Inhibitors of the Hepatitis C Virus RNA-Dependent RNA Polymerase NS5B

Abstract
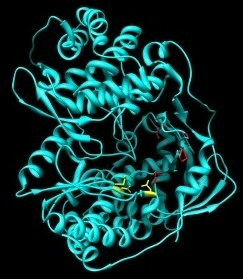
:
1. Introduction
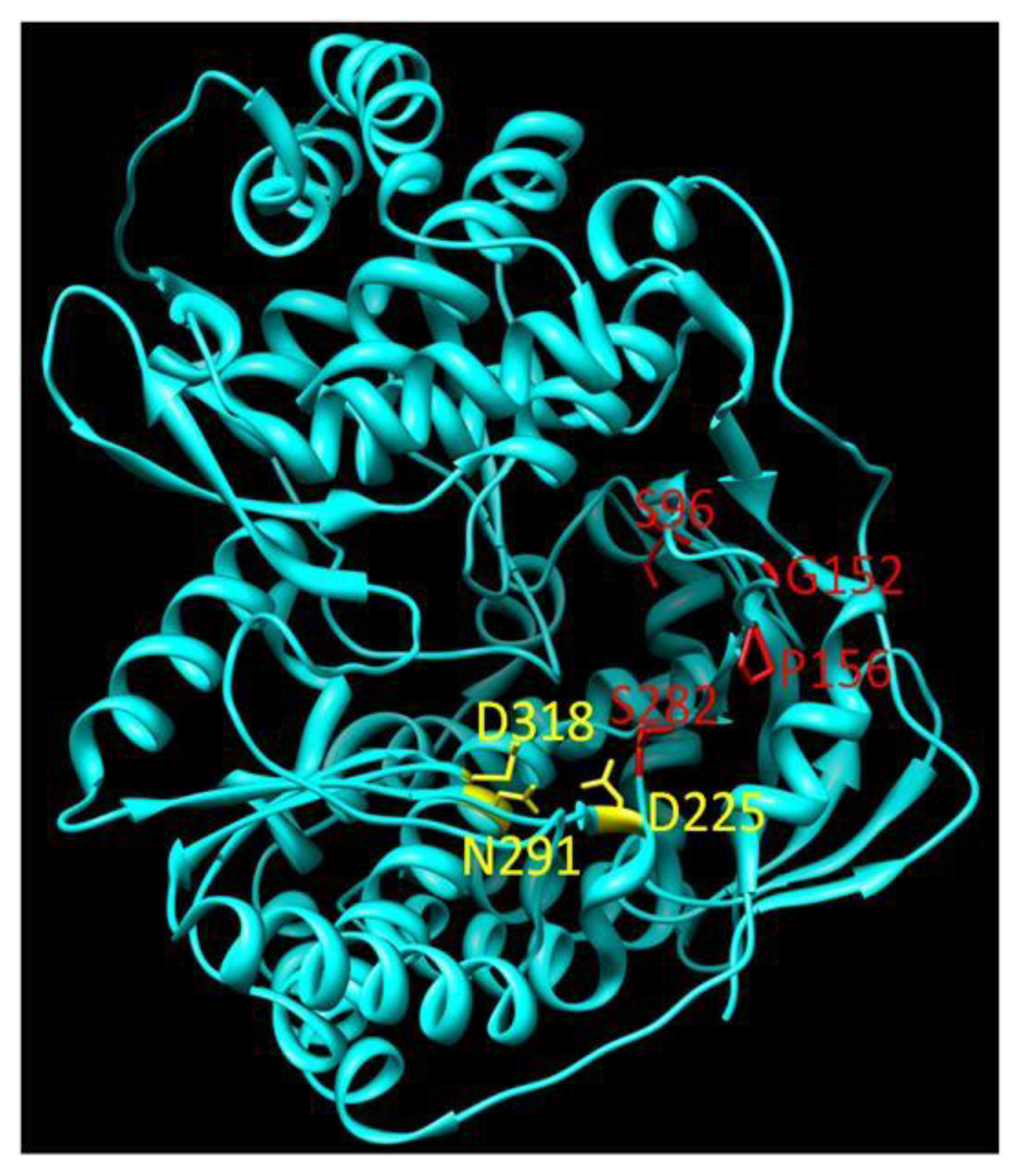
2. Structure and Function of HCV NS5B
3. Classes of Inhibitors Targeting NS5B
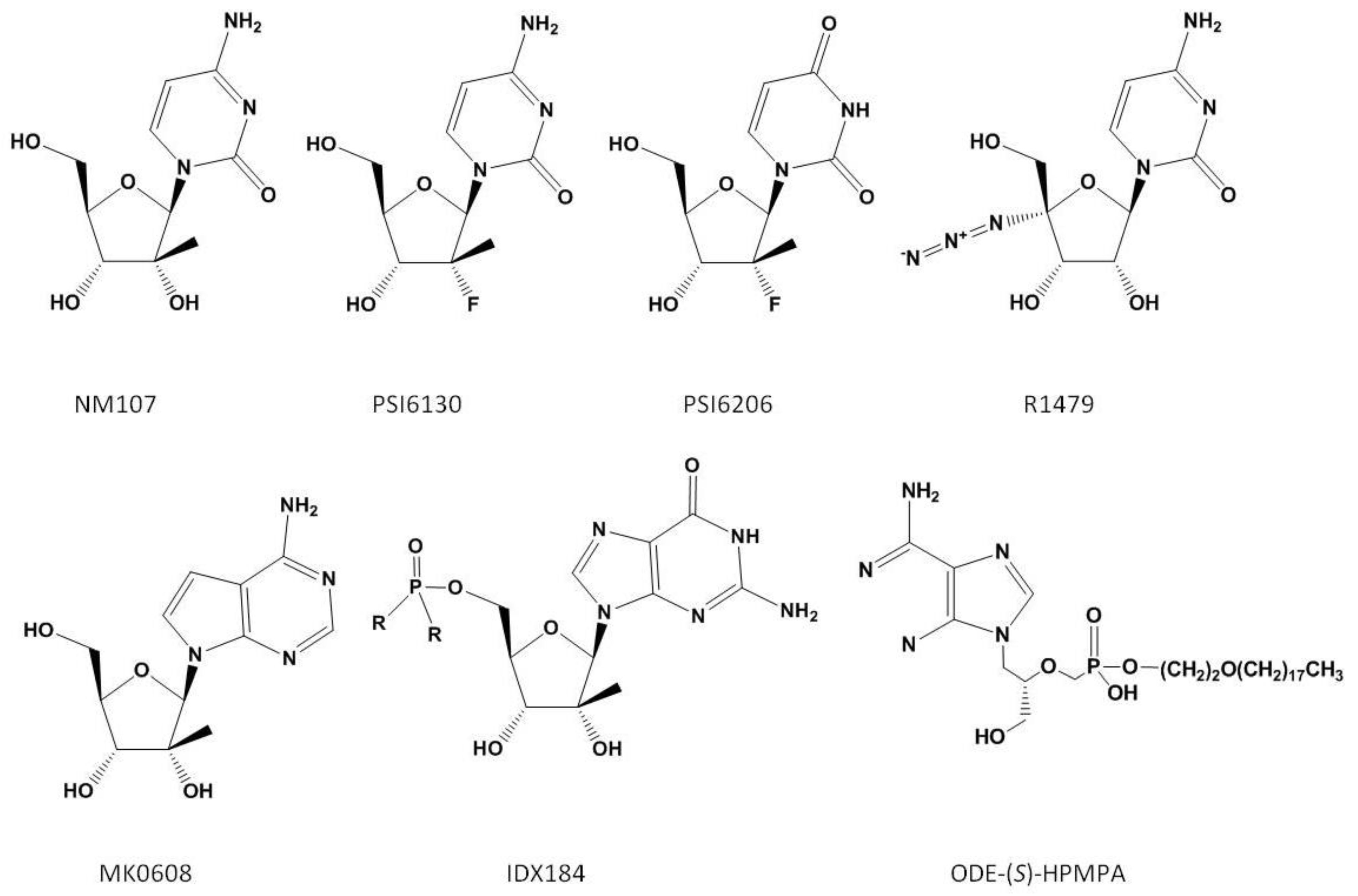
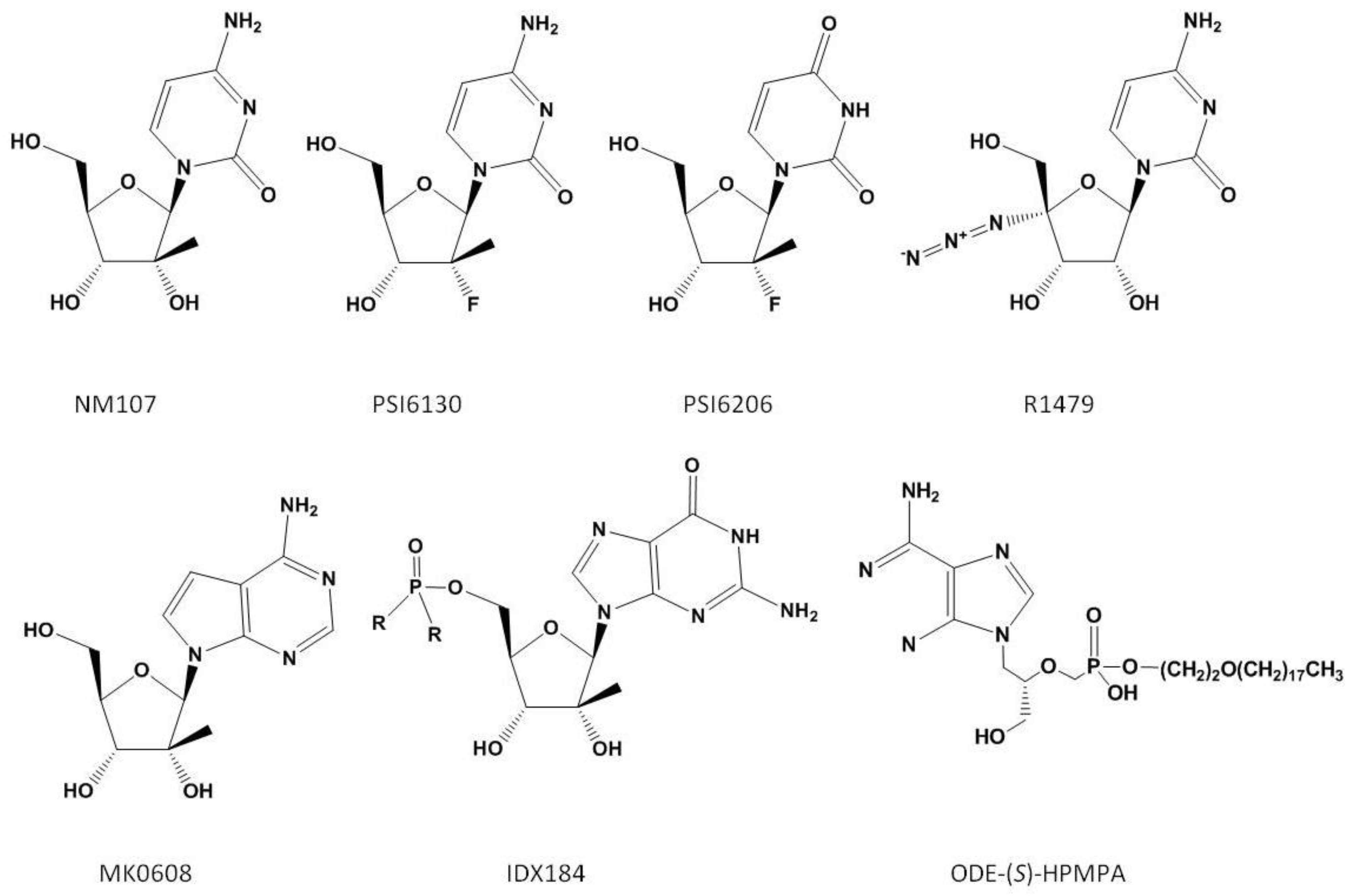
3.1. Nucleoside Analogues
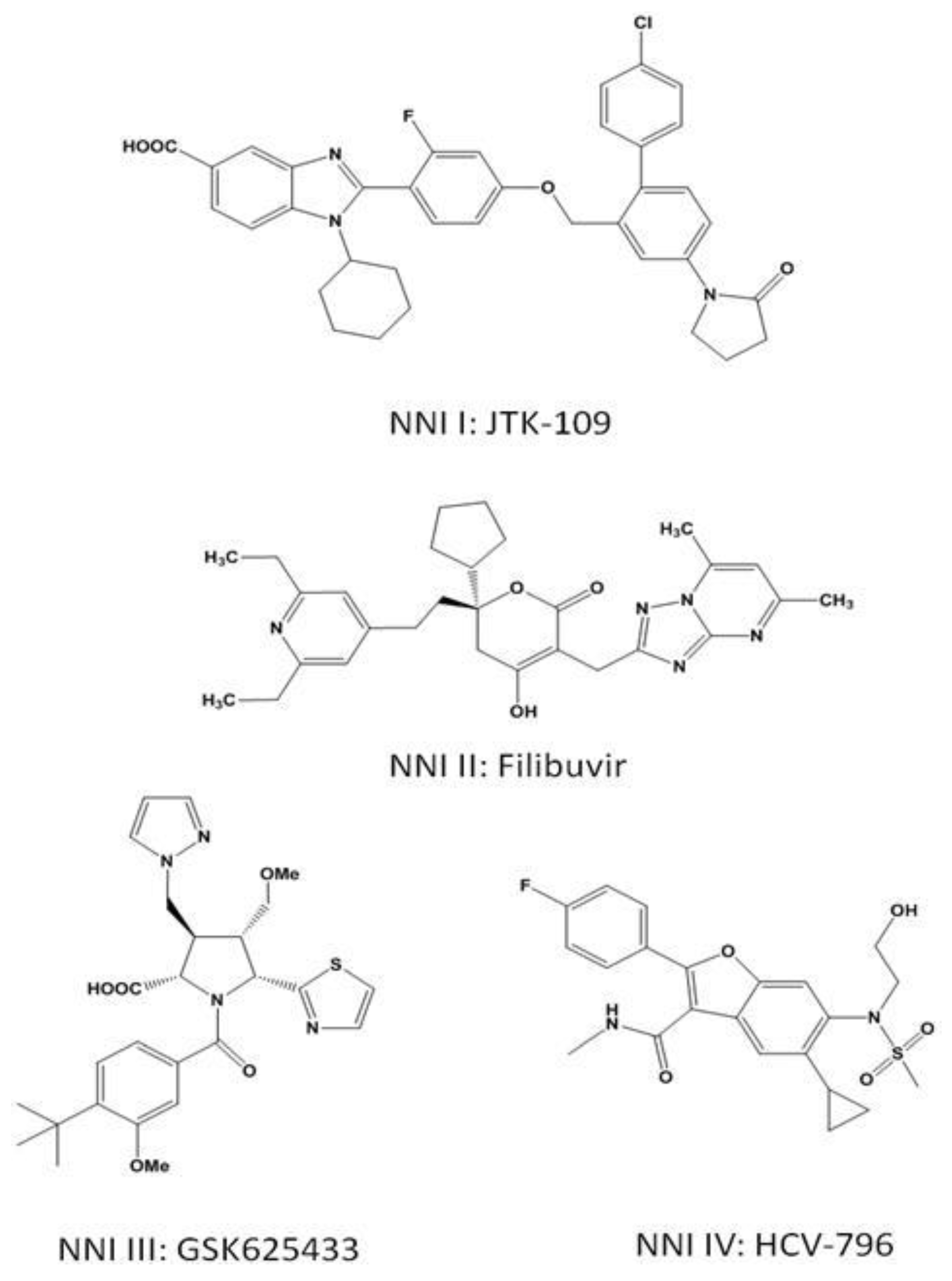
3.2. Non-Nucleoside Inhibitors
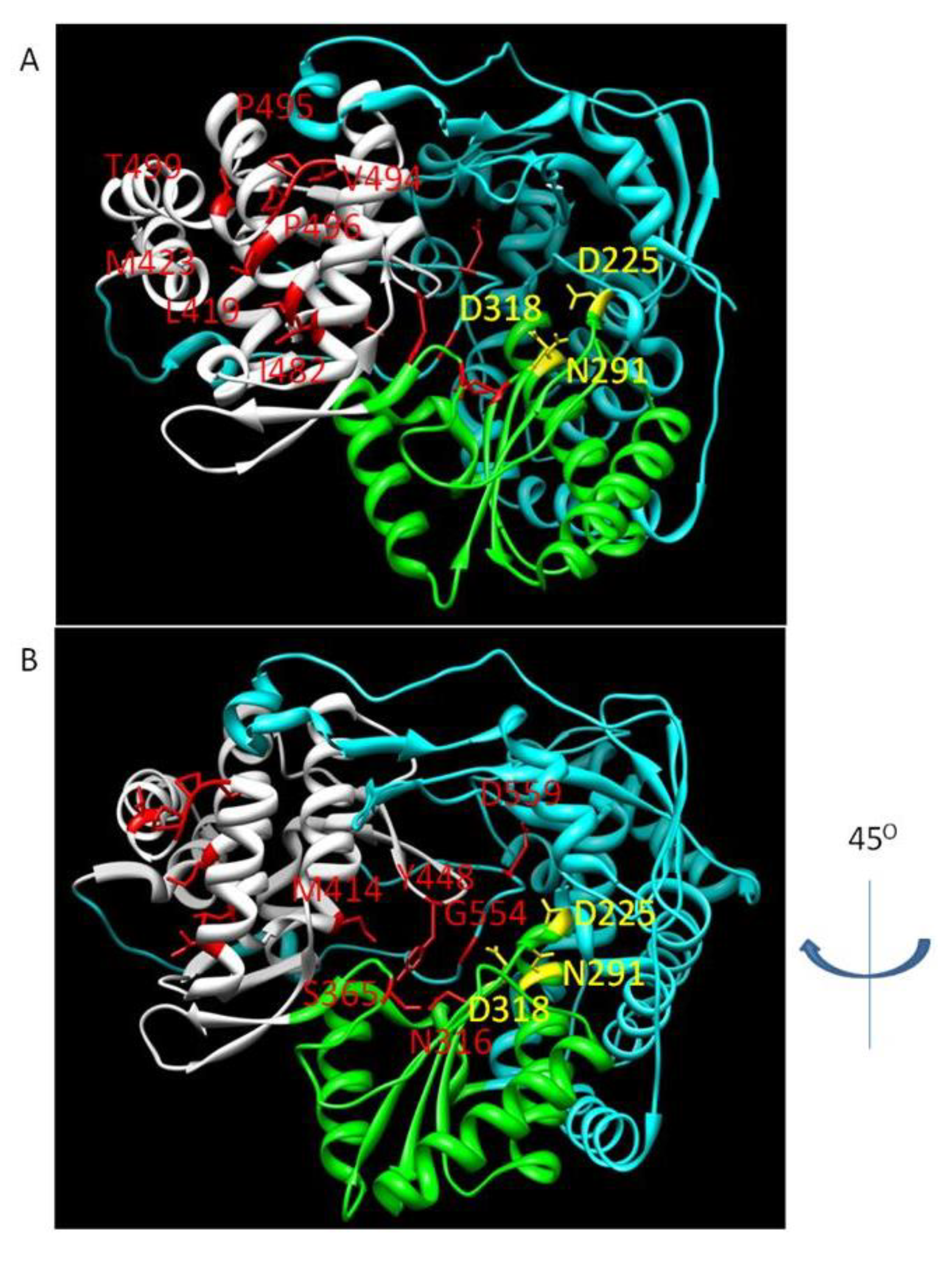
3.3. NNI Binding Sites
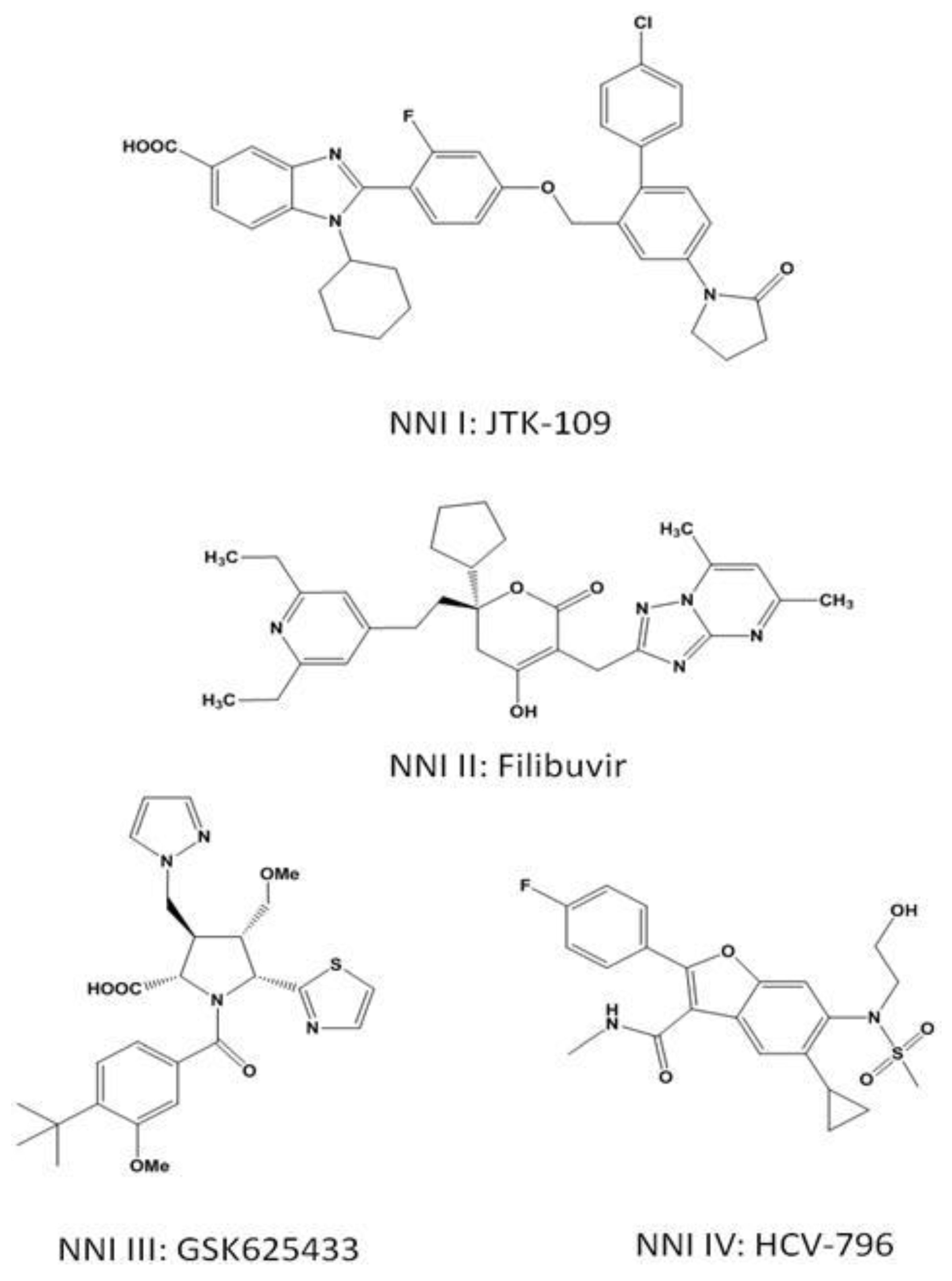
3.3.1. NNI I
Benzimidazoles and Indoles
3.3.2. NNI II
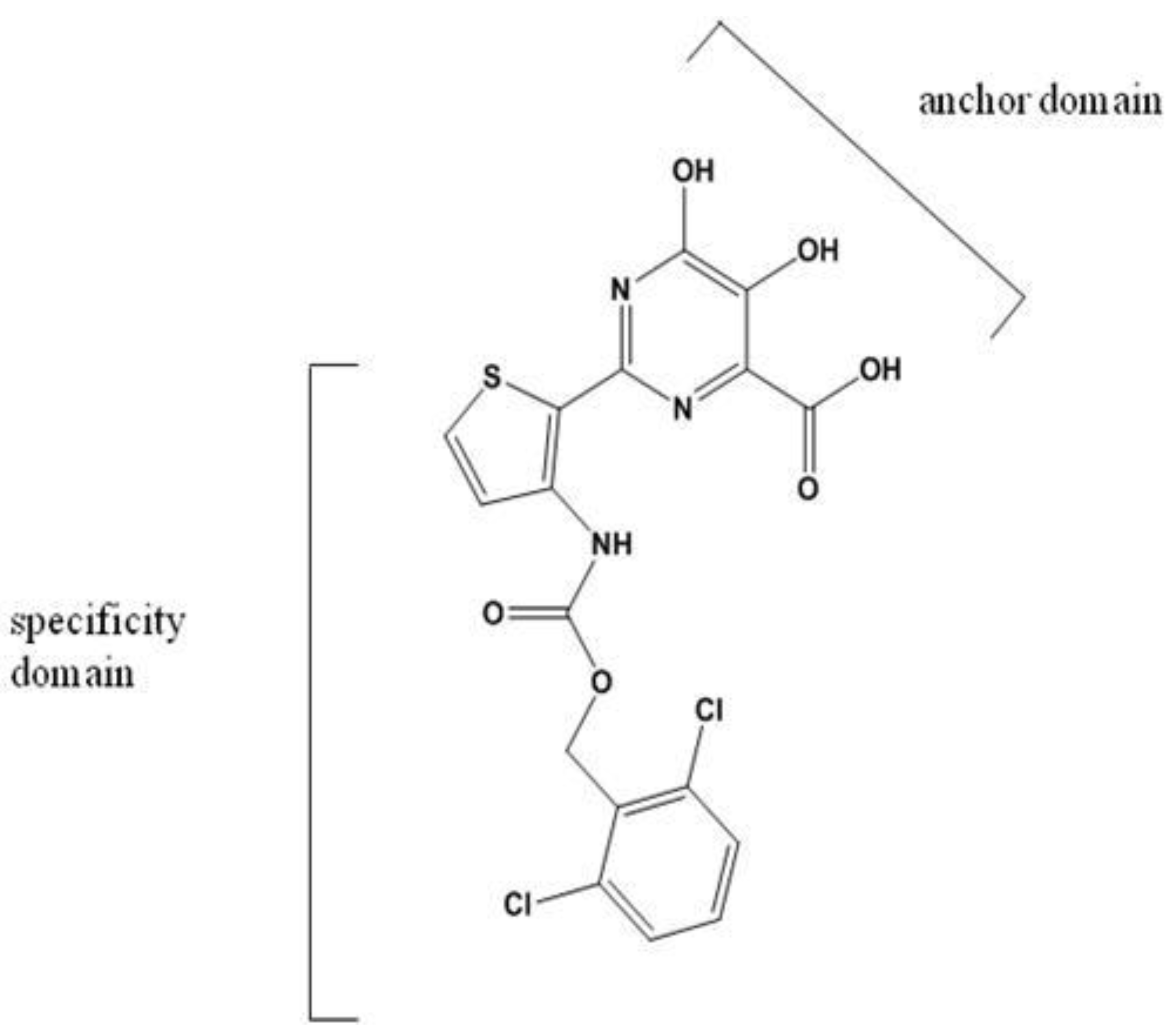
Thiophenes, Phenylalanines, Hydroxypyranones, and Pyranoindoles
3.3.3. NNI III
Benzothiadiazines and Acylpyrrolidines
3.3.4. NNI IV
Benzofurans
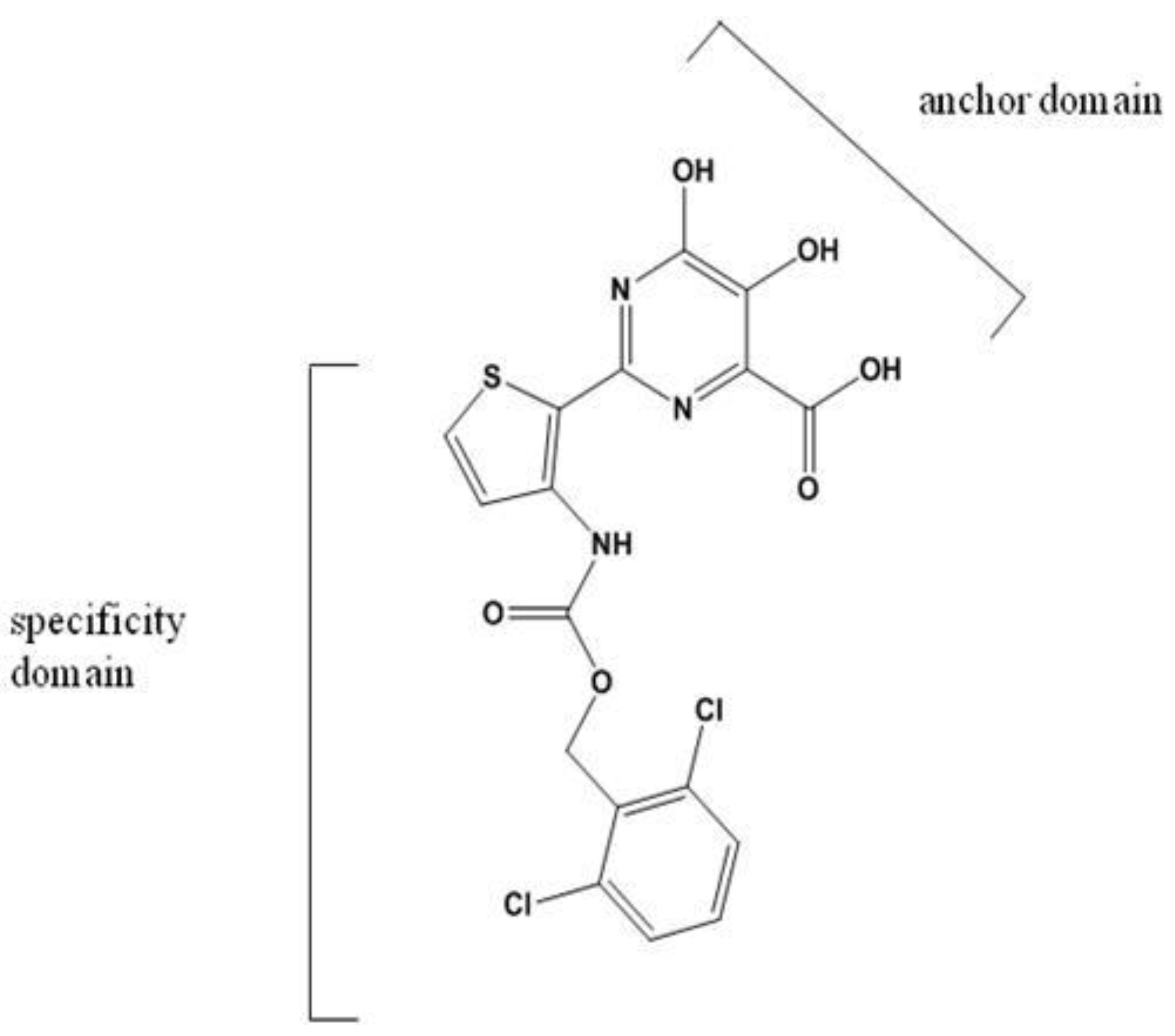
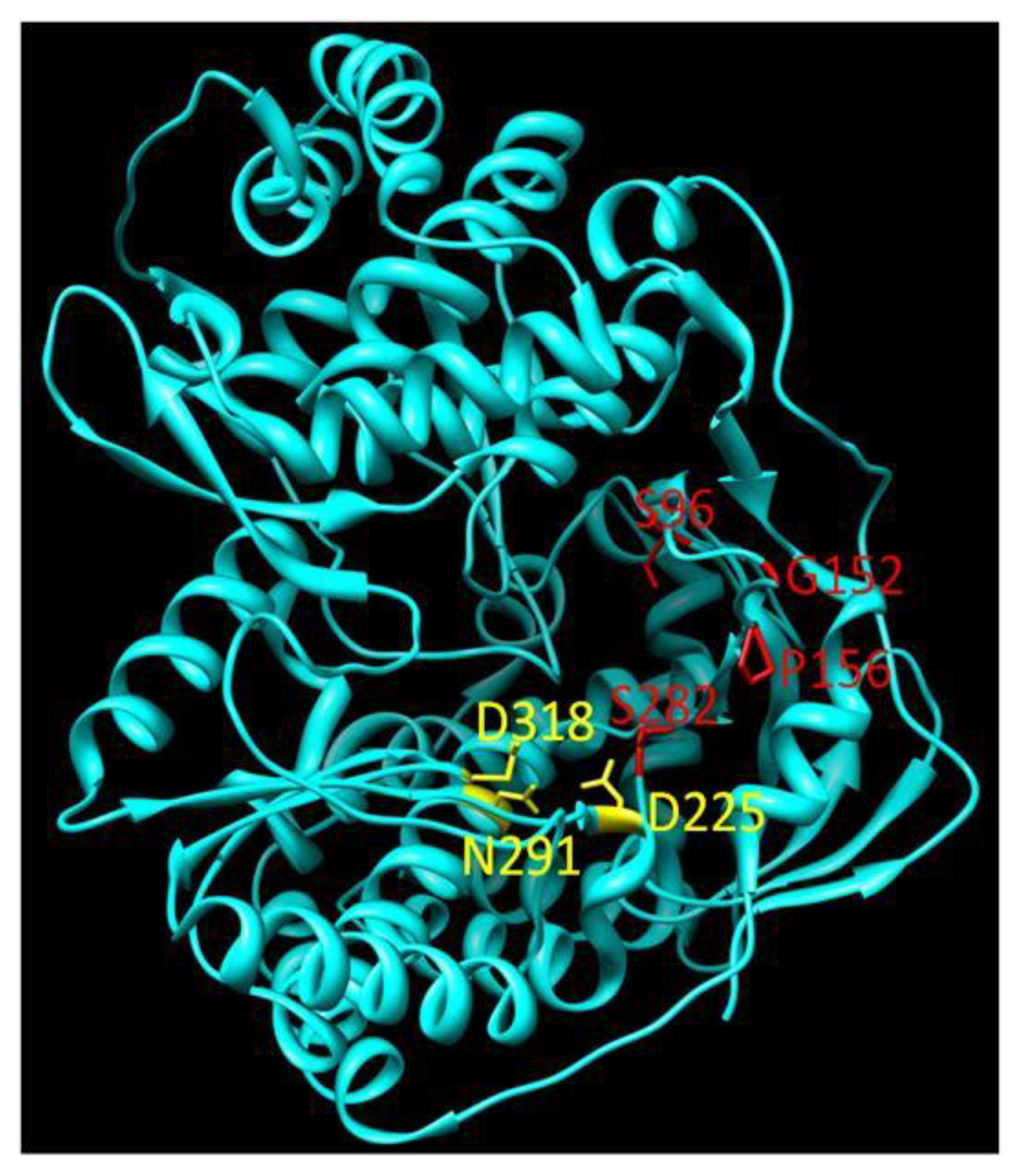
3.4. Pyrophosphate Analogues
4. Antiviral Resistance
5. Pre-Existing Mutations
6. Overcoming Resistance
7. Conclusions and Perspectives
Acknowledgements
References and Notes
- The Global Burden of Hepatitis C Working Group. Global Burden of Disease (GBD) for hepatitis C. J. Clin. Pharmacol. 2004, 44, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Chevaliez, S.; Pawlotsky, J.M. Interferon-based therapy of hepatitis C. Advan. Drug Delivery Rev. 2007, 59, 1222–1241. [Google Scholar] [CrossRef]
- Opar, A. Excitement grows for potential revolution in hepatitis C virus treatment. Nat. Rev. Drug Discov. 2010, 9, 501–503. [Google Scholar] [CrossRef]
- Flisiak, R.; Parfieniuk, A. Investigational drugs for hepatitis C. Expert Opin. Investig. Drugs 2010, 19, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; Mizokami, M.; Murphy, D.G.; Okamoto, H.; Pawlotsky, J.M.; Penin, F.; Sablon, E.; Shin, I.T.; Stuyver, L.J.; Thiel, H.J.; Viazov, S.; Weiner, A.J.; Widell, A. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Goncales, F.L., Jr.; Haussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; Craxi, A.; Lin, A.; Hoffman, J.; Yu, J. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Sette, H., Jr.; Morgan, T.R.; Balan, V.; Diago, M.; Marcellin, P.; Ramadori, G.; Bodenheimer, H., Jr.; Bernstein, D.; Rizzetto, M.; Zeuzem, S.; Pockros, P.J.; Lin, A.; Ackrill, A.M. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 2004, 140, 346–355. [Google Scholar] [CrossRef]
- Murakawa, K.; Esumi, M.; Kato, T.; Kambara, H.; Shikata, T. Heterogeneity within the nonstructural protein 5-encoding region of hepatitis C viruses from a single patient. Gene 1992, 117, 229–232. [Google Scholar] [CrossRef]
- Okamoto, H.; Kojima, M.; Okada, S.; Yoshizawa, H.; Iizuka, H.; Tanaka, T.; Muchmore, E.E.; Peterson, D.A.; Ito, Y.; Mishiro, S. Genetic drift of hepatitis C virus during an 8.2-year infection in a chimpanzee: Variability and stability. Virology 1992, 190, 894–899. [Google Scholar] [CrossRef]
- Ogata, N.; Alter, H.J.; Miller, R.H.; Purcell, R.H. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 3392–3396. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Dahari, H.; Ribeiro, R.M.; Perelson, A.S. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci. Transl. Med. 2010, 2, 30ra32. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.U.; Lam, N.P.; Dahari, H.; Gretch, D.R.; Wiley, T.E.; Layden, T.J.; Perelson, A.S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef]
- Kieffer, T.L.; Kwong, A.D.; Picchio, G.R. Viral resistance to specifically targeted antiviral therapies for hepatitis C (STAT-Cs). J. Antimicrob. Chemother. 2010, 65, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis c virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Lesburg, C.A.; Cable, M.B.; Ferrari, E.; Hong, Z.; Mannarino, A.F.; Weber, P.C. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 1999, 6, 937–943. [Google Scholar]
- Miller, R.H.; Purcell, R.H. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 2057–2061. [Google Scholar] [CrossRef]
- Doublie, S.; Tabor, S.; Long, A.M.; Richardson, C.C.; Ellenberger, T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature 1998, 391, 251–258. [Google Scholar] [CrossRef]
- Shim, J.H.; Larson, G.; Wu, J.Z.; Hong, Z. Selection of 3'-template bases and initiating nucleotides by hepatitis C virus NS5B RNA-dependent RNA polymerase. J. Virol. 2002, 76, 7030–7039. [Google Scholar] [CrossRef]
- Bressanelli, S.; Tomei, L.; Roussel, A.; Incitti, I.; Vitale, R. L.; Mathieu, M.; De Francesco, R.; Rey, F.A. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 13034–13039. [Google Scholar] [CrossRef]
- Ago, H.; Adachi, T.; Yoshida, A.; Yamamoto, M.; Habuka, N.; Yatsunami, K.; Miyano, M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 1999, 7, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Cameron, C.E.; Walker, M.P.; Castro, C.; Yao, N.; Lau, J.Y.; Zhong, W. A novel mechanism to ensure terminal initiation by hepatitis C virus NS5B polymerase. Virology 2001, 285, 6–11. [Google Scholar] [CrossRef]
- Kao, C.C.; Yang, X.; Kline, A.; Wang, Q.M.; Barket, D.; Heinz, B.A. Template requirements for RNA synthesis by a recombinant hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2000, 74, 11121–11128. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.W.; Ito, T.; Lai, M.M. A recombinant hepatitis C virus RNA-dependent RNA polymerase capable of copying the full-length viral RNA. J. Virol. 1999, 73, 7694–7702. [Google Scholar] [CrossRef] [PubMed]
- Ranjith-Kumar, C.T.; Gutshall, L.; Sarisky, R.T.; Kao, C.C. Multiple interactions within the hepatitis C virus RNA polymerase repress primer-dependent RNA synthesis. J. Mol. Biol. 2003, 330, 675–685. [Google Scholar] [CrossRef]
- Zhong, W.; Uss, A.S.; Ferrari, E.; Lau, J.Y.; Hong, Z. De novo initiation of RNA synthesis by hepatitis C virus nonstructural protein 5B polymerase. J. Virol. 2000, 74, 2017–2022. [Google Scholar] [CrossRef]
- Dutartre, H.; Boretto, J.; Guillemot, J.C.; Canard, B. A relaxed discrimination of 2'-O-methyl-GTP relative to GTP between de novo and Elongative RNA synthesis by the hepatitis C RNA-dependent RNA polymerase NS5B. J. Biol. Chem. 2005, 280, 6359–6368. [Google Scholar] [CrossRef]
- Harrus, D.; Ahmed-El-Sayed, N.; Simister, P.C.; Miller, S.; Triconnet, M.; Hagedorn, C.H.; Mahias, K.; Rey, F.A.; Astier-Gin, T.; Bressanelli, S. Further insights into the roles of GTP and the C-terminus of the Hepatitis C virus polymerase in the initiation of RNA synthesis. J. Biol. Chem. 2010. [CrossRef]
- Ferrari, E.; He, Z.; Palermo, R.E.; Huang, H.C. Hepatitis C virus NS5B polymerase exhibits distinct nucleotide requirements for initiation and elongation. J. Biol. Chem. 2008, 283, 33893–33901. [Google Scholar] [CrossRef]
- Tomei, L.; Altamura, S.; Bartholomew, L.; Bisbocci, M.; Bailey, C.; Bosserman, M.; Cellucci, A.; Forte, E.; Incitti, I.; Orsatti, L.; Koch, U.; De Francesco, R.; Olsen, D.B.; Carroll, S.S.; Migliaccio, G. Characterization of the inhibition of hepatitis C virus RNA replication by nonnucleosides. J. Virol. 2004, 78, 938–946. [Google Scholar] [CrossRef]
- Tomei, L.; Altamura, S.; Bartholomew, L.; Biroccio, A.; Ceccacci, A.; Pacini, L.; Narjes, F.; Gennari, N.; Bisbocci, M.; Incitti, I.; Orsatti, L.; Harper, S.; Stansfield, I.; Rowley, M.; De Francesco, R.; Migliaccio, G. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2003, 77, 13225–13231. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Johnston, V.K.; Gutshall, L.L.; Nguyen, T.T.; Gontarek, R.R.; Darcy, M.G.; Tedesco, R.; Dhanak, D.; Duffy, K.J.; Kao, C.C.; Sarisky, R.T. Arresting initiation of hepatitis C virus RNA synthesis using heterocyclic derivatives. J. Biol. Chem. 2003, 278, 16602–16607. [Google Scholar] [CrossRef] [PubMed]
- Summa, V.; Petrocchi, A.; Pace, P.; Matassa, V.G.; De Francesco, R.; Altamura, S.; Tomei, L.; Koch, U.; Neuner, P. Discovery of alpha, gamma-diketo acids as potent selective and reversible inhibitors of hepatitis C virus NS5b RNA-dependent RNA polymerase. J. Med. Chem. 2004, 47, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Oberg, B. Antiviral effects of phosphonoformate (PFA, foscarnet sodium). Pharmacol. Ther. 1982, 19, 387–415. [Google Scholar] [CrossRef]
- De Clercq, E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004, 30, 115–133. [Google Scholar] [CrossRef]
- Poijarvi-Virta, P. Prodrug approaches of nucleotides and oligonucleotides. Curr. Med. Chem. 2006, 13, 3441–3465. [Google Scholar] [CrossRef]
- Lavie, A.; Konrad, M. Structural requirements for efficient phosphorylation of nucleotide analogs by human thymidylate kinase. Mini-Rev. Med. Chem. 2004, 4, 351–359. [Google Scholar] [CrossRef]
- Deval, J.; Powdrill, M.H.; D'Abramo, C.M.; Cellai, L.; Gotte, M. Pyrophosphorolytic excision of nonobligate chain terminators by hepatitis C virus NS5B polymerase. Antimicrob. Agents. Chemother. 2007, 51, 2920–2928. [Google Scholar] [CrossRef]
- D'Abramo, C.M.; Cellai, L.; Gotte, M. Excision of incorporated nucleotide analogue chain-terminators can diminish their inhibitory effects on viral RNA-dependent RNA polymerases. J. Mol. Biol. 2004, 337, 1–14. [Google Scholar] [CrossRef]
- Meyer, P.R.; Matsuura, S.E.; Mian, A.M.; So, A.G.; Scott, W.A. A mechanism of AZT resistance: An increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell 1999, 4, 35–43. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Chen, Y.; Dutartre, H.; Sophys, M.; Morera, S.; Guerreiro, C.; Schneider, B.; Mulard, L.; Janin, J.; Veron, M.; Deville-Bonne, D.; Canard, B. Structural analysis of the activation of ribavirin analogs by NDP kinase: Comparison with other ribavirin targets. Mol. Pharmacol. 2003, 63, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Magee, W.C.; Aldern, K.A.; Hostetler, K.Y.; Evans, D.H. Cidofovir and (S)-9-[3-hydroxy-(2-phosphonomethoxy)propyl]adenine are highly effective inhibitors of vaccinia virus DNA polymerase when incorporated into the template strand. Antimicrob. Agents Chemother. 2008, 52, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Inocencio, N.; Cruz, J.E.; Cammack, N.; Klumpp, K. Incorporation and chain termination efficiencies can differ between nucleoside inhibitors of HCV replication at initiation adn elongation phases: Identification of a potential additional mechanism of action of R1479-TP Presented at the 16th International Symposium on Hepatitis C Virus and Related Viruses. Nice, France, October 3–7, 2009. [Google Scholar]
- Tchesnokov, E.P.; Obikhod, A.; Schinazi, R.F.; Gotte, M. Delayed chain termination protects the anti-hepatitis B virus drug entecavir from excision by HIV-1 reverse transcriptase. J. Biol. Chem. 2008, 283, 34218–34228. [Google Scholar] [CrossRef]
- Pierra, C.; Amador, A.; Benzaria, S.; Cretton-Scott, E.; D'Amours, M.; Mao, J.; Mathieu, S.; Moussa, A.; Bridges, E.G.; Standring, D.N.; Sommadossi, J.P.; Storer, R.; Gosselin, G. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2'-C-methylcytidine. J. Med. Chem. 2006, 49, 6614–6620. [Google Scholar] [CrossRef]
- Carroll, S.S.; Tomassini, J.E.; Bosserman, M.; Getty, K.; Stahlhut, M.W.; Eldrup, A.B.; Bhat, B.; Hall, D.; Simcoe, A.L.; LaFemina, R.; Rutkowski, C.A.; Wolanski, B.; Yang, Z.; Migliaccio, G.; De Francesco, R.; Kuo, L.C.; MacCoss, M.; Olsen, D. B. Inhibition of hepatitis C virus RNA replication by 2'-modified nucleoside analogs. J. Biol. Chem. 2003, 278, 11979–11984. [Google Scholar] [CrossRef] [PubMed]
- Sherman, K.E.; Lawitz, E.; O'Brien, C.B.; Godofsky, E.; Brown, N. Valopicitabine (NM283) plus peg-interferon for chronic hepatitis C in treatment naïve and nonresponder patients: Interim phase IIb results. J. Clin. Virol. 2006, 36, S24–S24. [Google Scholar] [CrossRef]
- Olsen, D.B.; Eldrup, A.B.; Bartholomew, L.; Bhat, B.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; Fay, J.F.; Flores, O.A.; Getty, K.L.; Grobler, J.A.; LaFemina, R.L.; Markel, E.J.; Migliaccio, G.; Prhavc, M.; Stahlhut, M.W.; Tomassini, J.E.; MacCoss, M.; Hazuda, D.J.; Carroll, S.S. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob. Agents Chemother. 2004, 48, 3944–3953. [Google Scholar] [CrossRef]
- Carroll, S.S.; Ludmerer, S.; Handt, L.; Koeplinger, K.; Zhang, N.R.; Graham, D.; Davies, M.E.; MacCoss, M.; Hazuda, D.; Olsen, D.B. Robust antiviral efficacy upon administration of a nucleoside analog to hepatitis C virus-infected chimpanzees. Antimicrob. Agents Chemother. 2009, 53, 926–934. [Google Scholar] [CrossRef]
- Migliaccio, G.; Tomassini, J.E.; Carroll, S.S.; Tomei, L.; Altamura, S.; Bhat, B.; Bartholomew, L.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; Cortese, R.; De Francesco, R.; Eldrup, A.B.; Getty, K.L.; Hou, X.S.; LaFemina, R.L.; Ludmerer, S.W.; MacCoss, M.; McMasters, D.R.; Stahlhut, M.W.; Olsen, D.B.; Hazuda, D.J.; Flores, O.A. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 2003, 278, 49164–49170. [Google Scholar] [CrossRef]
- Lalezari, J.; Azmuth, D.; Casiró, A.; Vargas, H.E.; Dubuc Patrick, G.; Lius, W.; Pietropaolo, K.; Zhou, X.; Sullivan-Bólyai, J.; Mayer, D.L. Antiviral Activity, Safety and Pharmacokinetics of IDX184, a Liver Targeted Nucleotide HCV Polymerase Inhibitor, in Patients with Chronic Hepatitis C. Hepatology 2009, 50, 228A. [Google Scholar]
- Lallos, L.; La Colla, M.; Serra, I.; Bilello, J.P.; Soubasakos, M.A.; Li, B.; Panzo, R.J.; Gillum, J.M.; Bonin, A.; Seifer, M.; Standring, D.N. Combination of IDX184, a nucleotide prodrug polymerase inhibitor, with other classes of HCV inhibitors is additive to synergistic in the HCV replicon in vitro. Hepatology 2009, 50, 1045A. [Google Scholar]
- Murakami, E.; Bao, H.; Ramesh, M.; McBrayer, T.R.; Whitaker, T.; Micolochick Steuer, H.M.; Schinazi, R.F.; Stuyver, L.J.; Obikhod, A.; Otto, M.J.; Furman, P.A. Mechanism of activation of beta-D-2'-deoxy-2'-fluoro-2'-c-methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob. Agents Chemother. 2007, 51, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Murakami, E.; Niu, C.; Bao, H.; Micolochick Steuer, H.M.; Whitaker, T.; Nachman, T.; Sofia, M.A.; Wang, P.; Otto, M.J.; Furman, P.A. The mechanism of action of beta-D-2'-deoxy-2'-fluoro-2'-C-methylcytidine involves a second metabolic pathway leading to beta-D-2'-deoxy-2'-fluoro-2'-C-methyluridine 5'-triphosphate, a potent inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 2008, 52, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Murakami, E.; Espiritu, C.; Steuer, H.M.; Niu, C.; Keilman, M.; Bao, H.; Zennou, V.; Bourne, N.; Julander, J.G.; Morrey, J.D.; Smee, D.F.; Frick, D.N.; Heck, J.A.; Wang, P.; Nagarathnam, D.; Ross, B.S.; Sofia, M.J.; Otto, M.J.; Furman, P.A. PSI-7851, a pronucleotide of beta-D-2'-deoxy-2'-fluoro-2'-C-methyluridine monophosphate, is a potent and pan-genotype inhibitor of hepatitis C virus replication. Antimicrob. Agents Chemother. 2010, 54, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Jiang, W.R.; Robledo, N.; Leveque, V.; Ali, S.; Lara-Jaime, T.; Masjedizadeh, M.; Smith, D.B.; Cammack, N.; Klumpp, K.; Symons, J. Characterization of the metabolic activation of hepatitis C virus nucleoside inhibitor beta-D-2'-Deoxy-2'-fluoro-2'-C-methylcytidine (PSI-6130) and identification of a novel active 5'-triphosphate species. J. Biol. Chem. 2007, 282, 29812–29820. [Google Scholar] [CrossRef]
- Rodriguez-Torres, M.; Lawitz, E.; Flach, S.; Denning, J.M.; Albanis, E.; Symonds, W.; Berrey, M.M. Antiviral Activity, Pharmacokinetics, Safety, and Tolerability of PSI-7851, a Novel Nucleotide Polymerase Inhibitor for HCV, Following Single and 3 Day Multiple Ascending Oral Doses in Healthy Volunteers and Patients with Chronic HCV Infection. Hepatology 2009, 50, 228A. [Google Scholar]
- Gane, E.J.; Roberts, S.K.; Stedman, C.A.; Angus, P.W.; Ritchie, B.; Elston, R.; Ipe, D.; Morcos, P.N.; Najera, I.; Chu, T.; Berrey, M.; Bradford, W.Z.; Laughlin, M.; Shulman, N.; Smith, P.F. Combination therapy with a nucleoside polymerase (R7128) and protease (R7227/ITMN-191) Inhibitor in HCV: Safety, pharmacokinetics, and virologic results from INFORM-1. Hepatology 2009, 50, 394A–395A. [Google Scholar]
- Le Pogam, S.; Chhabra, M.; Ali, S.; Yan, J.; Ilnicka, M.J.; Kang, H.; Wong, J.M.; Kosaka, A.; Ewing, A.; Seshaadri, A.; De La Rosas, A.; Bradford, W.Z.; Klumpp, K.; Shulman, N.; Smith, P.F.; Cammack, N.; Najera, I. Combination therapy with nucleoside polymerase R7128 and protease R7227/ITMN-191 inhibitors in genotype 1 HCV infected patients: Interim resistance analysis of INFORM-1 cohorts A-D. Hepatology 2009, 50, 1037A. [Google Scholar]
- Klumpp, K.; Leveque, V.; Le Pogam, S.; Ma, H.; Jiang, W.R.; Kang, H.; Granycome, C.; Singer, M.; Laxton, C.; Hang, J.Q.; Sarma, K.; Smith, D.B.; Heindl, D.; Hobbs, C.J.; Merrett, J.H.; Symons, J.; Cammack, N.; Martin, J.A.; Devos, R.; Najera, I. The novel nucleoside analog R1479 (4'-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J. Biol. Chem. 2006, 281, 3793–3799. [Google Scholar] [CrossRef]
- Le Pogam, S.; Jiang, W.R.; Leveque, V.; Rajyaguru, S.; Ma, H.; Kang, H.; Jiang, S.; Singer, M.; Ali, S.; Klumpp, K.; Smith, D.; Symons, J.; Cammack, N.; Najera, I. In vitro selected Con1 subgenomic replicons resistant to 2'-C-methyl-cytidine or to R1479 show lack of cross resistance. Virology 2006, 351, 349–359. [Google Scholar] [CrossRef]
- Perrone, P.; Luoni, G.M.; Kelleher, M.R.; Daverio, F.; Angell, A.; Mulready, S.; Congiatu, C.; Rajyaguru, S.; Martin, J.A.; Leveque, V.; Le Pogam, S.; Najera, I.; Klumpp, K.; Smith, D.B.; McGuigan, C. Application of the phosphoramidate ProTide approach to 4'-azidouridine confers sub-micromolar potency versus hepatitis C virus on an inactive nucleoside. J. Med. Chem. 2007, 50, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Pockros, P.J.; Nelson, D.; Godofsky, E.; Rodriguez-Torres, M.; Everson, G.T.; Fried, M.W.; Ghalib, R.; Harrison, S.; Nyberg, L.; Shiffman, M.L.; Najera, I.; Chan, A.; Hill, G. R1626 plus peginterferon Alfa-2a provides potent suppression of hepatitis C virus RNA and significant antiviral synergy in combination with ribavirin. Hepatology 2008, 48, 385–397. [Google Scholar] [CrossRef]
- De Clercq, E. Acyclic nucleoside phosphonates: A new dimension to the chemotherapy of DNA virus and retrovirus infections. J. Med. Microbiol. 1998, 47, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.L.; Kaihara, K.A.; Korba, B.E.; Schooley, R.T.; Beadle, J.R.; Hostetler, K.Y. The octadecyloxyethyl ester of (S)-9-[3-hydroxy-2-(phosphonomethoxy) propyl]adenine is a potent and selective inhibitor of hepatitis C virus replication in genotype 1A, 1B, and 2A replicons. Antimicrob. Agents Chemother. 2009, 53, 2660–2662. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.L.; Jones, K.A.; Valiaeva, N.; Beadle, J.; Hostetler, K.Y.; Schooley, R.T. Resistance profile of the novel hepatitis C virus (HCV) acyclic nucleoside phosphonate inhibitor ODE-S-MPMPA. Antiviral Therapy 2010, 15, A26. [Google Scholar]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Dutartre, H.; Bussetta, C.; Boretto, J.; Canard, B. General catalytic deficiency of hepatitis C virus RNA polymerase with an S282T mutation and mutually exclusive resistance towards 2'-modified nucleotide analogues. Antimicrob. Agents Chemother. 2006, 50, 4161–4169. [Google Scholar] [CrossRef]
- Kukolj, G.; McGibbon, G.A.; McKercher, G.; Marquis, M.; Lefebvre, S.; Thauvette, L.; Gauthier, J.; Goulet, S.; Poupart, M.A.; Beaulieu, P.L. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 2005, 280, 39260–39267. [Google Scholar] [CrossRef]
- Hirashima, S.; Suzuki, T.; Ishida, T.; Noji, S.; Yata, S.; Ando, I.; Komatsu, M.; Ikeda, S.; Hashimoto, H. Benzimidazole derivatives bearing substituted biphenyls as hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors: Structure-activity relationship studies and identification of a potent and highly selective inhibitor JTK-109. J. Med. Chem. 2006, 49, 4721–4736. [Google Scholar] [CrossRef]
- Di Marco, S.; Volpari, C.; Tomei, L.; Altamura, S.; Harper, S.; Narjes, F.; Koch, U.; Rowley, M.; De Francesco, R.; Migliaccio, G.; Carfi, A. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J. Biol. Chem. 2005, 280, 29765–29770. [Google Scholar] [CrossRef]
- Erhardt, A.; Deterding, K.; Benhamou, Y.; Reiser, M.; Forns, X.; Pol, S.; Calleja, J.L.; Ross, S.; Spangenberg, H.C.; Garcia-Samaniego, J.; Fuchs, M.; Enriquez, J.; Wiegand, J.; Stern, J.; Wu, K.; Kukolj, G.; Marquis, M.; Beaulieu, P.; Nehmiz, G.; Steffgen, J. Safety, pharmacokinetics and antiviral effect of BILB 1941, a novel hepatitis C virus RNA polymerase inhibitor, after 5 days oral treatment. Antivir. Ther. 2009, 14, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Larrey, D.; Lohse, A.; Ledinghen, V.d.; Trepo, C.; Gerlach, T.; Zarski, J. P.; Tran, A.; Mathurin, P.; Thimme, R.; Arastéh, K.; Trautwein, C.; Cerny, A.; Dikopoulos, N.; Schuchmann, M.; Heim, M.H.; Gerken, G.; Stern, J.; Wu, K.; Abdallah, N.; Girlich, B.; Josepf, S.; Wulf, B.; Berger, F.; Steffgen, J. 4 Week Therapy with the Non-nucleosidic Polymerase Inhibitor BI207127 in Combination with Peginterferon-alfa2A and Ribavirin in Treatment Naive and Treatment Experienced Chronic HCV GT1 Patients. J. Hepatol. 2010, 52, S466. [Google Scholar] [CrossRef]
- De Francesco, R. Robust antiviral efficacy of a “finger-loop” allosteric inhibitor of the HCV polymerase in HCV infected chimpanzees. Global Antiviral Journal 2007, 3, 25. [Google Scholar]
- Brainard, D.M.; Anderson, M.S.; Petry, A.; Van Dyck, K.; De Lepeleire, I.; Sneddon, K.; Cummings, C.E.; Nachbar, R.B.; Barnard, R.J.; Sun, P.; Panorchan, P.; Sanderson, J.B.; Udezue, E.; Wagner, F.; Iwamoto, M.; Chodakewitz, J.; Wagner, J.A. Safety and antiviral activity of NS5B polymerase inhibitor MK-3281, in treatment-naïve genotype 1A, 1B AND 3 HCV infected patients. Hepatology 2009, 50, 1026A. [Google Scholar]
- Wang, M.; Ng, K.K.; Cherney, M.M.; Chan, L.; Yannopoulos, C.G.; Bedard, J.; Morin, N.; Nguyen-Ba, N.; Alaoui-Ismaili, M.H.; Bethell, R.C.; James, M.N. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J. Biol. Chem. 2003, 278, 9489–9495. [Google Scholar] [CrossRef]
- Chan, L.; Pereira, O.; Reddy, T.J.; Das, S.K.; Poisson, C.; Courchesne, M.; Proulx, M.; Siddiqui, A.; Yannopoulos, C.G.; Nguyen-Ba, N.; Roy, C.; Nasturica, D.; Moinet, C.; Bethell, R.; Hamel, M.; L'Heureux, L.; David, M.; Nicolas, O.; Courtemanche-Asselin, P.; Brunette, S.; Bilimoria, D.; Bedard, J. Discovery of thiophene-2-carboxylic acids as potent inhibitors of HCV NS5B polymerase and HCV subgenomic RNA replication. Part 2: Tertiary amides. Bioorg. Med. Chem. Lett. 2004, 14, 797–800. [Google Scholar] [CrossRef]
- Chan, L.; Das, S.K.; Reddy, T.J.; Poisson, C.; Proulx, M.; Pereira, O.; Courchesne, M.; Roy, C.; Wang, W.; Siddiqui, A.; Yannopoulos, C.G.; Nguyen-Ba, N.; Labrecque, D.; Bethell, R.; Hamel, M.; Courtemanche-Asselin, P.; L'Heureux, L.; David, M.; Nicolas, O.; Brunette, S.; Bilimoria, D.; Bedard, J. Discovery of thiophene-2-carboxylic acids as potent inhibitors of HCV NS5B polymerase and HCV subgenomic RNA replication. Part 1: Sulfonamides. Bioorg. Med. Chem. Lett. 2004, 14, 793–796. [Google Scholar] [CrossRef]
- Biswal, B.K.; Cherney, M.M.; Wang, M.; Chan, L.; Yannopoulos, C.G.; Bilimoria, D.; Nicolas, O.; Bedard, J.; James, M.N. Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J. Biol.Chem. 2005, 280, 18202–18210. [Google Scholar] [CrossRef]
- Le Pogam, S.; Kang, H.; Harris, S.F.; Leveque, V.; Giannetti, A.M.; Ali, S.; Jiang, W.R.; Rajyaguru, S.; Tavares, G.; Oshiro, C.; Hendricks, T.; Klumpp, K.; Symons, J.; Browner, M.F.; Cammack, N.; Najera, I. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J. Virol. 2006, 80, 6146–6154. [Google Scholar] [CrossRef]
- Jacobson, I.; Pockros, P.; Lalezari, J.; Lawitz, E.; Rodriguez-Torres, M.; DeJesus, E.; Haas, F.; Martorell, C.; Pruitt, R.; Durham, K.; Srinivasan, S.; Rosario, M.; Jagannatha, S.; Hammond, J. Antiviral Activity of Filibuvir in Combination with Pegylated Interferon Alfa-2a and Ribavirin for 28 days in Treatment Naive Patients Chronically Infected with HCV Genotype 1. J. Hepatol. 2009, 50, S382–S383. [Google Scholar] [CrossRef]
- Cooper, C.; Lawitz, E.J.; Ghali, P.; Rodriguez-Torres, M.; Anderson, F.H.; Lee, S.S.; Bédard, J.; Chauret, N.; Thibert, R.; Boivin, I.; Nicolas, O.; Proulx, L. Evaluation of VCH-759 monotherapy in hepatitis C infection. J. Hepatol. 2009, 51, 39–46. [Google Scholar] [CrossRef]
- Tedesco, R.; Shaw, A.N.; Bambal, R.; Chai, D.; Concha, N.O.; Darcy, M.G.; Dhanak, D.; Fitch, D.M.; Gates, A.; Gerhardt, W.G.; Halegoua, D.L.; Han, C.; Hofmann, G.A.; Johnston, V.K.; Kaura, A.C.; Liu, N.; Keenan, R.M.; Lin-Goerke, J.; Sarisky, R.T.; Wiggall, K.J.; Zimmerman, M.N.; Duffy, K.J. 3-(1,1-dioxo-2H-(1,2,4)-benzothiadiazin-3-yl)-4-hydroxy-2(1H)-quinolinones, potent inhibitors of hepatitis C virus RNA-dependent RNA polymerase. J. Med. Chem. 2006, 49, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Rodriguez-Torres, M.; DeMicco, M.; Nguyen, T.; Godofsky, E.; Appleman, J.; Rahimy, M.; Crowley, C.; Freddo, J. Antiviral activity of ANA598, a potent non-nucleoside polymerase inhibitor, in chronic hepatitis C patients. J. Hepatol. 2009, 50, S384. [Google Scholar] [CrossRef]
- Lawitz, E.; Rodriquez-Torres, M.; Rustgi, V.K.; Hassanein, T.; Rahimy, M.H.; Crowley, C.A.; Freddo, J.L.; Muir, A.; McHutchison, J. Safety and antiviral activity of ANA598 in combination with pegylated interferon a2A plus ribavirin in treatment-naive genotype-1 chronic HCV patients. J. Hepatol. 2010, 52, S467. [Google Scholar] [CrossRef]
- Burton, G.; Ku, T.W.; Carr, T.J.; Kiesow, T.; Sarisky, R.T.; Lin-Goerke, J.; Hofmann, G.A.; Slater, M.J.; Haigh, D.; Dhanak, D.; Johnson, V.K.; Parry, N.R.; Thommes, P. Studies on acyl pyrrolidine inhibitors of HCV RNA-dependent RNA polymerase to identify a molecule with replicon antiviral activity. Bioorg. Med. Chem. Lett. 2007, 17, 1930–1933. [Google Scholar] [CrossRef]
- Gray, F.; Amphlett, E.; Bright, H.; Chambers, L.; Cheasty, A.; Fenwick, R.; Haigh, D.; Hartley, D.; Howes, P.; Jarvest, R.; Mirzai, F.; Nerozzi, F.; Parry, N.; Slater, M.; Smith, S.; Thommes, P.; Wilkinson, C.; Williams, E. GSK625433: A Novel and Highly Potent Inhibitor of the HCV NS5B Polymerase. J. Hepatol. 2007, 46, S225–S225. [Google Scholar] [CrossRef]
- Hang, J.Q.; Yang, Y.; Harris, S.F.; Leveque, V.; Whittington, H.J.; Rajyaguru, S.; Ao-Ieong, G.; McCown, M.F.; Wong, A.; Giannetti, A.M.; Le Pogam, S.; Talamas, F.; Cammack, N.; Najera, I.; Klumpp, K. Slow binding inhibition and mechanism of resistance of non-nucleoside polymerase inhibitors of hepatitis C virus. J. Biol. Chem. 2009, 284, 15517–15529. [Google Scholar] [CrossRef]
- Howe, A.Y.; Cheng, H.; Johann, S.; Mullen, S.; Chunduru, S.K.; Young, D.C.; Bard, J.; Chopra, R.; Krishnamurthy, G.; Mansour, T.; O'Connell, J. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 2008, 52, 3327–3338. [Google Scholar] [CrossRef]
- Nittoli, T.; Curran, K.; Insaf, S.; DiGrandi, M.; Orlowski, M.; Chopra, R.; Agarwal, A.; Howe, A.Y.; Prashad, A.; Floyd, M.B.; Johnson, B.; Sutherland, A.; Wheless, K.; Feld, B.; O'Connell, J.; Mansour, T.S.; Bloom, J. Identification of anthranilic acid derivatives as a novel class of allosteric inhibitors of hepatitis C NS5B polymerase. J. Med. Chem. 2007, 50, 2108–2116. [Google Scholar] [CrossRef]
- McCown, M.F.; Rajyaguru, S.; Kular, S.; Cammack, N.; Najera, I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob. Agents Chemother. 2009, 53, 2129–2132. [Google Scholar] [CrossRef]
- Middleton, T.; He, Y.; Beyer, J.; Menon, R.; Klein, C.; Cohen, D.; Collins, C. Resistance profile of ABT-333 and relationship to viral load decrease in patients treated in combination with peg-interferon and ribavirin for 28 days. J. Hepatol. 2010, 52, S296–S297. [Google Scholar] [CrossRef]
- Mathiesen, S.; Roge, B.T.; Weis, N.; Lundgren, J.D.; Obel, N.; Gerstoft, J. Foscarnet used in salvage therapy of HIV-1 patients harbouring multiple nucleotide excision mutations. AIDS 2004, 18, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Attenni, B.; Malancona, S.; Colarusso, S.; Conte, I.; Di Filippo, M.; Harper, S.; Pacini, B.; Giomini, C.; Thomas, S.; Incitti, I.; Tomei, L.; De Francesco, R.; Altamura, S.; Matassa, V.G.; Narjes, F. 2-(2-Thienyl)-5,6-dihydroxy-4-carboxypyrimidines as inhibitors of the hepatitis C virus NS5B polymerase: Discovery, SAR, modeling, and mutagenesis. J. Med. Chem. 2006, 49, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, R.; Tomei, L.; Altamura, S.; Summa, V.; Migliaccio, G. Approaching a new era for hepatitis C virus therapy: Inhibitors of the NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase. Antiviral. Res. 2003, 58, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dahlberg, M.E.; Benkovic, S.J. Kinetic mechanism of DNA polymerase I (Klenow fragment): Identification of a second conformational change and evaluation of the internal equilibrium constant. Biochemistry 1991, 30, 4835–4843. [Google Scholar] [CrossRef]
- Powdrill, M.H.; Deval, J.; Narjes, F.; De Francesco, R.; Gotte, M. Mechanism of hepatitis C virus RNA polymerase inhibition with dihydroxypyrimidines. Antimicrob. Agents Chemother. 2010, 54, 977–983. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, W.W.; Pratt, J.; Rockway, T.; Harris, K.; Vasavanonda, S.; Tripathi, R.; Pithawalla, R.; Kati, W.M. Mechanistic study of HCV polymerase inhibitors at individual steps of the polymerization reaction. Biochemistry 2006, 45, 11312–11323. [Google Scholar] [CrossRef]
- Tomei, L.; Altamura, S.; Paonessa, G.; De Francesco, R.; Migliaccio, G. HCV antiviral resistance: The impact of in vitro studies on the development of antiviral agents targeting the viral NS5B polymerase. Antiviv. Chem. Chemother. 2005, 16, 225–245. [Google Scholar] [CrossRef]
- Zhou, Y.; Muh, U.; Hanzelka, B.L.; Bartels, D.J.; Wei, Y.; Rao, B.G.; Brennan, D.L.; Tigges, A.M.; Swenson, L.; Kwong, A.D.; Lin, C. Phenotypic and structural analyses of hepatitis C virus NS3 protease Arg155 variants: Sensitivity to telaprevir (VX-950) and interferon alpha. J. Biol. Chem. 2007, 282, 22619–22628. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Everson, G.T.; Gordon, S.C.; Jacobson, I.M.; Sulkowski, M.; Kauffman, R.; McNair, L.; Alam, J.; Muir, A.J. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 2009, 360, 1827–1838. [Google Scholar] [CrossRef]
- Cooper, C.; Lawitz, E.J.; Ghali, P.; Rodriguez-Torres, M.; Anderson, F.H.; Lee, S.S.; Bedard, J.; Chauret, N.; Thibert, R.; Boivin, I.; Nicolas, O.; Proulx, L. Evaluation of VCH-759 monotherapy in hepatitis C infection. J. Hepatol. 2009, 51, 39–46. [Google Scholar] [CrossRef]
- Roberts, S.K.; Cooksley, G.; Dore, G.J.; Robson, R.; Shaw, D.; Berns, H.; Hill, G.; Klumpp, K.; Najera, I.; Washington, C. Robust antiviral activity of R1626, a novel nucleoside analog: A randomized, placebo-controlled study in patients with chronic hepatitis C. Hepatology 2008, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- McCown, M.F.; Rajyaguru, S.; Le Pogam, S.; Ali, S.; Jiang, W.R.; Kang, H.; Symons, J.; Cammack, N.; Najera, I. The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob. Agents Chemother. 2008, 52, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Leveque, V.; Le Pogam, S.; Ma, H.; Philipp, F.; Inocencio, N.; Smith, M.; Alker, A.; Kang, H.; Najera, I.; Klumpp, K.; Symons, J.; Cammack, N.; Jiang, W.R. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479. Antimicrob. Agents Chemother. 2008, 52, 4356–4369. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Gaudieri, S.; Rauch, A.; Pfafferott, K.; Barnes, E.; Cheng, W.; McCaughan, G.; Shackel, N.; Jeffrey, G.P.; Mollison, L.; Baker, R.; Furrer, H.; Gunthard, H.F.; Freitas, E.; Humphreys, I.; Klenerman, P.; Mallal, S.; James, I.; Roberts, S.; Nolan, D.; Lucas, M. Hepatitis C virus drug resistance and immune-driven adaptations: Relevance to new antiviral therapy. Hepatology 2009, 49, 1069–1082. [Google Scholar] [CrossRef]
- Bartels, D.J.; Zhou, Y.; Zhang, E.Z.; Marcial, M.; Byrn, R. A.; Pfeiffer, T.; Tigges, A.M.; Adiwijaya, B.S.; Lin, C.; Kwong, A.D.; Kieffer, T.L. Natural prevalence of hepatitis C virus variants with decreased sensitivity to NS3.4A protease inhibitors in treatment-naive subjects. J. Infect. Dis. 2008, 198, 800–807. [Google Scholar] [CrossRef]
- Cubero, M.; Esteban, J.I.; Otero, T.; Sauleda, S.; Bes, M.; Esteban, R.; Guardia, J.; Quer, J. Naturally occurring NS3-protease-inhibitor resistant mutant A156T in the liver of an untreated chronic hepatitis C patient. Virology 2008, 370, 237–245. [Google Scholar] [CrossRef]
- Kuntzen, T.; Timm, J.; Berical, A.; Lennon, N.; Berlin, A.M.; Young, S.K.; Lee, B.; Heckerman, D.; Carlson, J.; Reyor, L.L.; Kleyman, M.; McMahon, C.M.; Birch, C.; Schulze Zur Wiesch, J.; Ledlie, T.; Koehrsen, M.; Kodira, C.; Roberts, A.D.; Lauer, G.M.; Rosen, H.R.; Bihl, F.; Cerny, A.; Spengler, U.; Liu, Z.; Kim, A.Y.; Xing, Y.; Schneidewind, A.; Madey, M.A.; Fleckenstein, J.F.; Park, V.M.; Galagan, J.E.; Nusbaum, C.; Walker, B.D.; Lake-Bakaar, G.V.; Daar, E.S.; Jacobson, I.M.; Gomperts, E.D.; Edlin, B.R.; Donfield, S.M.; Chung, R.T.; Talal, A.H.; Marion, T.; Birren, B.W.; Henn, M.R.; Allen, T.M. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naive patients. Hepatology 2008, 48, 1769–1778. [Google Scholar] [CrossRef]
- Lu, L.; Mo, H.; Pilot-Matias, T.J.; Molla, A. Evolution of resistant M414T mutants among hepatitis C virus replicon cells treated with polymerase inhibitor A-782759. Antimicrob. Agents Chemother. 2007, 51, 1889–1896. [Google Scholar] [CrossRef]
- Kieffer, T.L.; Sarrazin, C.; Miller, J.S.; Welker, M.W.; Forestier, N.; Reesink, H.W.; Kwong, A.D.; Zeuzem, S. Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology 2007, 46, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O'Boyle, D.R., 2nd; Lemm, J.A.; Wang, C.; Knipe, J.O.; Chien, C.; Colonno, R.J.; Grasela, D.M.; Meanwell, N.A.; Hamann, L.G. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef]
- Delang, L.; Coelmont, L.; Neyts, J. Antiviral therapy for hepatitis C Virus: Beyond the standard of care. Viruses 2010, 2, 826–866. [Google Scholar] [CrossRef]
- Menéndez-Arias, L. Molecular basis of human immunodeficiency virus drug resistance: An update. Antivir. Res. 2010, 85, 210–231. [Google Scholar] [CrossRef] [PubMed]
- Marchand, B.; White, K.L.; Ly, J.K.; Margot, N.A.; Wang, R.; McDermott, M.; Miller, M.D.; Gotte, M. Effects of the translocation status of human immunodeficiency virus type 1 reverse transcriptase on the efficiency of excision of tenofovir. Antimicrob. Agents Chemother. 2007, 51, 2911–2919. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N.; Arion, D.; Parikh, U.; Koontz, D.; Schinazi, R.F.; Mellors, J.W.; Parniak, M.A. The 3'-azido group is not the primary determinant of 3'-azido-3'-deoxythymidine (AZT) responsible for the excision phenotype of AZT-resistant HIV-1. J. Biol. Chem. 2005, 280, 29047–29052. [Google Scholar] [CrossRef]
- Smith, A.J.; Scott, W.A. The influence of natural substrates and inhibitors on the nucleotide-dependent excision activity of HIV-1 reverse transcriptase in the infected cell. Curr. Pharm. Des. 2006, 12, 1827–1841. [Google Scholar] [CrossRef]
- Das, K.; Bauman, J.D.; Clark, A.D., Jr.; Frenkel, Y.V.; Lewi, P.J.; Shatkin, A.J.; Hughes, S.H.; Arnold, E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1466–1471. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Company | Target | Progress |
|---|---|---|---|
| NM283 | Idenix/Novartis | Active site | Stopped |
| R7128 | Roche/Pharmasset | Active site | Phase II |
| IDX184 | Idenix | Active site | Phase II |
| R1626 | Roche/Pharmasset | Active site | Stopped |
| PSI7851 | Pharmasset | Active site | Phase II |
| MK0608 | Merck | Active site | Unknown |
| HCV-796 | ViroPharma/Wyeth | NNI IV | Stopped |
| PF-868554, Filibuvir | Pfizer | NNI II | Phase II |
| ABT-333 | Abbott | NNI IV | Phase II |
| ABT-072 | Abbott | NNI IV | Phase II |
| VCH-759 | Vertex | NNI II | Phase II |
| VCH-916 | Vertex | NNI II | Phase II |
| BILB-1941 | Boehringer Ingelheim | NNI I | Stopped |
| IDX375 | Idenix | Palm | Phase I |
| GS9190 | Gilead | NNI IV | Phase II |
| MK-3281 | Merck | NNI I | Phase I |
| BI207127 | Boehringer Ingelheim | NNI I | Phase I |
| VCH-222 | Vertex | NNI II | Phase II |
| ANA598 | Anadys | NNI III | Phase II |
| JTK-109 | Japan Tobacco | NNI I | Stopped |
| GSK625433 | GlaxoSmithKline | NNI III | Phase I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powdrill, M.H.; Bernatchez, J.A.; Götte, M. Inhibitors of the Hepatitis C Virus RNA-Dependent RNA Polymerase NS5B. Viruses 2010, 2, 2169-2195. https://doi.org/10.3390/v2102169
Powdrill MH, Bernatchez JA, Götte M. Inhibitors of the Hepatitis C Virus RNA-Dependent RNA Polymerase NS5B. Viruses. 2010; 2(10):2169-2195. https://doi.org/10.3390/v2102169
Chicago/Turabian StylePowdrill, Megan H., Jean A. Bernatchez, and Matthias Götte. 2010. "Inhibitors of the Hepatitis C Virus RNA-Dependent RNA Polymerase NS5B" Viruses 2, no. 10: 2169-2195. https://doi.org/10.3390/v2102169
APA StylePowdrill, M. H., Bernatchez, J. A., & Götte, M. (2010). Inhibitors of the Hepatitis C Virus RNA-Dependent RNA Polymerase NS5B. Viruses, 2(10), 2169-2195. https://doi.org/10.3390/v2102169