Oncogenic Potential of Hepatitis C Virus Proteins



{kind=link}
{kind=link}
Abstract
:1. Introduction
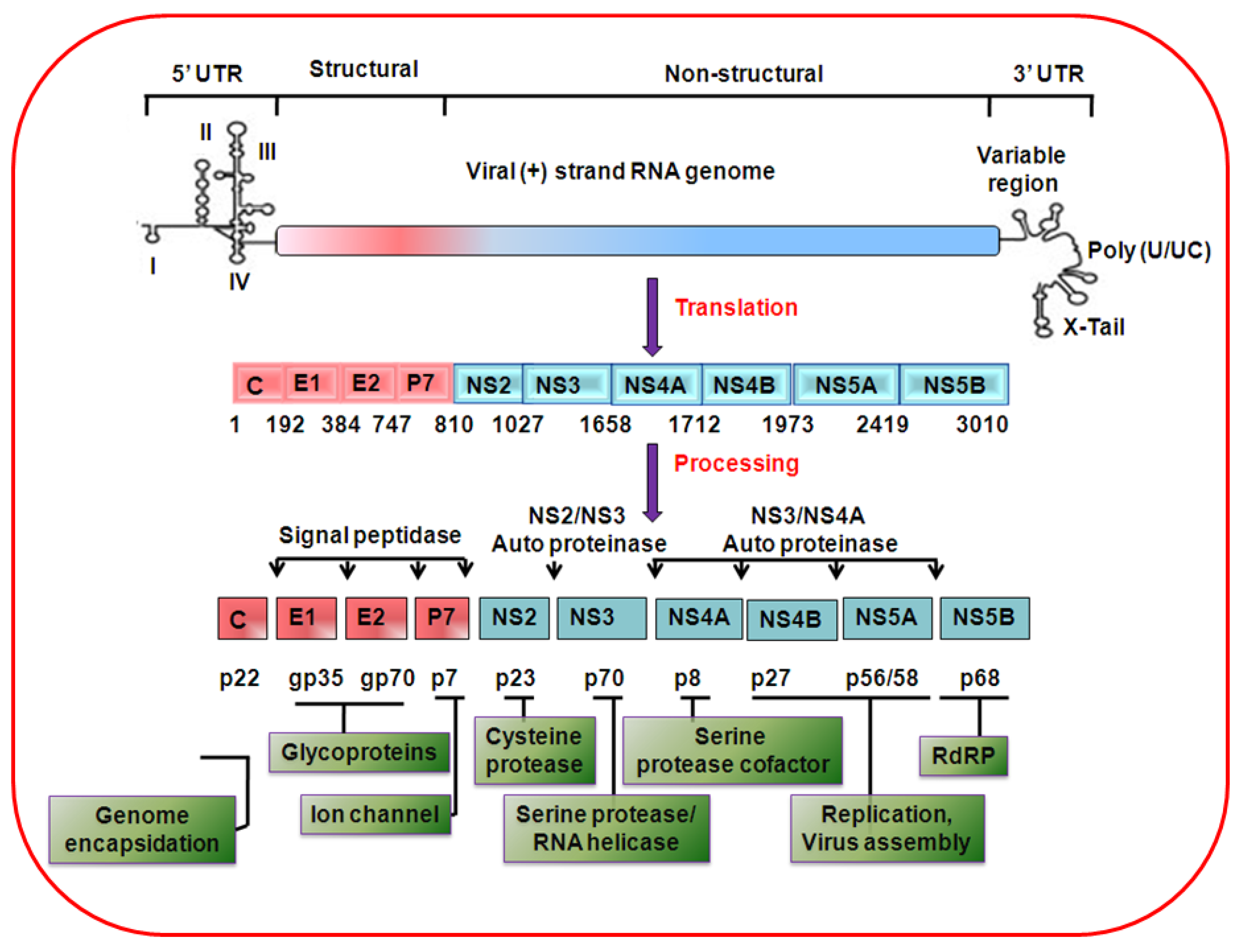
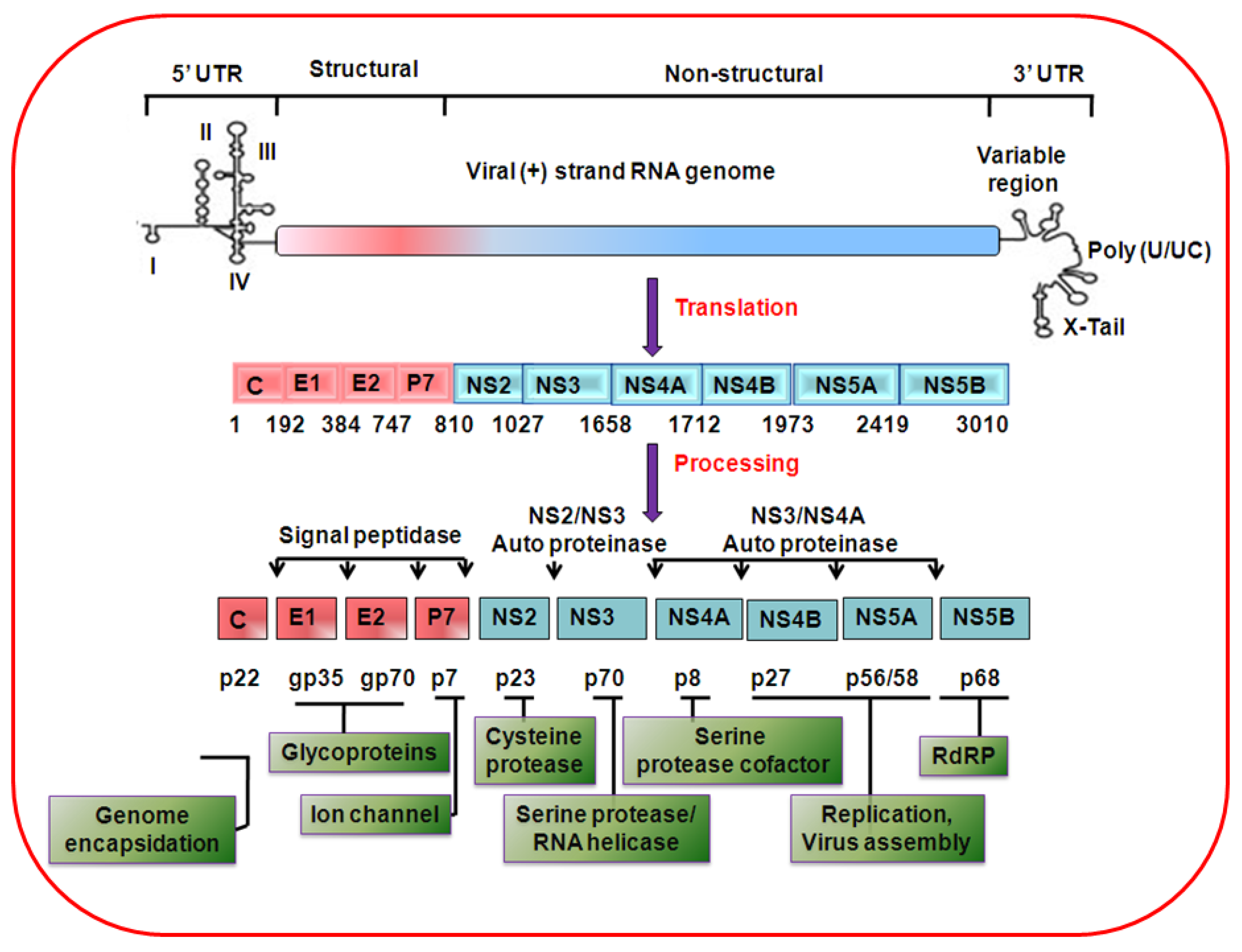
2. HCV genome organization, protein synthesis and life cycle
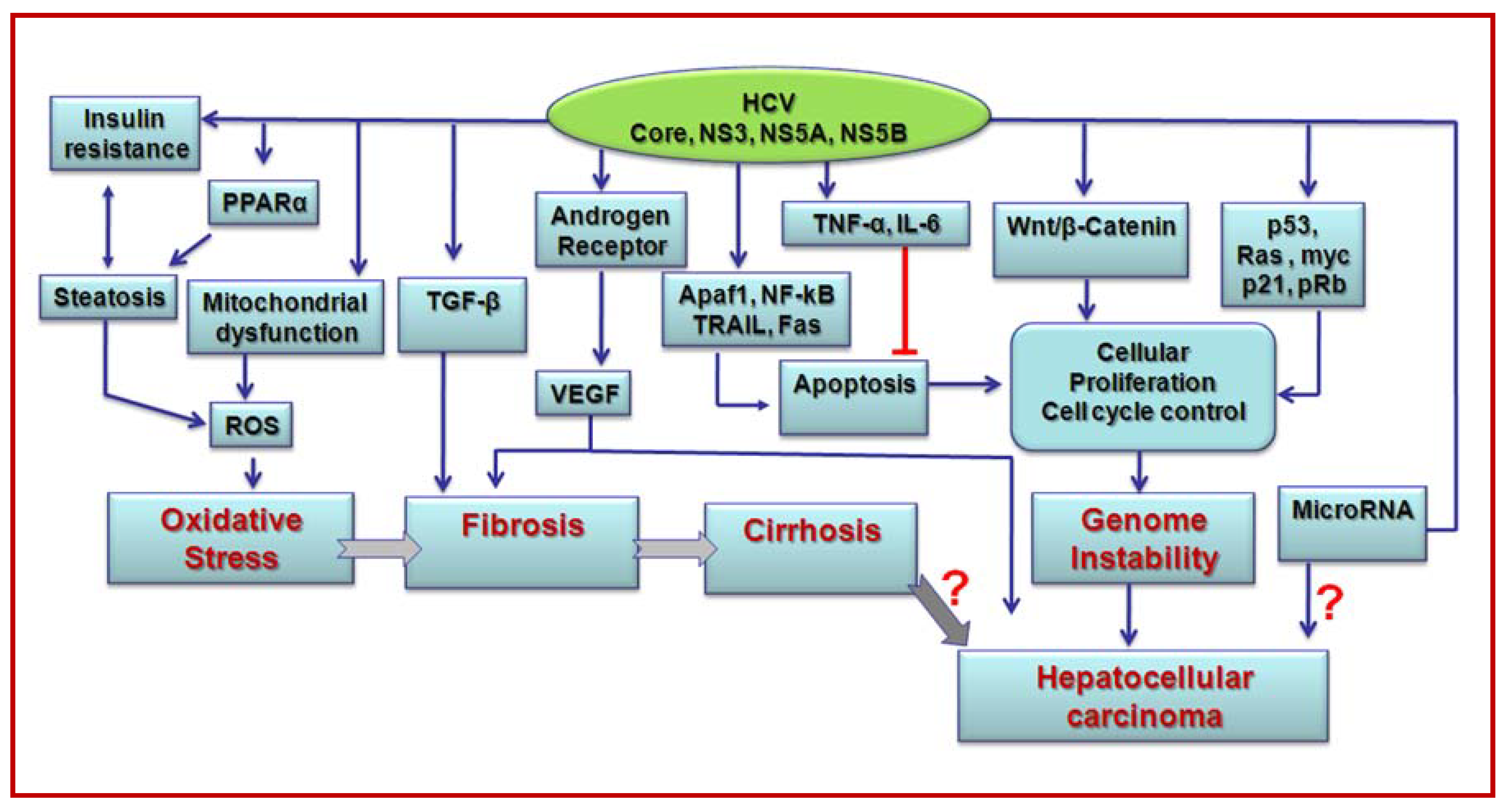
3. Transcriptional modulation and oncogene regulation by HCV
4. Hepatocyte growth regulation by HCV proteins
5. Role of HCV proteins in cytokine modulation
6. Role of HCV in oxidative stress and apoptosis
7. HCV associated metabolic disorders and liver disease progression
8. Induction of miRNAs by HCV
9. Cooperative interactions of HCV and other agents in promoting liver disease
10. Summary
Acknowledgments
References and Notes
- El-Serag, H.B. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology 2004, 127, S27–S34. [Google Scholar] [CrossRef] [PubMed]
- Kiyosawa, K.; Umemura, T.; Ichijo, T.; Matsumoto, A.; Yoshizawa, K.; Gad, A.; Tanaka, E. Hepatocellular carcinoma: recent trends in Japan. Gastroenterology 2004, 127, S17–S26. [Google Scholar] [CrossRef]
- Umemura, T.; Ichijo, T.; Yoshizawa, K.; Tanaka, E.; Kiyosawa, K. Epidemiology of hepatocellular carcinoma in Japan. J. Gastroenterol. 2009, 44, 102–107. [Google Scholar] [CrossRef]
- Gearhart, T.L.; Bouchard, M.J. The hepatitis B virus X protein modulates hepatocyte proliferation pathways to stimulate viral replication. J. Virol. 2010, 84, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, N.; Oka, M.; Yamada-Okabe, H.; Mori, N.; Tamesa, T.; Okada, T.; Takemoto, N.; Tangoku, A.; Hamada, K.; Nakayama, H.; Miyamoto, T.; Uchimura, S.; Hamamoto, Y. Comparison of gene expression profiles between hepatitis B virus- and hepatitis C virus-infected hepatocellular carcinoma by oligonucleotide microarray data on the basis of a supervised learning method. Cancer Res. 2002, 62, 3939–3944. [Google Scholar] [PubMed]
- Yoon, S.Y.; Kim, J.M.; Oh, J.H.; Jeon, Y.J.; Lee, D.S.; Kim, J.H.; Choi, J.Y.; Ahn, B.M.; Kim, S.; Yoo, H.S.; Kim, Y.S.; Kim, N.S. Gene expression profiling of human HBV- and/or HCV-associated hepatocellular carcinoma cells using expressed sequence tags. Int. J. Oncol. 2006, 29, 315–327. [Google Scholar] [CrossRef]
- De Giorgi, V.; Monaco, A.; Worchech, A.; Tornesello, M.; Izzo, F.; Buonaguro, L.; Marincola, F.M.; Wang, E.; Buonaguro, F.M. Gene profiling, biomarkers and pathways characterizing HCV-related hepatocellular carcinoma. J. Transl. Med. 2009, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, Y.; Guidotti, L.G.; Kuhlen, C.V.; Fowler, P.; Chisari, F.V. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 1998, 188, 341–350. [Google Scholar] [CrossRef]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef]
- Bartsch, H.; Nair, J. Oxidative stress and lipid peroxidation-derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect. Prev. 2004, 28, 385–391. [Google Scholar] [CrossRef]
- Ray, R.B.; Lagging, L.M.; Meyer, K.; Ray, R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J. Virol. 1996, 70, 4438–4443. [Google Scholar] [CrossRef] [PubMed]
- Gale, M., Jr.; Kwieciszewski, B.; Dossett, M.; Nakao, H.; Katze, M.G. Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J. Virol. 1999, 73, 6506–6516. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Yang, J.M.; Min, M.K. Hepatitis C virus nonstructural protein NS4B transforms NIH3T3 cells in cooperation with the Ha-ras oncogene. Biochem. Biophys. Res. Commun. 2000, 267, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.Y.; Weinman, S.A.; Lemon, S.M. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef]
- Friebe, P.; Lohmann, V.; Krieger, N.; Bartenschlager, R. Sequences in the 5’nontranslated region of hepatitis C virus required for RNA replication. J. Virol. 2001, 75, 12047–12057. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Lemon, S.M. 3’ nontranslated RNA signals required for replication of hepatitis C virus RNA. J. Virol. 2003, 77, 3557–3568. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Virology of hepatitis C virus. Clin. Ther. 1996, 18, 9–36. [Google Scholar] [CrossRef]
- Simmonds, P. The origin and evolution of hepatitis viruses in humans. J. Gen. Virol. 2001, 82, 693–712. [Google Scholar] [CrossRef]
- Harada, S.; Watanabe, Y.; Takeuchi, K.; Suzuki, T.; Katayama, T.; Takebe, Y.; Saito, I.; Miyamura, T. Expression of processed core protein of hepatitis C virus in mammalian cells. J. Virol. 1991, 65, 3015–3021. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Lagging, L.M.; Meyer, K.; Steele, R.; Ray, R. Transcriptional regulation of cellular and viral promoters by the hepatitis C virus core protein. Virus Res. 1995, 37, 209–220. [Google Scholar] [CrossRef]
- Liu, Q.; Tackney, C.; Bhat, R.A.; Prince, A.M.; Zhang, P. Regulated processing of hepatitis C virus core protein is linked to subcellular localization. J. Virol. 1997, 71, 657–662. [Google Scholar] [CrossRef]
- Yasui, K.; Wakita, T.; Tsukiyama-Kohara, K.; Funahashi, S.I.; Ichikawa, M.; Kajita, T.; Moradpour, D.; Wands, J.R.; Kohara, M. The native form and maturation process of hepatitis C virus core protein. J. Virol. 1998, 72, 6048–6055. [Google Scholar] [CrossRef]
- Xu, Z.; Choi, J.; Yen, T.S.; Lu, W.; Strohecker, A.; Govindarajan, S.; Chien, D.; Selby, M.J.; Ou, J. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 2001, 20, 3840–3848. [Google Scholar] [CrossRef]
- Walewski, J.L.; Keller, T.R.; Stump, D.D.; Branch, A.D. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA 2001, 7, 710–721. [Google Scholar] [CrossRef]
- Varaklioti, A.; Vassilaki, N.; Georgopoulou, U.; Mavromara, P. Alternate translation occurs within the core coding region of the hepatitis C viral genome. J. Biol. Chem. 2002, 277, 17713–17721. [Google Scholar] [CrossRef] [PubMed]
- Vassilaki, N.; Mavromara, P. Two alternative translation mechanisms are responsible for the expression of the HCV ARFP/F/core+1 coding open reading frame. J. Biol. Chem. 2003, 278, 40503–40513. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Steele, R.; Ray, R.; Ray, R.B. Functional properties of a 16 kDa protein translated from an alternative open reading frame of the core-encoding genomic region of hepatitis C virus. J. Gen. Virol. 2004, 85, 2299–2306. [Google Scholar] [CrossRef]
- Ray, R.B.; Ray, R. Hepatitis C virus core protein: intriguing properties and functional relevance. FEMS Microbiol. Lett. 2001, 202, 149–156. [Google Scholar] [CrossRef]
- Meyer, K.; Basu, A.; Ray, R. Functional features of hepatitis C virus glycoproteins for pseudotype virus entry into mammalian cells. Virology 2000, 276, 214–226. [Google Scholar] [CrossRef]
- Meyer, K.; Beyene, A.; Bowlin, T.L.; Basu, A.; Ray, R. Coexpression of hepatitis C virus E1 and E2 chimeric envelope glycoproteins displays separable ligand sensitivity and increases pseudotype infectious titer. J. Virol. 2004, 78, 12838–12847. [Google Scholar] [CrossRef]
- Lohmann, V.; Körner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Tomei, L.; Failla, C.; Santolini, E.; De Francesco, R.; La Monica, N. NS3 is a serine protease required for processing of hepatitis C virus polyprotein. J. Virol. 1993, 67, 4017–4026. [Google Scholar] [CrossRef]
- Suzich, J.A.; Tamura, J.K.; Palmer-Hill, F.; Warrener, P.; Grakoui, A.; Rice, C.M.; Feinstone, S.M.; Collett, M.S. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J. Virol. 1993, 67, 6152–6158. [Google Scholar] [CrossRef]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Bhattacherjee, V.; Prescott, L.E.; Pike, I.; Rodgers, B.; Bell, H.; El-Zayadi, A.R.; Kew, M.C.; Conradie, J.; Lin, C.K.; Marsden, H.; et al. Use of NS-4 peptides to identify type-specific antibody to hepatitis C virus genotypes 1, 2, 3, 4, 5 and 6. J. Gen. Virol. 1995, 76, 1737–1748. [Google Scholar] [CrossRef]
- Hugle, T.; Fehrmann, F.; Bieck, E.; Kohara, M.; Krausslich, H.G.; Rice, C.M.; Blum, H.E.; Moradpour, D. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology 2001, 284, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Aizaki, H.; He, J.W.; Lai, M.M. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 2004, 78, 3480–3488. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Sakuma, I.; Asahina, Y.; Kurosaki, M.; Murakami, T.; Yamamoto, C.; Ogura, Y.; Izumi, N.; Marumo, F.; Sato, C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N. Engl. J. Med. 1996, 334, 77–81. [Google Scholar] [CrossRef]
- Gale, M.J., Jr.; Korth, M.J.; Tang, N.M.; Tan, S.L.; Hopkins, D.A.; Dever, T.E.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 1997, 230, 217–227. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 28, e1000035. [Google Scholar] [CrossRef]
- Behrens, S.E.; Tomei, L.; De Francesco, R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996, 15, 12–22. [Google Scholar] [CrossRef]
- Lohmann, V.; Roos, A.; Körner, F.; Koch, J.O.; Bartenschlager, R. Biochemical and kinetic analyses of NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Virology 1998, 249, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Lohmann, V.; Kaul, A.; Krieger, N.; Rinck, G.; Rutter, G.; Strand, D.; Bartenschlager, R. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 2002, 76, 4008–4021. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9294–9299. [Google Scholar] [CrossRef]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, C.; Chang, K.S.; Jiang, J.; Ahn, B.C.; Wakita, T.; Liang, T.J.; Luo, G. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J. Virol. 2005, 79, 13963–13973. [Google Scholar] [CrossRef] [PubMed]
- Heller, T.; Saito, S.; Auerbach, J.; Williams, T.; Moreen, T.R.; Jazwinski, A.; Cruz, B.; Jeurkar, N.; Sapp, R.; Luo, G.; Liang, T.J. An in vitro model of hepatitis C virion production. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 2579–2583. [Google Scholar] [CrossRef]
- Kanda, T.; Basu, A.; Steele, R.; Wakita, T.; Ryerse, J.S.; Ray, R.; Ray, R.B. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J. Virol. 2006, 80, 4633–4639. [Google Scholar] [CrossRef]
- Yi, M.; Villanueva, R.A.; Thomas, D.L.; Wakita, T.; Lemon, S.M. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2310–2315. [Google Scholar] [CrossRef]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Liang, C.; Chen, W.L.; Jung, J.U.; Ou, J.H. Perturbation of autophagic pathway by hepatitis C virus. Autophagy 2008, 4, 830–831. [Google Scholar] [CrossRef]
- Dreux, M.; Chisari, F.V. Autophagy proteins promote hepatitis C virus replication. Autophagy 2009, 5, 1224–1225. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef]
- Hsieh, T.Y.; Matsumoto, M.; Chou, H.C.; Schneider, R.; Hwang, S.B.; Lee, A.S.; Lai, M.M. Hepatitis C virus core protein interacts with heterogeneous nuclear ribonucleoprotein K. J. Biol. Chem. 1998, 273, 17651–17659. [Google Scholar] [CrossRef]
- Jin, D.Y.; Wang, H.L.; Zhou, Y.; Chun, A.C.; Kibler, K.V.; Hou, Y.D.; Kung, H.; Jeang, K.T. Hepatitis C virus core protein-induced loss of LZIP function correlates with cellular transformation. EMBO J. 2000, 19, 729–740. [Google Scholar] [CrossRef]
- Aoki, H.; Hayashi, J.; Moriyama, M.; Arakawa, Y.; Hino, O. Hepatitis C virus core protein interacts with 14-3-3 protein and activates the kinase Raf-1. J. Virol. 2000, 74, 1736–1741. [Google Scholar] [CrossRef]
- You, L.R.; Chen, C.M.; Lee, Y.H. Hepatitis C virus core protein enhances NF-kappaB signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J. Virol. 1999, 73, 1672–1681. [Google Scholar] [CrossRef]
- Ray, R.B.; Steele, R.; Meyer, K.; Ray, R. Transcriptional repression of p53 promoter by hepatitis C virus core protein. J. Biol. Chem. 1997, 272, 10983–10986. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Steele, R.; Meyer, K.; Ray, R. Hepatitis C virus core protein represses p21WAF1/Cip1/Sid1 promoter activity. Gene 1998, 208, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yoshida, I.; Takamatsu, M.; Ishido, S.; Fujita, T.; Oka, K.; Hotta, H. Complex formation between hepatitis C virus core protein and p21Waf1/Cip1/Sdi1. Biochem. Biophys. Res. Commun. 2000, 273, 479–484. [Google Scholar] [CrossRef]
- Liao, Q.J.; Ye, L.B.; Timani, K.A.; She, Y.L.; Yang, X.J.; Ye, L.; Wu, Z.H. Hepatitis C virus non-structural 5A protein can enhance full-length core protein-induced nuclear factor-kappaB activation. World J. Gastroenterol. 2005, 11, 6433–6439. [Google Scholar] [CrossRef]
- Mamiya, N.; Worman, H.J. Hepatitis C virus core protein binds to a DEAD box RNA helicase. J. Biol. Chem. 1999, 274, 15751–15756. [Google Scholar] [CrossRef]
- Owsianka, A.M.; Patel, A.H. Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology 1999, 257, 330–340. [Google Scholar] [CrossRef]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; Okamoto, E.; Hayashi, N.; Hori, M. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar] [CrossRef]
- Shrivastava, A.; Manna, S.K.; Ray, R.; Aggarwal, B.B. Ectopic expression of hepatitis C virus core protein differentially regulates nuclear transcription factors. J. Virol. 1998, 72, 9722–9728. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kuroki, M.; Abe, K.; Dansako, H.; Ikeda, M.; Wakita, T.; Kato, N. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J. Virol. 2007, 81, 13922–13926. [Google Scholar] [CrossRef] [PubMed]
- Angus, A.G.; Dalrymple, D.; Boulant, S.; McGivern, D.R.; Clayton, R.F.; Scott, M.J.; Adair, R.; Graham, S.; Owsianka, A.M.; Targett-Adams, P.; Li, K.; Wakita, T.; McLauchlan, J.; Lemon, S.M.; Patel, A.H. Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. J. Gen. Virol. 2010, 91, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Wagayama, H.; Shiraki, K.; Sugimoto, K.; Ito, T.; Fujikawa, K.; Yamanaka, T.; Takase, K.; Nakano, T. High expression of p21WAF1/CIP1 is correlated with human hepatocellular carcinoma in patients with hepatitis C virus-associated chronic liver diseases. Hum. Pathol. 2002, 33, 429–434. [Google Scholar] [CrossRef]
- Kwun, H.J.; Jang, K.L. Dual effects of hepatitis C virus Core protein on the transcription of cyclin-dependent kinase inhibitor p21 gene. J. Viral. Hepat. 2003, 10, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Ludwig, S.; Ehrhardt, C.; Albrecht, U.; Erhardt, A.; Schaper, F.; Heinrich, P.C.; Häussinger, D. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003, 17, 488–490. [Google Scholar] [CrossRef]
- Vlotides, G.; Sörensen, A.S.; Kopp, F.; Zitzmann, K.; Cengic, N.; Brand, S.; Zachoval, R.; Auernhammer, C.J. SOCS-1 and SOCS-3 inhibit IFN-alpha-induced expression of the antiviral proteins 2,5-OAS and MxA. Biochem. Biophys. Res. Commun. 2004, 320, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Choe, W.H.; Hiasa, Y.; Kamegaya, Y.; Blackard, J.T.; Schmidt, E.V.; Chung, R.T. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology 2005, 128, 1034–1041. [Google Scholar] [CrossRef]
- Fukutomi, T.; Zhou, Y.; Kawai, S.; Eguchi, H.; Wands, J.R.; Li, J. Hepatitis C virus core protein stimulates hepatocyte growth: correlation with upregulation of wnt-1 expression. Hepatology 2005, 41, 1096–1105. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; McCormick, C.; Harris, M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 2005, 79, 5006–5016. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Choi, S.H.; Kang, S.M.; Kang, J.I.; Ahn, B.Y.; Kim, H.; Jung, G.; Choi, K.Y.; Hwang, S.B. Nonstructural 5A protein activates beta-catenin signaling cascades: implication of hepatitis C virus-induced liver pathogenesis. J. Hepatol. 2009, 51, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Hanada, T.; Tokuhisa, T.; Kosai, K.; Sata, M.; Kohara, M.; Yoshimura, A. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J. Exp. Med. 2002, 196, 641–653. [Google Scholar] [CrossRef]
- Ray, R.B.; Ray, R. Perspective in medical virology. In Virus and liver cancer; Tabor, E., Ed.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2002; Volume 6, pp. 85–92. [Google Scholar]
- Bergqvist, A.; Rice, C.M. Transcriptional activation of the interleukin-2 promoter by hepatitis C virus core protein. J. Virol. 2001, 75, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.M.; Ware, C.F. Hepatitis C virus core protein: possible roles in viral pathogenesis. Curr. Top. Microbiol. Immunol. 2000, 242, 117–134. [Google Scholar] [PubMed]
- Basu, A.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus core protein is necessary for the maintenance of immortalized human hepatocytes. Virology 2002, 298, 53–62. [Google Scholar] [CrossRef]
- Lin, J.; Tang, H.; Jin, X.; Jia, G.; Hsieh, J.T. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene 2002, 21, 3082–3088. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Turkson, J.; Hassanein, T.; Siddiqui, A. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication. J. Virol. 2005, 79, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Sarcar, B.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A mediated STAT3 activation requires co-operation of Jak1 kinase. Virology 2004, 322, 51–60. [Google Scholar] [CrossRef]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef]
- Agarwal, M.L.; Agarwal, A.; Taylor, W.R.; Stark, G.R. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 8493–8497. [Google Scholar] [CrossRef]
- Harrington, E.A.; Bruce, J.L.; Harlow, E.; Dyson, N. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 11945–11950. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef]
- Lan, K.H.; Sheu, M.L.; Hwang, S.J.; Yen, S.H.; Chen, S.Y.; Wu, J.C.; Wang, Y.J.; Kato, N.; Omata, M.; Chang, F.Y.; Lee, S.D. HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 2002, 21, 4801–4811. [Google Scholar] [CrossRef]
- Qadri, I.; Iwahashi, M.; Simon, F. Hepatitis C virus NS5A protein binds TBP and p53, inhibiting their DNA binding and p53 interactions with TBP and ERCC3. Biochim. Biophys. Acta. 2002, 1592, 193–204. [Google Scholar] [CrossRef]
- Wu, S.C.; Chang, S.C.; Wu, H.Y.; Liao, P.J.; Chang, M.F. Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J. Biol. Chem. 2008, 283, 29396–29404. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Lo, S.Y.; Chen, M.; Wu, K.; Fung, Y.K.; Ou, J.H. Activation of p53 tumor suppressor by hepatitis C virus core protein. Virology 1999, 264, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Giambartolomei, S.; Cupelli, F.; Merlo, P.; Fontemaggi, G.; Spaziani, A.; Balsano, C. Physical and functional interaction between HCV core protein and the different p73 isoforms. Oncogene 2003, 22, 2573–2580. [Google Scholar] [CrossRef]
- Cho, J.; Baek, W.; Yang, S.; Chang, J.; Sung, Y.C.; Suh, M. HCV core protein modulates Rb pathway through pRb down-regulation and E2F-1 up-regulation. Biochim. Biophys. Acta. 2001, 1538, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Munakata, T.; Liang, Y.; Kim, S.; McGivern, D.R.; Huibregtse, J.; Nomoto, A.; Lemon, S.M. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 2007, 3, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Liu, J.C.; McNamara, G.; Levine, A.; Duan, L.; Lai, M.M. Hepatitis C virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. J. Virol. 2009, 83, 12590–12600. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, C.N.; Bosco, E.E.; Fox, S.R.; Okaya, T.; Tarapore, P.; Schwemberger, S.J.; Babcock, G.F.; Lentsch, A.B.; Fukasawa, K.; Knudsen, E.S. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005, 65, 4568–4577. [Google Scholar] [CrossRef]
- Miyamoto, H.; Moriishi, K.; Moriya, K.; Murata, S.; Tanaka, K.; Suzuki, T.; Miyamura, T.; Koike, K.; Matsuura, Y. Involvement of the PA28gamma-dependent pathway in insulin resistance induced by hepatitis C virus core protein. J. Virol. 2007, 81, 1727–1735. [Google Scholar] [CrossRef]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Gonzalez, F.J.; Aoyama, T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Invest. 2008, 118, 683–694. [Google Scholar] [PubMed]
- Koike, K. Steatosis, liver injury, and hepatocarcinogenesis in hepatitis C viral infection. J. Gastroenterol. 2009, 44, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Meyer, K.; Ray, R. Hepatitis C virus core protein promotes immortalization of primary human hepatocytes. Virology 2000, 271, 197–204. [Google Scholar] [CrossRef]
- Basu, A.; Meyer, K.; Lai, K.K.; Saito, K.; Di Bisceglie, A.M.; Grosso, L.E.; Ray, R.B.; Ray, R. Microarray analyses and molecular profiling of Stat3 signaling pathway induced by hepatitis C virus core protein in human hepatocytes. Virology 2006, 349, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Sakamuro, D.; Furukawa, T.; Takegami, T. Hepatitis C virus nonstructural protein NS3 transforms NIH 3T3 cells. J. Virol. 1995, 69, 3893–3896. [Google Scholar] [CrossRef] [PubMed]
- Zemel, R.; Gerechet, S.; Greif, H.; Bachmatove, L.; Birk, Y.; Golan-Goldhirsh, A.; Kunin, M.; Berdichevsky, Y.; Benhar, I.; Tur-Kaspa, R. Cell transformation induced by hepatitis C virus NS3 serine protease. J. Viral. Hepat. 2001, 8, 96–102. [Google Scholar] [CrossRef]
- He, Q.Q.; Cheng, R.X.; Sun, Y.; Feng, D.Y.; Chen, Z.C.; Zheng, H. Hepatocyte transformation and tumor development induced by hepatitis C virus NS3 c-terminal deleted protein. World J. Gastroenterol. 2003, 9, 474–478. [Google Scholar] [CrossRef]
- Ishido, S.; Fujita, T.; Hotta, H. Complex formation of NS5B with NS3 and NS4A proteins of hepatitis C virus. Biochem. Biophys. Res. Commun. 1998, 244, 35–40. [Google Scholar] [CrossRef]
- Zekri, A.R.; Ashour, M.S.; Alam, El-Din. H.M.; Khaled, H.M.; Abu-Shady, M. Cytokines as markers for disease progression in HCV associated liver diseases. World J. Gastroenterol. 2005, 11, 6624–6630. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.T.; MacDonald, A.J.; Rowan, A.G.; Mills, K.H. Hepatitis C virus non-structural protein 4 suppresses Th1 responses by stimulating IL-10 production from monocytes. Eur. J. Immunol. 2003, 33, 3448–3457. [Google Scholar] [CrossRef]
- Saito, K.; Ait-Goughoulte, M.; Truscott, S.M.; Meyer, K.; Blazevic, A.; Abate, G.; Ray, R.B.; Hoft, D.F.; Ray, R. Hepatitis C virus inhibits cell surface expression of HLA-DR, prevents dendritic cell maturation, and induces interleukin-10 production. J. Virol. 2008, 82, 3320–3328. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.R.; Bahnassy, A.A.; Abdel-Wahab, S.A.; Khafagy, M.M.; Loutfy, S.A.; Radwan, H.; Shaarawy, S.M. Expression of pro- and anti-inflammatory cytokines in relation to apoptotic genes in Egyptian liver disease patients associated with HCV-genotype-4. J. Gastroenterol. Hepatol. 2009, 24, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; Jessup, J.M.; Shetty, K.; Zasloff, M.; Mishra, B.; Reddy, E.P.; Johnson, L.; Mishra, L. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2445–2450. [Google Scholar] [CrossRef]
- Malaguarnera, M.; Di Fazio, I.; Ferlito, L.; Pistone, G.; Laurino, A.; Vinci, E.; Mazzoleni, G. Increase of serum beta2-microglobulin in patients affected by HCV correlated hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2000, 12, 937–939. [Google Scholar] [CrossRef]
- Ait-Goughoulte, M.; Banerjee, A.; Meyer, K.; Mazumdar, B.; Saito, K.; Ray, R.B.; Ray, R. Hepatitis C virus core protein interacts with fibrinogen-beta and attenuates cytokine stimulated acute-phase response. Hepatology 2010, 51, 1505–1513. [Google Scholar] [CrossRef]
- Tilg, H.; Wilmer, A.; Vogel, W.; Herold, M.; Nolchen, B.; Judmaier, G.; Huber, C. Serum levels of cytokines in chronic liver diseases. Gastroenterology 1992, 103, 264–274. [Google Scholar] [CrossRef]
- Torre, D.; Zeroli, M.; Giola, G.; Ferrario, G.; Fiori, P.; Bonetta, G.; Tambini, R. Serum levels of interleukin-1 alpha, interleukin-1 beta, interleukin-6, and tumor necrosis factor in patients with acute viral hepatitis. Clin. Infect. Dis. 1994, 18, 194–198. [Google Scholar] [CrossRef]
- Taniguchi, H.; Kato, N.; Otsuka, M.; Goto, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein upregulates transforming growth factor-beta 1 transcription. J. Med. Virol. 2004, 72, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Hur, W.; Wang, J.S.; Jang, J.W.; Kim, C.W.; Bae, S.H.; Jang, S.K.; Yang, S.H.; Sung, Y.C.; Kwon, O.J.; Yoon, S.K. HCV core protein promotes liver fibrogenesis via up-regulation of CTGF with TGF-beta1. Exp. Mol. Med. 2005, 37, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Hwang, S.B. Modulation of the transforming growth factor-beta signal transduction pathway by hepatitis C virus nonstructural 5A protein. J. Biol. Chem. 2006, 281, 7468–7478. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; Okazaki, K.; Seki, T. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef]
- Battaglia, S.; Benzoubir, N.; Nobilet, S.; Charneau, P.; Samuel, D.; Zignego, A.L.; Atfi, A.; Bréchot, C.; Bourgeade, M.F. Liver cancer-derived hepatitis C virus core proteins shift TGF-beta responses from tumor suppression to epithelial-mesenchymal transition. PLoS One 2009, 4, e4355. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Selimovic, D.; Ghozlan, H.; Abdel-kader, O. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 2009, 49, 1469–1482. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus core protein augments androgen receptor-mediated signaling. J. Virol. 2008, 82, 11066–11072. [Google Scholar] [CrossRef]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005, 13, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007, 21, 2474–2485. [Google Scholar] [CrossRef]
- Machida, K.; Cheng, K.T.; Sung, V.M.; Lee, K.J.; Levine, A.M.; Lai, M.M. Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J. Virol. 2004, 78, 8835–8843. [Google Scholar] [CrossRef]
- Castello, G.; Scala, S.; Palmieri, G.; Curley, S.A.; Izzo, F. HCV-related hepatocellular carcinoma: From chronic inflammation to cancer. Clin. Immunol. 2010, 134, 237–250. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Fan, J.; Ren, H.; Jia, N.; Fei, E.; Zhou, T.; Jiang, P.; Wu, M.; Wang, G. DJ-1 decreases Bax expression through repressing p53 transcriptional activity. J. Biol. Chem. 2008, 283, 4022–4030. [Google Scholar] [CrossRef]
- Thompson, C.B. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef]
- Canbay, A.; Higuchi, H.; Bronk, S.F.; Taniai, M.; Sebo, T.J.; Gores, G.J. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology 2002, 123, 1323–1330. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Pianko, S.; Patella, S.; Ostapowicz, G.; Desmond, P.; Sievert, W. Fas-mediated hepatocyte apoptosis is increased by hepatitis C virus infection and alcohol consumption, and may be associated with hepatic fibrosis: mechanisms of liver cell injury in chronic hepatitis C virus infection. J. Viral. Hepat. 2001, 8, 406–413. [Google Scholar] [CrossRef]
- Mundt, B.; Wirth, T.; Zender, L.; Waltemathe, M.; Trautwein, C.; Manns, M.P.; Kühnel, F.; Kubicka, S. Tumour necrosis factor related apoptosis inducing ligand (TRAIL) induces hepatic steatosis in viral hepatitis and after alcohol intake. Gut 2005, 54, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Riordan, S.M.; Skinner, N.A.; Kurtovic, J.; Locarnini, S.; McIver, C.J.; Williams, R.; Visvanathan, K. Tolllike receptor expression in chronic hepatitis C: correlation with proinflammatory cytokine levels and liver injury. Inflamm. Res. 2006, 55, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Tsukiyama-Kohara, K.; Seike, E.; Toné, S.; Shibasaki, F.; Shimizu, M.; Takahashi, H.; Hayashi, Y.; Funata, N.; Taya, C.; Yonekawa, H.; Kohara, M. Inhibition of cytochrome c release in Fas-mediated signaling pathway in transgenic mice induced to express hepatitis C viral proteins. J. Biol. Chem. 2001, 276, 12140–12146. [Google Scholar] [CrossRef]
- Hara, Y.; Hino, K.; Okuda, M.; Furutani, T.; Hidaka, I.; Yamaguchi, Y.; Korenaga, M.; Li, K.; Weinman, S.A.; Lemon, S.M.; Okita, K. Hepatitis C virus core protein inhibits deoxycholic acid-mediated apoptosis despite generating mitochondrial reactive oxygen species. J. Gastroenterol. 2006, 41, 257–268. [Google Scholar] [CrossRef]
- Chou, A.H.; Tsai, H.F.; Wu, Y.Y.; Hu, C.Y.; Hwang, L.H.; Hsu, P.I.; Hsu, P.N. Hepatitis C virus core protein modulates TRAIL-mediated apoptosis by enhancing Bid cleavage and activation of mitochondria apoptosis signaling pathway. J. Immunol. 2005, 174, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Zhou, X.Y.; Phillips, N.J.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein impairs TNF-mediated hepatic apoptosis, but not by an anti-FAS antibody, in transgenic mice. Virology 2002, 294, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Gorke, S.; Rau, S.J.; Zeisel, M.B.; Hildt, E.; Himmelsbach, K.; Carvajal-Yepes, M.; Huber, R.; Wakita, T.; Schmitt-Graeff, A.; Royer, C.; Blum, H.E.; Fischer, R.; Baumert, T.F. Hepatitis C virus infection sensitizes human hepatocytes to TRAIL-induced apoptosis in a caspase 9-dependent manner. J. Immunol. 2008, 181, 4926–4935. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Basu, A.; Saito, K.; Ray, R.B.; Ray, R. Inhibition of hepatitis C virus core protein expression in immortalized human hepatocytes induces cytochrome c-independent increase in Apaf-1 and caspase-9 activation for cell death. Virology 2005, 336, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Meyer, K.; Steele, R.; Shrivastava, A.; Aggarwal, B.B.; Ray, R. Inhibition of tumor necrosis factor (TNF-alpha)-mediated apoptosis by hepatitis C virus core protein. J. Biol. Chem. 1998, 273, 2256–2259. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Meyer, K.; Warner, R.; Basu, A.; Ray, R.B.; Ray, R. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J. Virol. 2006, 80, 4372–4379. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Huang, H.J.; Cavenee, W.K. The potential of TRAIL for cancer chemotherapy. Apoptosis 2001, 6, 191–197. [Google Scholar] [CrossRef]
- Meurette, O.; Huc, L.; Rebillard, A.; Le Moigne, G.; Lagadic-Gossmann, D.; Dimanche-Boitrel, M.T. TRAIL (TNF-related apoptosis-inducing ligand) induces necrosis-like cell death in tumor cells at acidic extracellular pH. Ann. NY. Acad. Sci. 2005, 1056, 379–387. [Google Scholar] [CrossRef]
- Ray, R.B.; Meyer, K.; Ray, R. Suppression of apoptotic cell death by hepatitis C virus core protein. Virology 1996, 226, 176–182. [Google Scholar] [CrossRef]
- Banerjee, A.; Saito, K.; Meyer, K.; Banerjee, S.; Ait-Goughoulte, M.; Ray, R.B.; Ray, R. Hepatitis C virus core protein and cellular protein HAX-1 promote 5-fluorouracil-mediated hepatocyte growth inhibition. J. Virol. 2009, 83, 9663–9671. [Google Scholar] [CrossRef]
- Gong, G.Z.; Jiang, Y.F.; He, Y.; Lai, L.Y.; Zhu, Y.H.; Su, X.S. HCV NS5A abrogates p53 protein function by interfering with p53-DNA binding. World J. Gastroenterol. 2004, 10, 2223–2227. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kato, N.; Lan, K.; Yoshida, H.; Kato, J.; Goto, T.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J. Biol. Chem. 2000, 275, 34122–34130. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.H.; Sheu, M.L.; Hwang, S.J.; Yen, S.H.; Chen, S.Y.; Wu, J.C.; Wang, Y.J.; Kato, N.; Omata, M.; Chang, F.Y.; Lee, S.D. HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 2002, 21, 4801–4811. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L.; Sheu, M.L.; Yen, S.H. Hepatitis C virus NS5A as a potential viral Bcl-2 homologue interacts with Bax and inhibits apoptosis in hepatocellular carcinoma. Int. J. Cancer. 2003, 107, 65–73. [Google Scholar] [CrossRef]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; Paterlini-Bréchot, P. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef]
- Chan, S.W.; Egan, P. A Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005, 19, 1510–1512. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Tsukamoto, K.; Kimura, S.; Moriya, K.; Koike, K. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology 2004, 126, 840–848. [Google Scholar] [CrossRef]
- Knobler, H.; Zhornicky, T.; Sandler, A.; Haran, N.; Ashur, Y.; Schattner, A. Tumor necrosis factor-alpha-induced insulin resistance may mediate the hepatitis C virus-diabetes association. Am. J. Gastroenterol. 2003, 98, 2751–2756. [Google Scholar] [CrossRef]
- Sheikh, M.Y.; Choi, J.; Qadri, I.; Friedman, J.E.; Sanyal, A.J. Hepatitis C virus infection: molecular pathways to metabolic syndrome. Hepatology 2008, 47, 2127–2133. [Google Scholar] [CrossRef]
- Moucari, R.; Asselah, T.; Cazals-Hatem, D.; Voitot, H.; Boyer, N.; Ripault, M.P.; Sobesky, R.; Martinot-Peignoux, M.; Maylin, S.; Nicolas-Chanoine, M.H.; Paradis, V.; Vidaud, M.; Valla, D.; Bedossa, P.; Marcellin, P. Insulin resistance in chronic hepatitis C: association with genotypes 1 and 4, serum HCV RNA level, and liver fibrosis. Gastroenterology 2008, 134, 416–423. [Google Scholar] [CrossRef]
- Banerjee, S.; Saito, K.; Ait-Goughoulte, M.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J. Virol. 2008, 82, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Wang, J.H.; Hu, T.H.; Chen, C.H.; Chang, K.C.; Yen, Y.H.; Kuo, Y.H.; Tsai, M.C.; Lu, S.N.; Lee, C.M. Insulin resistance is associated with hepatocellular carcinoma in chronic hepatitis C infection. World J. Gastroenterol. 2010, 16, 2265–2271. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Meyer, K.; Mazumdar, B.; Ray, R.B.; Ray, R. Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin induced metabolic gene expression. J. Virol. 2010, 84, 5936–5946. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, V.; Berenguer, M.; Rayón, J.M.; Carrasco, D.; Berenguer, J. Contribution of obesity to hepatitis C-related fibrosis progression. Am. J. Gastroenterol. 2002, 97, 2408–2414. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Manzini, P.; D'Antico, S.; Vanni, E.; Longo, F.; Leone, N.; Massarenti, P.; Piga, A.; Marchesini, G.; Rizzetto, M. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology 2004, 39, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Koike, K.; Moriya, K. Metabolic aspects of hepatitis C viral infection: steatohepatitis resembling but distinct from NASH. J. Gastroenterol. 2005, 40, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Clément, S.; Pascarella, S.; Conzelmann, S.; Gonelle-Gispert, C.; Guilloux, K.; Negro, F. The hepatitis C virus core protein indirectly induces alpha-smooth muscle actin expression in hepatic stellate cells via interleukin-8. J. Hepatol. 2010, 52, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Fujie, H.; Yotsuyanagi, H.; Moriya, K.; Shintani, Y.; Tsutsumi, T.; Takayama, T.; Makuuchi, M.; Matsuura, Y.; Miyamura, T.; Kimura, S.; Koike, K. Steatosis and intrahepatic hepatitis C virus in chronic hepatitis. J. Med. Virol. 1999, 59, 141–145. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic fibrosis -- overview. Toxicology 2008, 254, 120–129. [Google Scholar] [CrossRef]
- Nieto, N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology 2006, 44, 1487–1501. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Junfang, Ji.; Wang, X.W. New kids on the block Diagnostic and prognostic microRNAs in hepatocellular carcinoma. Cancer Biology & Therapy 2009, 8, 1683–1690. [Google Scholar]
- Varnholt, H.; Drebber, U.; Schulze, F.; Wedemeyer, I.; Schirmacher, P.; Dienes, H.P.; Odenthal, M. MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology 2008, 47, 1223–1232. [Google Scholar] [CrossRef]
- Ura, S.; Honda, M.; Yamashita, T.; Ueda, T.; Takatori, H.; Nishino, R.; Sunakozaka, H.; Sakai, Y.; Horimoto, K.; Kaneko, S. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology 2009, 49, 1098–1112. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Kutay, H.; Bai, S.; Datta, J.; Motiwala, T.; Pogribny, I.; Frankel, W.; Jacob, S.T.; Ghoshal, K. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J. Cell. Biochem. 2006, 99, 671–678. [Google Scholar] [CrossRef]
- Jopling, C.L.; Norman, K.; Sarnow, P. Positive and negative modulation of viral and cellular mRNAs by liver-specific microRNA miR-122. Cold Spring Harb Symp. Quant. Biol. 2006, 71, 369–376. [Google Scholar] [CrossRef]
- Randall, G.; Panis, M.; Cooper, J.D.; Tellinghuisen, T.L.; Sukhodolets, K.E.; Pfeffer, S.; Landthaler, M.; Landgraf, P.; Kan, S.; Lindenbach, B.D.; Chien, M.; Weir, D.B.; Russo, J.J.; Ju, J.; Brownstein, M.J.; Sheridan, R.; Sander, C.; Zavolan, M.; Tuschl, T.; Rice, C.M. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12884–12889. [Google Scholar] [CrossRef]
- Lin, C.J.; Gong, H.Y.; Tseng, H.C.; Wang, W.L.; Wu, J.L. miR-122 targets an anti-apoptotic gene, Bcl-w, in human hepatocellular carcinoma cell lines. Biochem. Biophys. Res. Commun. 2008, 375, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; Zaret, K.S.; Taylor, J.M. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Wakita, T.; Yang, W. Systematic identification of microRNA and messenger RNA profiles in hepatitis C virus-infected human hepatoma cells. Virology 2010, 398, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Li, Y.; Walters, K.A.; Rosenzweig, E.R.; Lederer, S.L.; Aicher, L.D.; Proll, S.; Katze, M.G. Computational identification of hepatitis C virus associated microRNA-mRNA regulatory modules in human livers. BMC Genomics 2009, 10, 373–383. [Google Scholar] [CrossRef]
- Wang, B.; Majumder, S.; Nuovo, G.; Kutay, H.; Volinia, S.; Patel, T.; Schmittgen, T.D.; Croce, C.; Ghoshal, K.; Jacob, S.T. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology 2009, 50, 1152–1161. [Google Scholar] [CrossRef]
- Shan, Y.; Zheng, J.; Lambrecht, R.W.; Bonkovsky, H.L. Reciprocal effects of micro-RNA-122 on expression of heme oxygenase-1 and hepatitis C virus genes in human hepatocytes. Gastroenterology 2007, 133, 1166–1174. [Google Scholar] [CrossRef]
- Madhoun, M.F.; Fazili, J.; Bright, B.C.; Bader, T.; Roberts, D.N.; Bronze, M.S. Hepatitis C prevalence in patients with hepatocellular carcinoma without cirrhosis. Am. J. Med. Sci. 2010, 339, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Benvegnù, L.; Gios, M.; Boccato, S.; Alberti, A. Natural history of compensated viral cirrhosis: a prospective study on the incidence and hierarchy of major complications. Gut 2004, 53, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Koike, K. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol. 2007, 22, S108–111. [Google Scholar] [CrossRef]
- Jin, D.Y. Molecular pathogenesis of hepatitis C virus-associated hepatocellular carcinoma. Front. Biosci. 2007, 12, 222–233. [Google Scholar] [CrossRef]
- Blonski, W.; Reddy, K.R. Hepatitis C virus infection and hepatocellular carcinoma. Clin. Liver Dis. 2008, 12, 661–674. [Google Scholar] [CrossRef]
- Kalra, M.; Mayes, J.; Assefa, S.; Kaul, A.K.; Kaul, R. Role of sex steroid receptors in pathobiology of hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 5945–5961. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Matsuzaki, Y.; Abei, M.; Shoda, J.; Aikawa, T.; Tanaka, N.; Osuga, T. Multivariate analysis of risk factors for hepatocellular carcinoma in patients with hepatitis C virus-related liver cirrhosis. J. Gastroenterol. 1996, 31, 552–558. [Google Scholar] [CrossRef]
- Donato, F.; Boffetta, P.; Puoti, M. A meta-analysis of epidemiological studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma. Int. J. Cancer 1998, 75, 347–354. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 2004, 127, S35–50. [Google Scholar] [CrossRef] [PubMed]
- Garcìa-Samaniego, J.; Rodrìquez, M.; Berenguer, J.; Rodrìguez-Rosado, R.; Carbò, J.; Asensi, V.; Soriano, V. Hepatocellular carcinoma in HIV-infected patients with chronic hepatitis C. Am. J. Gastroenterol. 2001, 96, 179–183. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.; Richardson, P.A.; Everhart, J.E. The role of diabetes in hepatocellular carcinoma: a case-control study among United States Veterans. Am. J. Gastroenterol. 2001, 96, 2462–2467. [Google Scholar] [CrossRef]
- Donato, F.; Tagger, A.; Gelatti, U.; Parrinello, G.; Boffetta, P.; Albertini, A.; Decarli, A.; Travisi, P.; Ribero, M.L.; Martelli, C.; Porru, S.; Nardi, G. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am. J. Epidemiol. 2002, 155, 323–331. [Google Scholar] [CrossRef]
- Hassan, M.M.; Hwang, L.Y.; Hatten, C.J.; Swaim, M.; Li, D.; Abbruzzese, J.L.; Beasley, P.; Patt, Y.Z. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002, 36, 1206–1213. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2010 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/3.0/).
Share and Cite
Banerjee, A.; Ray, R.B.; Ray, R. Oncogenic Potential of Hepatitis C Virus Proteins. Viruses 2010, 2, 2108-2133. https://doi.org/10.3390/v2092108
Banerjee A, Ray RB, Ray R. Oncogenic Potential of Hepatitis C Virus Proteins. Viruses. 2010; 2(9):2108-2133. https://doi.org/10.3390/v2092108
Chicago/Turabian StyleBanerjee, Arup, Ratna B. Ray, and Ranjit Ray. 2010. "Oncogenic Potential of Hepatitis C Virus Proteins" Viruses 2, no. 9: 2108-2133. https://doi.org/10.3390/v2092108
APA StyleBanerjee, A., Ray, R. B., & Ray, R. (2010). Oncogenic Potential of Hepatitis C Virus Proteins. Viruses, 2(9), 2108-2133. https://doi.org/10.3390/v2092108