA StyleLim, S. M., Koraka, P., Osterhaus, A. D. M. E., & Martina, B. E. E. (2011). West Nile Virus: Immunity and Pathogenesis. Viruses, 3(6), 811-828. https://doi.org/10.3390/v3060811