Interactions of Host Proteins with the Murine Leukemia Virus Integrase


Abstract
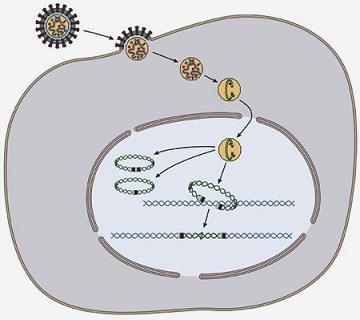
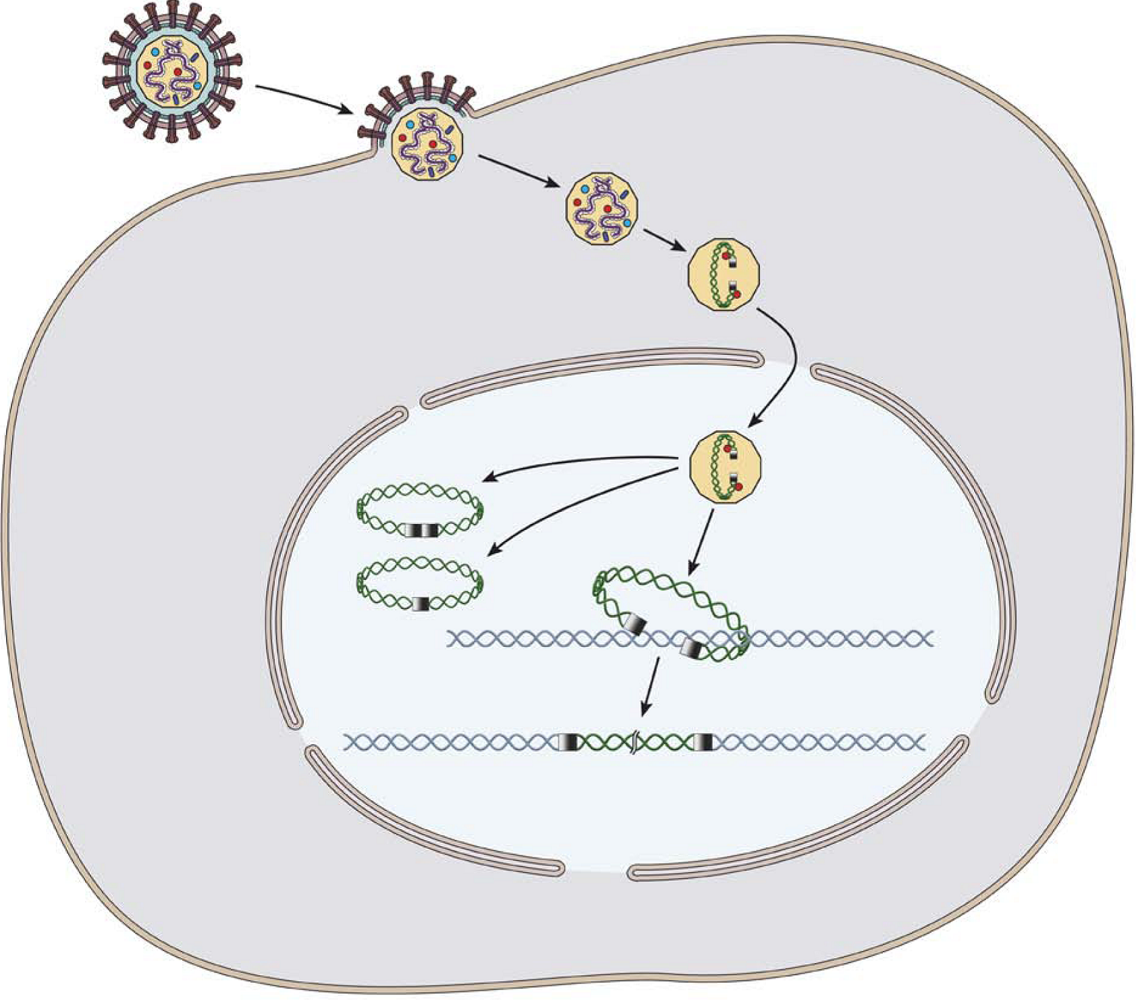
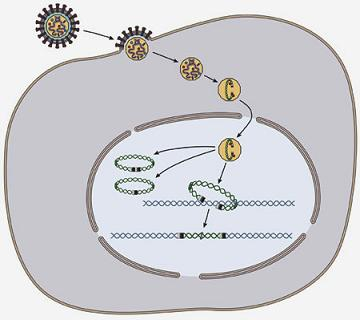
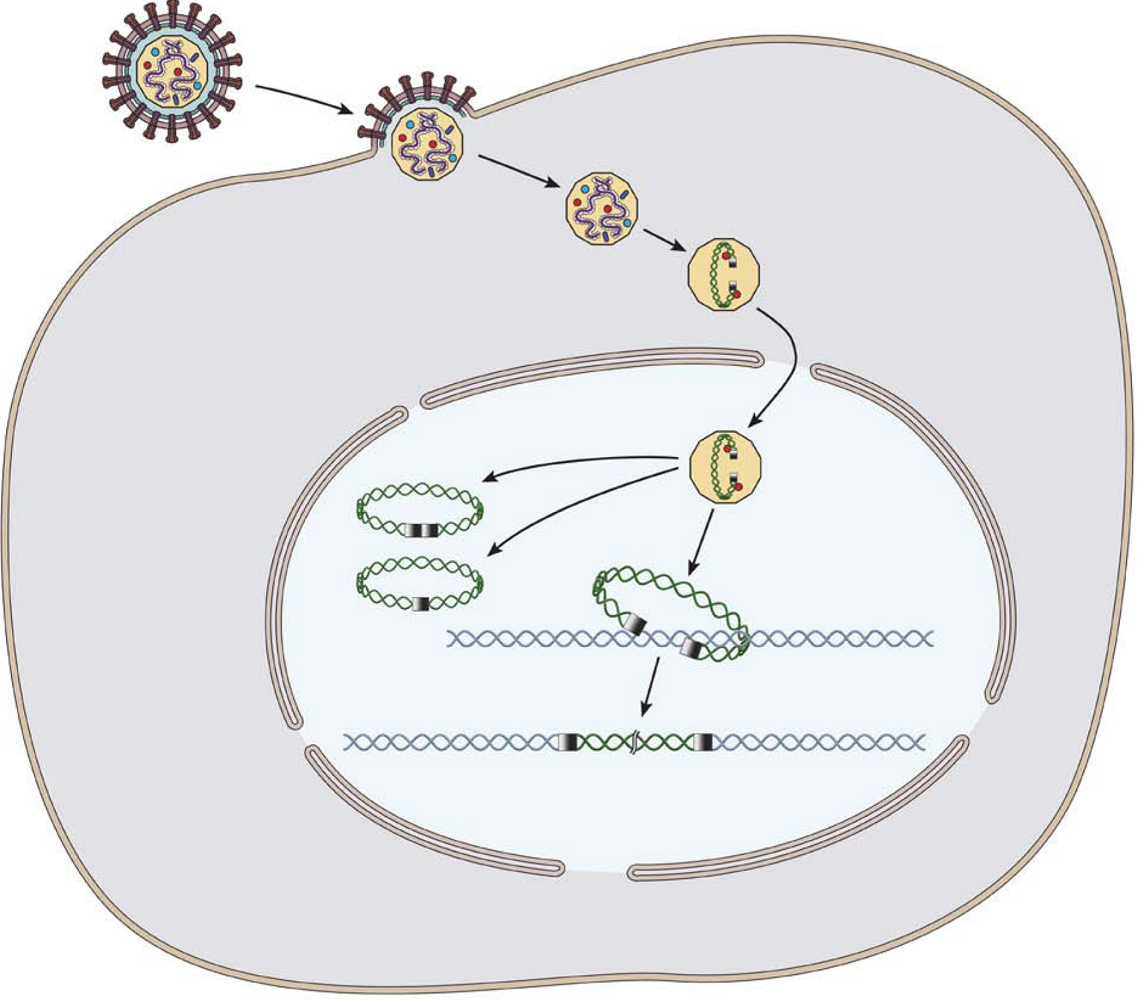
:
1. Introduction
2. Retrotransposon and phage integration
3. Identifying and isolating interacting factors
4. Why Study MoMLV Integrase?
5. MoMLV Integrase Interacting Factors
6. Perspectives and challenges

{kind=link}
{kind=link}
| Host Factors implicated in MLV PIC or IN interactions | Cellular Function | Proposed Function in viral life cycle |
|---|---|---|
| Emerin, BAF, Lap2α | Nuclear envelope; stabilization of actin cortical network | PIC importation, chromatin targeting of PIC; BAF inhibits autointegration of viral cDNA |
| Transcription factor IIE, beta subunit (TFIIE-β) | Subunit of RNA Pol II holoenzyme; recruits TFIIH to the Pol II-TFIIB-TFIID complex; stimulates RNA Pol II kinase and TFIIH DNA-dependent ATPase activities | Unknown; other subunits of basal Pol II complex isolated in other screens |
| Enhancer of zeste homolog 1 (Enx-1/Ezh2) | Polycomb Group 2 subunit complex with Eed and Suz12; chromatin structure maintenance and transcriptional regulation; Histone methyltransferase (H3K27 and H1K26) | Eed interacts with HIV-1 IN, Nef, and MA. Activity unknown in MLV |
| Flap endonuclease-1 (Fen1) | Removes 5’ initiator tRNA from Okazaki fragments; DNA repair in NHEJ and V(D)J recombination; 5’ to 3’ exonuclease and RNase H actvities | Resolution of free 5’ PO4 ends of viral DNA? |
| Ku70/XRCC6 | NHEJ, chromosome and telomere maintenance, 70 kD subunit of Ku86 heterodimer, with Ku80 subunit of DNA-PKcs | Repair of gaps generated by IN cleavage at host/viral DNA junctions? |
| Tata binding protein ABT1 (ABT1) | Associates with Tata binding protein and activates basal transcription of class II promoters | Unknown |
| B-Activating transcription factor (B-ATF) | AP-1/ATF superfamily; Basic Leucine zipper transcription factor; blocks transformation by H-Ras and v-Fos; negative regulator of AP-1 mediated transcription by binding to Jun proteins | Unknown |
| All1 fused translocated to Chromosome 9 (AF9)/mixed lineage-leukemia translocated to 3 (Mllt3) | H3 hypermethylation; contains one YEATS domain (YNL107w/ ENL/AF-9/ and TFIIF small subunit); interacts with BCOR and MPc3 (Polycomb 3 homolog, component of PRC1) | Unknown |
| Bromodomain containing protein 2 (Brd2/Fsrg-1/RING3) | Bromodomain repeat-containing protein; mitogen-activated kinase activity; homolog of Drosophila female sterile homeotic gene; homodimer; interacts with histone H4 acetylated at lysine 13 | Brd2 interacts with Latency-associated nuclear antigen (LANA-1) of KHSV |
| Zinc finger p38 (Znfp38) | Strong transcriptional activator; transactivation via its SCAN domain | Unknown |
| Peroxisome proliferative activated receptor, gamma, coactivator-1 related (PRC) | Serum-inducible coactivator of nuclear respiratory factor 1- dependent transcription from RNA pol II promoters; interacts with CREB1; stress response protein | Unknown |
| Ankyrin rep domain 49 (Ankrd49) | Putative transcription factor; contains acidic activation domain; Ankyrin repeat domain is similar to that of SWI6 | Unknown |
| Splicing factor 3b, subunit 2 (SF3b2) | Putative DNA-binding (bihelical) motif predicted to be involved in chromosomal organization; SAP domain; basic domain in HLH proteins of MYOD family; component of spliceosome C complex; phosphorylated by ATM or ATR in response to DNA damage | Interacts with HIV-1 Vpr; identified in two or more studies |
| Splicing factor 3a, subunit 3 (SF3a3) | C2H2-type Zinc finger; mRNA processing; component of SF3A; associates with SF3B and 12S RNA unit to form U2 snRNP complex | Unknown |
| U2 auxiliary factor 26 (U2AF26 ) | Pre-RNA splicing factor; can replace U2AF1 in constitutive and enhancer dependent splicing activities; can replace U2AF35in vitro; enhances U2AF2 binding to weak Pyrimidine tracts. | Unknown |
| U5 small nuclear ribonucleoprotein (U5 snRNP) | Transcriptional regulation; SNF2 N-terminal domain; GTP binding factor; ortholog of S. cerevisiae splicing factor Prp8p; component of spliceosome C complex; interacts with Ddx5 | Unknown |
| Step II Splicing factor SLU7 | Pre mRNA splicing; required for 3’ splice-site choice by association with the spliceosome prior to recognition of the splice site in step II | Unknown |
| Survival of motor neuron (SMN) | Essential role in snRNP assembly; component of import snRNP complex containing Gemins 2, 3, 4, 5, 6 and 7; contains one Tudor domain; deficiency leads to apoptosis | Gemin 2 interacts with HIV-1 IN |
| Dead box p68 (Ddx68/Ddx5) | Component of spliceosome C complex; RNA-dependent helicase and ATPase activity; stimulated by ss-RNA; interacts with HDAC1 | Identified in two or more genome wide studies (Bushman et al. 2009) |
| Ran binding protein 10 (RanBP10) | Competes with RanBP9 for MET binding; interacts with MET via its SPRY domain; interacts with Ran in vitro; does not interact with Sos nor activate Ras pathway | Several Ran family members identified in various studies (RanBP2, RanBP17); RanBP9 interacts with phosphorylated HIV-1 IN |
| Radixin | Member of ezrin, radixin, moesin family of actin binding proteins. Binds directly to barbed ends of actin filaments in plasma membrane. | ERM family member Moesin implicated in MLV and HIV viral trafficking |
Acknowledgments
References
- Brown, P.O. Integration. In Retroviruses; Coffin, J., Hughes, S.H., Varmus, H.E., Eds.; 1997; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA. [Google Scholar]
- Hindmarsh, P.; Leis, J. Retroviral DNA integration. Microbiol. Mol. Biol. Rev. 1999, 63, 836–843. [Google Scholar] [PubMed]
- Goff, S.P. Intracellular trafficking of retroviral genomes during the early phases of infection: viral exploitation of cellular pathways. J. Gene Med. 2001, 3, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Risco, C.; Menendez-Arias, L.; Copeland, T.D.; Pinto da Silva, P.; Oroszlan, S. Intracellular transport of the murine leukemia virus during acute infection of NIH 3T3 cells: nuclear import of nucleocapsid protein and integrase. J. Cell Sci. 1995, 108, 3039–3050. [Google Scholar] [PubMed]
- Farnet, C.M.; Haseltine, W.A. Determination of viral proteins present in the human immunodeficiency virus type 1 preintegration complex. J. Virol. 1991, 65, 1910–1915. [Google Scholar] [PubMed]
- Fassati, A.; Goff, S.P. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 2001, 75, 3626–3635. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Craigie, R. The road to chromatin - nuclear entry of retroviruses. Nat. Rev. Microbiol. 2007, 5, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.I.; Sharova, N.; McDonald, T.L.; Pushkarskaya, T.; Tarpley, W.G.; Stevenson, M. Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6125–6129. [Google Scholar] [CrossRef] [PubMed]
- Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993, 12, 2099–2108. [Google Scholar] [PubMed]
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar] [PubMed]
- Jolicoeur, P.; Baltimore, D. Effect of Fv-1 gene product on proviral DNA formation and integration in cells infected with murine leukemia viruses. Proc. Natl. Acad. Sci. U. S. A. 1976, 73, 2236–2240. [Google Scholar] [CrossRef] [PubMed]
- DesGroseillers, L.; Jolicoeur, P. Physical mapping of the Fv-1 tropism host range determinant of BALB/c murine leukemia viruses. J. Virol. 1983, 48, 685–696. [Google Scholar] [PubMed]
- Yamashita, M.; Perez, O.; Hope, T.J.; Emerman, M. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog. 2007, 3, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Emerman, M. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J. Virol. 2004, 78, 5670–5678. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.; Hensel, M.; Emerman, M. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J. 1992, 11, 3053–3058. [Google Scholar] [PubMed]
- Bowerman, B.; Brown, P.O.; Bishop, J.M.; Varmus, H.E. A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev. 1989, 3, 469–478. [Google Scholar] [CrossRef]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [PubMed]
- Mulky, A.; Cohen, T.V.; Kozlov, S.V.; Korbei, B.; Foisner, R.; Stewart, C.L.; KewalRamani, V.N. The LEM domain proteins emerin and LAP2alpha are dispensable for human immunodeficiency virus type 1 and murine leukemia virus infections. J. Virol. 2008, 82, 5860–5868. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.C.; Daigle, J.E.; Vandegraaff, N.; Engelman, A. Wild-type levels of human immunodeficiency virus type 1 infectivity in the absence of cellular emerin protein. J. Virol. 2007, 81, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Yang, H.; Craigie, R. LAP2alpha and BAF collaborate to organize the Moloney murine leukemia virus preintegration complex. EMBO J. 2004, 23, 4670–4678. [Google Scholar] [CrossRef] [PubMed]
- Farnet, C.M.; Bushman, F.D. HIV-1 cDNA integration: requirement for HMG I(Y) protein for function of preintegration complexes in vitro. Cell 1997, 88, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yoder, K.; Hansen, M.S.; Olvera, J.; Miller, M.D.; Bushman, F.D. Retroviral cDNA integration: stimulation by HMG I family proteins. J. Virol. 2000, 74, 10965–10974. [Google Scholar] [CrossRef] [PubMed]
- Jeanson, L.; Subra, F.; Vaganay, S.; Hervy, M.; Marangoni, E.; Bourhis, J.; Mouscadet, J.-F. Effect of Ku80 depletion on the preintegrative steps of HIV-1 replication in human cells. Virology 2002, 300, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An essential role for LEDGF/p75 in HIV integration. Science 2006, 314, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Juretzek, T.; Holm, T.; Gartner, K.; Kanzler, S.; Lindemann, D.; Herchenroder, O.; Picard-Maureau, M.; Rammling, M.; Heinkelein, M.; Rethwilm, A. Foamy Virus Integration. J. Virol. 2004, 78, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Enssle, J.; Moebes, A.; Heinkelein, M.; Panhuysen, M.; Mauer, B.; Schweizer, M.; Neumann-Haefelin, D.; Rethwilm, A. An active foamy virus integrase is required for virus replication. J. Gen. Virol. 1999, 80, 1445–1452. [Google Scholar] [PubMed]
- Roth, M.J.; Schwartzberg, P.L.; Goff, S.P. Structure of the termini of DNA intermediates in the integration of retroviral DNA: dependence on IN function and terminal DNA sequence. Cell 1989, 58, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Aiyar, A.; Hindmarsh, P.; Skalka, A.M.; Leis, J. Concerted integration of linear retroviral DNA by the avian sarcoma virus integrase in vitro: dependence on both long terminal repeat termini. J. Virol. 1996, 70, 3571–3580. [Google Scholar] [PubMed]
- Jonsson, C.B.; Donzella, G.A.; Gaucan, E.; Smith, C.M.; Roth, M.J. Functional domains of Moloney murine leukemia virus integrase defined by mutation and complementation analysis. J. Virol. 1996, 70, 4585–4597. [Google Scholar] [PubMed]
- Vink, C.; Yeheskiely, E.; van der Marel, G.A.; van Boom, J.H.; Plasterk, R.H. Site-specific hydrolysis and alcoholysis of human immunodeficiency virus DNA termini mediated by the viral integrase protein. Nucleic Acids Res. 1991, 19, 6691–6698. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Vanegas, M.; Fregoso, O.; Saenz, D.; Chung, S.; Peretz, M.; Poeschla, E.M. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 2004, 78, 9524–9537. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in Lentiviral Infectivity and Integration Targeting. PLoS ONE 2007, 2, e1340. [Google Scholar] [CrossRef] [PubMed]
- Pryciak, P.M.; Muller, H.P.; Varmus, H.E. Simian virus 40 minichromosomes as targets for retroviral integration in vivo. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9237–9241. [Google Scholar] [CrossRef] [PubMed]
- Pryciak, P.M.; Varmus, H.E. Nucleosomes, DNA-binding, proteins, and DNA sequence modulate retroviral integration target site selection. Cell 1992, 69, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Pruss, D.; Bushman, F.D.; Wolffe, A.P. Human immunodeficiency virus integrase directs integration to sites of severe DNA distortion within the nucleosome core. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 5913–5917. [Google Scholar] [CrossRef] [PubMed]
- Pruss, D.; Reeves, R.; Bushman, F.D.; Wolffe, A.P. The influence of DNA and nucleosome structure on integration events directed by HIV integrase. J. Biol. Chem. 1994, 269, 25031–25041. [Google Scholar] [PubMed]
- Muller, H.P.; Varmus, H.E. DNA bending creates favored sites for retroviral integration: an explanation for preferred insertion sites in nucleosomes. EMBO J. 1994, 13, 4704–4714. [Google Scholar] [PubMed]
- Withers-Ward, E.S.; Kitamura, Y.; Barnes, J.P.; Coffin, J.M. Distribution of targets for avian retrovirus DNA integration in vivo. Genes Dev. 1994, 8, 1473–1487. [Google Scholar] [CrossRef]
- Shih, C.-C.; Stoye, J.; Coffin, J.M. Highly preferred targets for retrovirus integration. Cell 1988, 53, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Voytas, D.F.; Boeke, J.D. Yeast retrotransposons and tRNAs. Trends Genet. 1993, 9, 421–427. [Google Scholar] [CrossRef]
- Sandmeyer, S. Yeast retrotransposons. Curr. Opin. Genet. Dev. 1992, 2, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Sandmeyer, S. Integration by design. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5586–5588. [Google Scholar] [CrossRef] [PubMed]
- Bryk, M.; Banerjee, M.; Conte, D.; Curcio, M.J. The Sgs1 helicase of Saccharomyces cerevisiae inhibits retrotransposition of Ty1 multimeric arrays. Mol. Cell. Biol. 2001, 21, 5374–5388. [Google Scholar] [CrossRef] [PubMed]
- Chalker, D.; Sandmeyer, S.B. Ty3 integrates within the region of RNA polymerase III transcription initiation. Genes Dev. 1994, 6, 117–128. [Google Scholar] [CrossRef]
- Chalker, D.; Sandmeyer, S.B. Transfer RNA genes are genomic targets for de novo transposition of the yeast retrotransposon Ty3. Genetics 1990, 126, 837–850. [Google Scholar] [PubMed]
- Zhu, Y.; Zou, S.; Wright, D.A.; Voytas, D.F. Tagging chromatin with retrotransposons: target specificity of the Saccharomyces Ty5 retrotransposon changes with the chromosomal localization of Sir3p and Sir4p. Genes Dev. 1999, 13, 2738–2749. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dai, J.; Fuerst, P.G.; Voytas, D.F. Controlling integration specificity of a yeast retrotransposon. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5891–5895. [Google Scholar] [CrossRef] [PubMed]
- Khan, E.; Mack, J.P.G.; Katz, R.A.; Kulkosky, J.; Skalka, A.M. Retroviral integrase domains: DNA binding and the recognition of LTR sequences. Nucleic Acids Res. 1991, 19, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S.; Eickbush, T.H. Modular Evolution of the Integrase Domain in the Ty3/Gypsy Class of LTR Retrotransposons. J. Virol. 1999, 73, 5186–5190. [Google Scholar] [PubMed]
- Rice, P.; Mizuuchi, K. Structure of the bacteriophage Mu transposase core: A common structural motif for DNA transposition and retroviral integration. Cell 1995, 82, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Segall, A.; Goodman, S.; Nash, H. Architectural elements in nucleoprotein complexes: interchangeability of specific and non-specific DNA binding proteins. EMBO J. 1994, 13, 4536–4548. [Google Scholar] [PubMed]
- Groth, A.C.; Calos, M.P. Phage Integrases: Biology and Applications. J. Mol. Biol. 2004, 335, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Surette, M.G.; Chaconas, G. A protein factor which reduces the negative supercoiling requirement in the Mu DNA strand transfer reaction is Escherichia coli integration host factor. J. Biol. Chem. 1989, 264, 3028–3034. [Google Scholar] [PubMed]
- Mizuuchi, M.; Baker, T.A.; Mizuuchi, K. Assembly of phage Mu transpososomes: Cooperative transitions assisted by protein and DNA scaffolds. Cell 1995, 83, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Craigie, R.; Davies, D.R. Retroviral integrases and their cousins. Curr. Opin. Struct. Biol. 1996, 6, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D.; Malani, N.; Fernandes, J.; D'Orso, I.; Cagney, G.; Diamond, T.L.; Zhou, H.; Hazuda, D.J.; Espeseth, A.S.; Konig, R.; Bandyopadhyay, S.; Ideker, T.; Goff, S.P.; Krogan, N.J.; Frankel, A.D.; Young, J.A.; Chanda, S.K. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009, 5, e1000437. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: Viral and Cellular Determinants of Target-Site Selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Felice, B.; Cattoglio, C.; Cittaro, D.; Testa, A.; Miccio, A.; Ferrari, G.; Luzi, L.; Recchia, A.; Mavilio, F. Transcription factor binding sites are genetic determinants of retroviral integration in the human genome. PLoS One 2009, 4, e4571. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Narezkina, A.; Taganov, K.D.; Litwin, S.; Stoyanova, R.; Hayashi, J.; Seeger, C.; Skalka, A.M.; Katz, R.A. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 2004, 78, 11656–11663. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Song, O.K. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Orlova, M.; Yueh, A.; Leung, J.; Goff, S.P. Reverse transcriptase of Moloney murine leukemia virus binds to eukaryotic release factor 1 to modulate suppression of translational termination. Cell 2003, 115, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Katz, R.A.; Ishov, A.M.; Maul, G.G.; Skalka, A.M. The cellular protein Daxx interacts with avian sarcoma virus integrase and viral DNA to repress viral transcription. J. Virol. 2005, 79, 4610–4618. [Google Scholar] [CrossRef] [PubMed]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Kalpana, G.V.; Marmon, S.; Wang, W.; Crabtree, G.R.; Goff, S.P. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science 1994, 266, 2002–2006. [Google Scholar] [PubMed]
- Hamamoto, S.; Nishitsuji, H.; Amagasa, T.; Kannagi, M.; Masuda, T. Identification of a novel human immunodeficiency virus type 1 integrase interactor, Gemin2, that facilitates efficient viral cDNA synthesis in vivo. J. Virol. 2006, 80, 5670–5677. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Thys, W.; De Rijck, J.; Gijsbers, R.; Albanese, A.; Arosio, D.; Emiliani, S.; Rain, J.C.; Benarous, R.; Cereseto, A.; Debyser, Z. Transportin-SR2 Imports HIV into the Nucleus. Curr. Biol. 2008, 18, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, G.L.; Zhang, J.Q.; Tian, L.; Xue, J.L.; Chen, J.Z.; Jia, W. Daxx interacts with HIV-1 integrase and inhibits lentiviral gene expression. Biochem. Biophys. Res. Commun. 2008, 373, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Yung, E. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nature Med. 2001, 7, 920–926. [Google Scholar] [CrossRef]
- Ariumi, Y.; Serhan, F.; Turelli, P.; Telenti, A.; Trono, D. The integrase interactor 1 (INI1) proteins facilitate Tat-mediated human immunodeficiency virus type 1 transcription. Retrovirology 2006, 3, 47. [Google Scholar] [CrossRef]
- Sorin, M.; Cano, J.; Das, S.; Mathew, S.; Wu, X.; Davies, K.P.; Shi, X.; Cheng, S.W.; Ott, D.; Kalpana, G.V. Recruitment of a SAP18-HDAC1 complex into HIV-1 virions and its requirement for viral replication. PLoS Pathog. 2009, 5, e1000463. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Turlure, F.; Devroe, E.; Silver, P.A.; Engelman, A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 2004, 9, 3187–3208. [Google Scholar] [CrossRef] [PubMed]
- Emiliani, S.; Mousnier, A.; Busschots, K.; Maroun, M.; Van Maele, B.; Tempé, D.; Vandekerckhove, L.; Moisant, F.; Ben-Slama, L.; Witvrouw, M.; Christ, F.; Rain, J.-C.; Dargemont, C.; Debyser, Z.; Benarous, R. Integrase Mutants Defective for Interaction with LEDGF/p75 Are Impaired in Chromosome Tethering and HIV-1 Replication. J. Biol. Chem. 2005, 280, 25517–25523. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J.; Vandekerckhove, L.; Gijsbers, R.; Hombrouck, A.; Hendrix, J.; Vercammen, J.; Engelborghs, Y.; Christ, F.; Debyser, Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J. Virol. 2006, 80, 11498–11509. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Llano, M.; Poeschla, E.M.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Hombrouck, A.; De Rijck, J.; Hendrix, J.; Vandekerckhove, L.; Voet, A.; Maeyer, M.D.; Witvrouw, M.; Engelborghs, Y.; Christ, F.; Gijsbers, R.; Debyser, Z. Virus Evolution Reveals an Exclusive Role for LEDGF/p75 in Chromosomal Tethering of HIV. PLoS Pathog. 2007, 3, e47. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar] [CrossRef] [PubMed]
- Busschots, K.; Vercammen, J.; Emiliani, S.; Benarous, R.; Engelborghs, Y.; Christ, F.; Debyser, Z. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 2005, 280, 17841–17847. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, R.; Ronen, K.; Vets, S.; Malani, N.; De Rijck, J.; McNeely, M.; Bushman, F.D.; Debyser, Z. LEDGF Hybrids Efficiently Retarget Lentiviral Integration Into Heterochromatin. Mol. Ther. 2010, 18, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Mulder, L.C.F.; Chakrabarti, L.A.; Muesing, M.A. Interaction of HIV-1 Integrase with DNA Repair Protein hRad18. J. Biol. Chem. 2002, 277, 27489–27493. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.G.; Tateishi, S.; Bieniasz, P.D.; Muesing, M.A.; Yamaizumi, M.; Mulder, L.C.F. Effect of DNA Repair Protein Rad18 on Viral Infection. PLoS Pathog. 2006, 2, e40. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Kao, G.; Taganov, K.; Greger, J.G.; Favorova, O.; Merkel, G.; Yen, T.J.; Katz, R.A.; Skalka, A.M. Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 4778–4783. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Katz, R.A.; Skalka, A.M. A role for DNA-PK in retroviral DNA integration. Science 1999, 284, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Baekelandt, V.; Claeys, A.; Cherepanov, P.; De Clercq, E.; De Strooper, B.; Nuttin, B.; Debyser, Z. DNA-Dependent Protein Kinase Is Not Required for Efficient Lentivirus Integration. J. Virol. 2000, 74, 11278–11285. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Turelli, P.; Masutani, M.; Trono, D. DNA damage sensors ATM, ATR, DNA-PKcs, and PARP-1 are dispensable for human immunodeficiency virus type 1 integration. J. Virol. 2005, 79, 2973–2978. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Olvera, J.M.; Yoder, K.E.; Mitchell, R.S.; Butler, S.L.; Lieber, M.; Martin, S.L.; Bushman, F.D. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 2001, 20, 3272–3281. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Komatsu, K.; Agematsu, K.; Matsuoka, M. DNA double strand break repair enzymes function at multiple steps in retroviral infection. Retrovirology 2009, 6, 114. [Google Scholar] [CrossRef]
- Yoder, K.; Sarasin, A.; Kraemer, K.; McIlhatton, M.; Bushman, F.; Fishel, R. The DNA repair genes XPB and XPD defend cells from retroviral infection. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4622–4627. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.Y.; Velazquez-Dones, A.L.; Lyman, S.K.; Fu, X.D. Phosphorylation-dependent and -independent nuclear import of RS domain-containing splicing factors and regulators. J. Biol. Chem. 2003, 278, 18050–18055. [Google Scholar] [CrossRef] [PubMed]
- Gallay, P.; Hope, T.; Chin, D.; Trono, D. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 9825–9830. [Google Scholar] [CrossRef] [PubMed]
- Hearps, A.C.; Jans, D.A. HIV-1 integrase is capable of targeting DNA to the nucleus via an Importin α/β-dependent mechanism. Biochem. J. 2006, 398, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Matreyek, K.A.; Oztop, I.; Lee, K.; Tipper, C.H.; Li, X.; Dar, M.J.; KewalRamani, V.N.; Engelman, A. The Requirement for Cellular Transportin 3 (TNPO3 or TRN-SR2) during Infection Maps to Human Immunodeficiency Virus Type 1 Capsid and Not Integrase. J. Virol. 2010, 84, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Zaitseva, L.; Cherepanov, P.; Leyens, L.; Wilson, S.J.; Rasaiyaah, J.; Fassati, A. HIV-1 exploits importin 7 to maximize nuclear import of its DNA genome. Retrovirology 2009, 6, 11. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Wang, J.J.; Li, W.J.; Huang, L.; Tian, L.; Xue, J.L.; Chen, J.Z.; Jia, W. Cellular protein TTRAP interacts with HIV-1 integrase to facilitate viral integration. Biochem. Biophys. Res. Commun. 2009, 387, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Allouch, A.; Cereseto, A. Identification of cellular factors binding to acetylated HIV-1 integrase. Amino Acids 2009. [Google Scholar]
- Li, L.; Farnet, C.M.; Anderson, W.F.; Bushman, F.D. Modulation of activity of Moloney murine leukemia virus preintegration complexes by host factors in vitro. J. Virol. 1998, 72, 2125–2131. [Google Scholar] [PubMed]
- Lee, M.S.; Craigie, R. A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 1528–1533. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, M.K.; Bisgrove, D.; Shinn, P.; Chen, H.; Hoffmann, C.; Hannenhalli, S.; Verdin, E.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 2005, 79, 6610–6619. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.; Chiang, C.Y.; Berry, C.; Bushman, F. Global analysis of cellular transcription following infection with an HIV-based vector. Mol. Ther. 2003, 8, 674–687. [Google Scholar] [CrossRef]
- Corbeil, J.; Sheeter, D.; Genini, D.; Rought, S.; Leoni, L.; Du, P.; Ferguson, M.; Masys, D.R.; Welsh, J.B.; Fink, J.L.; Sasik, R.; Huang, D.; Drenkow, J.; Richman, D.D.; Gingeras, T. Temporal gene regulation during HIV-1 infection of human CD4+ T cells. Genome Res. 2001, 11, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- van 't Wout, A.B.; Lehrman, G.K.; Mikheeva, S.A.; O'Keeffe, G.C.; Katze, M.G.; Bumgarner, R.E.; Geiss, G.K.; Mullins, J.I. Cellular gene expression upon human immunodeficiency virus type 1 infection of CD4(+)-T-cell lines. J. Virol. 2003, 77, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.P. Genetics of retroviral integration. Annu. Rev. Genet. 1992, 26, 527–544. [Google Scholar] [PubMed]
- Urisman, A.; Molinaro, R.J.; Fischer, N.; Plummer, S.J.; Casey, G.; Klein, E.A.; Malathi, K.; Magi-Galluzzi, C.; Tubbs, R.R.; Ganem, D.; Silverman, R.H.; DeRisi, J.L. dentification of a Novel Gammaretrovirus in Prostate Tumors of Patients Homozygous for R462Q RNASEL Variant. PLoS Pathog. 2006, 2, e25. [Google Scholar] [CrossRef] [PubMed]
- Denner, J. Detection of a gammaretrovirus, XMRV, in the human population: Open questions and implications for xenotransplantation. Retrovirology 2010, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Studamire, B.; Goff, S.P. Host proteins interacting with the Moloney murine leukemia virus integrase: multiple transcriptional regulators and chromatin binding factors. Retrovirology 2008, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Cavalli, G. Recruitment of Polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 2009, 136, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- van Lohuizen, M.; Tijms, M.; Voncken, J.W.; Schumacher, A.; Magnuson, T.; Wientjens, E. Interaction of mouse polycomb-group (Pc-G) proteins Enx1 and Enx2 with Eed: indication for separate Pc-G complexes. Mol. Cell. Biol. 1998, 18, 3572–3579. [Google Scholar] [PubMed]
- Simon, J.A.; Tamkun, J.W. Programming off and on states in chromatin: mechanisms of Polycomb and trithorax group complexes. Curr. Opin. Genet. Dev. 2002, 12, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome Regulation by Polycomb and Trithorax Proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; Bollen, M.; Esteller, M.; Di Croce, L.; de Launoit, Y.; Fuks, F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Kuzmichev, A.; Jenuwein, T.; Tempst, P.; Reinberg, D. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol. Cell 2004, 14, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; Rubin, M.A.; Chinnaiyan, A.M. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; Sabel, M.S.; Livant, D.; Weiss, S.J.; Rubin, M.A.; Chinnaiyan, A.M. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Liu, Y.; Hsu, Y.-J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.-C.; Orkin, S.H. EZH1 Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Peytavi, R.; Hong, S.S.; Gay, B.; d'Angeac, A.D.; Selig, L.; Benichou, S.; Benarous, R.; Boulanger, P. HEED, the product of the human homolog of the murine eed gene, binds to the matrix protein of HIV-1. J. Biol. Chem. 1999, 274, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- Violot, S.; Hong, S.S.; Rakotobe, D.; Petit, C.; Gay, B.; Moreau, K.; Billaud, G.; Priet, S.; Sire, J.; Schwartz, O.; Mouscadet, J.F.; Boulanger, P. The human polycomb group EED protein interacts with the integrase of human immunodeficiency virus type 1. J. Virol. 2003, 77, 12507–12522. [Google Scholar] [CrossRef] [PubMed]
- Witte, V.; Laffert, B.; Rosorius, O.; Lischka, P.; Blume, K.; Galler, G.; Stilper, A.; Willbold, D.; D'Aloja, P.; Sixt, M.; Kolanus, J.; Ott, M.; Kolanus, W.; Schuler, G.; Baur, A.S. HIV-1 Nef mimics an integrin receptor signal that recruits the polycomb group protein Eed to the plasma membrane. Mol. Cell 2004, 13, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Rakotobe, D.; Tardy, J.C.; Andre, P.; Hong, S.S.; Darlix, J.L.; Boulanger, P. Human Polycomb group EED protein negatively affects HIV-1 assembly and release. Retrovirology 2007, 4, 37. [Google Scholar] [CrossRef]
- Grimaud, C.; Nègre, N.; Cavalli, G. From genetics to epigenetics: the tale of Polycomb group and trithorax group genes. Chromosome Res. 2006, 14, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Crabtree, G.R. An EZ Mark to Miss. Cell Stem Cell 2008, 3, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Collins, E.C.; Appert, A.; Ariza-McNautghton, L.; Pannell, R.; Yamada, Y.; Rabbitts, T.H. Mouse Af9 is a controller of embryo patterning, like Mll, whose human homologue fuses with AF9 after chromosomal translocation in leukemia. Mol. Cell. Biol. 2002, 22, 7313–7324. [Google Scholar] [CrossRef] [PubMed]
- von Bergh, A.R.; Wijers, P.M.; Groot, A.J.; van Zelderen-Bhola, S.; Falkenburg, J.H.; Kluin, P.M.; Schuuring, E. Identification of a novel RAS GTPase-activating protein (RASGAP) gene at 9q34 as an MLL fusion partner in a patient with de novo acute myeloid leukemia. Genes Chromosomes Cancer 2004, 39, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, C.S.; de Erkenez, A.C.; Gould, G.C. The polycomb protein MPc3 interacts with AF9, an MLL fusion partner in t(9;11)(p22;q23) acute leukemias. Oncogene 2001, 20, 3798–3805. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.S.; de Erkenez, A.C.; Hemenway, C.S. The mixed lineage leukemia fusion partner AF9 binds specific isoforms of the BCL-6 corepressor. Oncogene 2003, 22, 3395–3406. [Google Scholar] [CrossRef] [PubMed]
- Mimori, T.; Akizuki, M.; Yamagata, H.; Inada, S.; Yoshida, S.; Homma, M. Characterization of a high molecular weight acidic nuclear protein recognized by autoantibodies in sera from patients with polymyositis-scleroderma overlap. J. Clin. Invest. 1981, 68, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Reeves, W.H.; Sthoeger, Z.M. Molecular cloning of cDNA encoding the p70 (Ku) lupus autoantigen. J. Biol. Chem. 1989, 264, 5047–5052. [Google Scholar] [PubMed]
- Burgers, P.M. Eukaryotic DNA polymerases in DNA replication and repair. Chromosoma 1998, 107, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Featherstone, C.; Jackson, S.P. Ku, a DNA repair protein with multiple cellular functions? Mutat. Res./DNA Repair 1999, 434, 3–15. [Google Scholar] [CrossRef]
- Gellert, M. V(D)J Recombination: Rag proteins, repair factors, and regulation. Annu. Rev. Biochem. 2002, 71, 101–132. [Google Scholar] [CrossRef] [PubMed]
- Celli, G.B.; Denchi, E.L.; de Lange, T. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat. Cell Biol. 2006, 8, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Kiechle, M.; Friedl, A.A.; Manivasakam, P.; Eckardt-Schupp, F.; Schiestl, R.H. DNA integration by Ty integrase in yku70 mutant Saccharomyces cerevisiae. Mol. Cell. Biol. 2000, 20, 8836–8844. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A.; Jackson, S.P. Involvement of DNA end-binding protein Ku in Ty element retrotransposition. Mol. Cell. Biol. 1999, 19, 6260–6288. [Google Scholar] [PubMed]
- Masson, C.; Bury-Mone, S.; Guiot, E.; Saez-Cirion, A.; Schoevaert-Brossault, D.; Brachet-Ducos, C.; Delelis, O.; Subra, F.; Jeanson-Leh, L.; Mouscadet, J.F. Ku80 participates in the targeting of retroviral transgenes to the chromatin of CHO cells. J. Virol. 2007, 81, 7924–7932. [Google Scholar] [CrossRef] [PubMed]
- van Gent, D.C.; Mizuuchi, K.; Gellert, M. Similarities between initiation of V(D)J recombination and retroviral integration. Science 1996, 271, 1592–1594. [Google Scholar] [PubMed]
- Liu, Y.; Kao, H.I.; Bambara, R.A. Flap endonuclease 1: a central component of DNA metabolism. Annu. Rev. Biochem. 2004, 73, 589–615. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Stucki, M.; Hassa, P.O.; Imhof, R.; Gehrig, P. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol. Cell 2001, 7, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Cereseto, A.; Manganaro , L.; Gutierrez , M.I.; Terreni , M.; Fittipladi , A.; Lusic , M.; Marcello , A.; Giacca , M. Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J. 2005, 24, 3070–3081. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Singh, P.; Liu, R.; Qiu, J.; Zheng, L.; Finger, L.D.; Alas, S. Multiple but dissectible functions of FEN-1 nucleases in nucleic acid processing, genome stability and diseases. BioEssays 2005, 27, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006, 5, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Tamkun, J.W.; Deuring, R.; Scott, M.P.; Kissinger, M.; Pattatucci, A.M.; Kaufman, T.C.; Kennison, J.A. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 1992, 68, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; Green, M.R. A novel, mitogen-activated nuclear kinase is related to a Drosophila developmental regulator. Genes Dev. 1996, 10, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Crowley, T.E.; Kaine, E.M.; Yoshida, M.; Nandi, A.; Wolgemuth, D.J. Reproductive cycle regulation of nuclear import, euchromatic localization, and association with components of Pol II mediator of a mammalian double-bromodomain protein. Mol. Endocrinol. 2002, 16, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Viejo-Borbolla, A.; Ottinger, M.; Bruning, E.; Burger, A.; Konig, R.; Kati, E.; Sheldon, J.A.; Schulz, T.F. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi's Sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 2005, 79, 13618–13629. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.H.; Hamana, N.; Nezu, J.; Shimane, M. A novel family of bromodomain genes. Genomics 2000, 63, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, M.J.; Tae, H.J.; Sollenberger, K.G.; Mascarenhas, N.T.; Johansen, L.M.; Taparowsky, E.J. B-ATF: a novel human bZIP protein that associates with members of the AP-1 transcription factor family. Oncogene 1995, 11, 2255–2265. [Google Scholar] [PubMed]
- Echlin, D.R.; Tae, H.J.; Mitin, N.; Taparowsky, E.J. B-ATF functions as a negative regulator of AP-1 mediated transcription and blocks cellular transformation by Ras and Fos. Oncogene 2000, 19, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.H.; Sorensen, A.B.; Morris, D.W.; Dutra, J.C.; Engelhard, E.K.; Wang, C.L.; Schmidt, J.; Pedersen, F.S. Tumor model-specific proviral insertional mutagenesis of the Fos/Jdp2/Batf locus. Virology 2005, 337, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.M.; Deppmann, C.D.; Erickson, K.D.; Coffin III, W.F.; Thornton, T.M.; Humphrey, S.E.; Martin, J.M.; Taparowsky, E.J. EBNA2 and Activated Notch Induce Expression of BATF. J. Virol. 2003, 77, 6029–6040. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, L.C.; Collins, T. The SCAN domain family of zinc finger transcription factors. Gene 2005, 359, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.; Goulding, M.; Walther, C.; Imai, K.; Fickenscher, H. The ubiquitous transactivator Zfp-38 is upregulated during spermatogenesis with differential transcription. Mech. Dev. 1992, 39, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Blacklow, S.C.; Collins, T. The zinc finger-associated SCAN box is a conserved oligomerization domain. Mol. Cell. Biol. 1999, 19, 8526–8535. [Google Scholar] [PubMed]
- Yang, X.W.; Wynder, C.; Doughty, M.L.; Heintz, N. BAC-mediated gene-dosage analysis reveals a role for Zipro1 (Ru49/Zfp38) in progenitor cell proliferation in cerebellum and skin. Nat. Genet. 1999, 22, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Scarpulla, R.C. PGC-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol. 2001, 21, 3738–3749. [Google Scholar] [CrossRef] [PubMed]
- Lelliott, C.; Medina-Gomez, G.; Petrovic , N.; Kis , A.; Feldmann , H.M.; Bjursell , N.P.; Curtis , K.; Campbell , M.; Hu , P.; Zhang , D.; Litwin , ]S.E.; Zaha , V.G.; Fountain , K.T.; Boudina , S.; Jimenez-Linan , M.; Blount , M.; Lopez , M.; Meirhaeghe , A.; Bohlooly-Y, M.; Storlien , L.; Stromstedt , M.; Snaith , M.; Oresic , M.; Abel , E.D.; Cannon, B.; Vidal-Puig , A. A Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 2006, 4, 2042–2056. [Google Scholar] [CrossRef]
- Monsalve, M.; Wu, Z.; Adelmant, G.; Puigserver, P.; Fan, M.; Spiegelman, B.M. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol. Cell 2000, 6, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Nagy, P.L.; Zalkin, H. Role of NRF-1 in bidirectional transcription of the human GPAT-AIRC purine biosynthesis locus. Nucleic Acids Res. 1997, 25, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Wegner, S.A.; Ehrenberg, P.K.; Chang, G.; Dayhoff, D.E.; Sleeker, A.L.; Michael, N.L. Genomic organization and functional characterization of the chemokine receptor CXCR4, a major entry co-receptor for human immunodeficiency virus type 1. J. Biol. Chem. 1998, 273, 4754–4760. [Google Scholar] [CrossRef] [PubMed]
- Solecki, D.; Bernhardt, G.; Lipp, M.; Wimmer, E. Identification of a nuclear respiratory factor-1 binding site within the core promoter of the human polio virus receptor/CD155 gene. J. Biol. Chem. 2000, 275, 12453–12462. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, A. Regulation of cortical structure by the ezrin-radixin-moesin protein family. Curr. Opin. Cell Biol. 1999, 11, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Funayama, N.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. A gene family consisting of ezrin, radixin and moesin. Its specific localization at actin filament/plasma membrane association sites. J. Cell Sci. 1992, 103, 131–143. [Google Scholar] [PubMed]
- Hoeflich, K.P.; Ikura, M. Radixin: cytoskeletal adopter and signaling protein. Int. J. Biochem. Cell Biol. 2004, 36, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, A.; Edwards, K.; Fehon, R.G. ERM proteins and merlin: integrators at the cell cortex. Mol. Cell. Biol. 2002, 3, 586–599. [Google Scholar]
- Naghavi, M.H.; Valente, S.; Hatziioannou, T.; de Los Santos, K.; Wen, Y.; Mott, C.; Gundersen, G.G.; Goff, S.P. Moesin regulates stable microtubule formation and limits retroviral infection in cultured cells. EMBO J. 2007, 26, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Janket, M.L.; DeRicco, J.S.; Borowski, L.; Ayyavoo, V. Human immunodeficiency virus (HIV-1) Vpr induced downregulation of NHE1 induces alteration in intracellular pH and loss of ERM complex in target cells. Virus Res. 2007, 126, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Haedicke, J.; de Los Santos, K.; Goff, S.P.; Naghavi, M.H. The Ezrin-radixin-moesin family member ezrin regulates stable microtubule formation and retroviral infection. J. Virol. 2008, 82, 4665–4670. [Google Scholar] [CrossRef] [PubMed]
- Barrero-Villar, M.; Cabrero, J.R.; Gordon-Alonso, M.; Barroso-Gonzalez, J.; Alvarez-Losada, S.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F.; Valenzuela-Fernandez, A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 2009, 122, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Yoshii, H.; Kamiyama, H.; Tominaga, C.; Tanaka, Y.; Sato, H.; Yamamoto, N. Ezrin, Radixin, and Moesin (ERM) proteins function as pleiotropic regulators of human immunodeficiency virus type 1 infection. Virology 2008, 375, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.L.; Arora, V.K.; Raney, A.; Kuo, L.S.; Xiao, G.H.; O'Neill, E.; Testa, J.R.; Foster, J.L.; Garcia, J.V. Activation of p21-activated kinase 2 by human immunodeficiency virus type 1 Nef induces merlin phosphorylation. J. Virol. 2005, 79, 14976–14980. [Google Scholar] [CrossRef] [PubMed]
- Battle, D.J.; Kasim, M.; Yong, J.; Lotti, F.; Lau, C.K.; Mouaikel, J.; Zhang, Z.; Han, K.; Wan, L.; Dreyfuss, G. The SMN complex: an assembly machine for RNPs. Cold Spring Harb. Symp. Quant. Biol. 2006, LXXI, 313–320. [Google Scholar] [CrossRef]
- Schrank, B.; Gotz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Gubitz, A.K.; Wan, L.; Battle, D.J.; Dostie, J.; Golembe, T.J.; Dreyfuss, G. Gemins modulate the expression and activity of the SMN complex. Hum. Mol. Genet. 2005, 14, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Golembe, T.J.; Yong, J.; Dreyfuss, G. Specific sequence features, recognized by the SMN complex, identify snRNAs and determine their fate as snRNPs. Mol. Cell. Biol. 2005, 25, 10989–11004. [Google Scholar] [CrossRef] [PubMed]
- Otter, S.; Grimmler, M.; Neuenkirchen, N.; Chari, A.; Sickmann, A.; Fischer, U. A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J. Biol. Chem. 2007, 282, 5825–5833. [Google Scholar] [CrossRef] [PubMed]
- Golembe, T.J.; Yong, J.; Battle, D.J.; Feng, W.; Wan, L.; Dreyfuss, G. Lymphotropic Herpesvirus saimiri Uses the SMN Complex To Assemble Sm Cores on Its Small RNAs. Mol. Cell. Biol. 2005, 25, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 2001, 13, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Mulhauser, F.; Wersig, C.; Groning, K.; Bilbe, G. Mammalian splicing factor SF3a120 represents a new member of the SURP family of proteins and is homologous to the essential splicing factor PRP21p of Saccharomyces cerevisiae. RNA 1995, 1, 260–272. [Google Scholar] [PubMed]
- Brosi, R.; Groning, K.; Behrens, S.E.; Luhrmann, R.; Kramer, A. Interaction of mammalian splicing factor SF3a with U2 snRNP and relation of its 60-kD subunit to yeast PRP9. Science 1993, 262, 102–105. [Google Scholar] [PubMed]
- Bates, G.J.; Nicol, S.M.; Wilson, B.J.; Jacobs, A.M.; Bourdon, J.C.; Wardrop, J.; Gregory, D.J.; Lane, D.P.; Perkins, N.D.; Fuller-Pace, F.V. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J. 2005, 24, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Iggo, R.D.; Lane, D.P. Nuclear protein p68 is an RNA-dependent ATPase. EMBO J. 1989, 8, 1827–1831. [Google Scholar] [PubMed]
- Wilson, B.J.; Bates, G.J.; Nicol, S.M.; Gregory, D.J.; Perkins, N.D.; Fuller-Pace, F.V. The p68 and p72 DEAD box RNA helicases interact with HDAC1 and repress transcription in a promoter-specific manner. BMC Mol. Biol. 2004, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Fuller-Pace, F.V. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006, 34, 4206–4215. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Gattoni, R.; Carrascal, M.; Abian, J.; Stevenin, J.; Bach-Elias, M. Roles of hnRNP A1, SR proteins, and p68 helicase in c-H-ras alternative splicing regulation. Mol. Cell. Biol. 2003, 23, 2927–2941. [Google Scholar] [CrossRef] [PubMed]
- Rossow, K.L.; Janknecht, R. Synergism between p68 RNA helicase and the transcriptional coactivators CBP and p300. Oncogene 2003, 22, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Spradling, A.C. The Drosophila P68 RNA helicase regulates transcriptional deactivation by promoting RNA release from chromatin. Genes Dev. 2006, 20, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, Z.; Schoen, S.R.; Messing, E.M.; Wu, G. A novel MET-interacting protein shares high sequence similarity with RanBPM, but fails to stimulate MET-induced Ras/Erk signaling. Biochem. Biophys. Res. Commun. 2004, 313, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; De Villartay, J.P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; Fischer, A.; Davies, E.G.; Kuis, W.; Leiva, L.; Cavazzana-Calvo, M. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; Asnafi, V.; MacIntyre, E.; Dal Cortivo, L.; Radford, I.; Brousse, N.; Sigaux, F.; Moshous, D.; Hauer, J.; Borkhardt, A.; Belohradsky, B.H.; Wintergerst, U.; Velez, M.C.; Leiva, L.; Sorensen, R.; Wulffraat, N.; Blanche, S.; Bushman, F.D.; Fischer, A.; Cavazzana-Calvo, M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; Gilmour, K.C.; Adams, S.; Thornhill, S.I.; Parsley, K.L.; Staal, F.J.; Gale, R.E.; Linch, D.C.; Bayford, J.; Brown, L.; Quaye, M.; Kinnon, C.; Ancliff, P.; Webb, D.K.; Schmidt, M.; von Kalle, C.; Gaspar, H.B.; Thrasher, A.J. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Uren, A.G.; Kool, J.; Berns, A.; van Lohuizen, M. Retroviral insertional mutagenesis: past, present and future. Oncogene 2005, 24, 7656–7672. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Studamire, B.; Goff, S.P. Interactions of Host Proteins with the Murine Leukemia Virus Integrase. Viruses 2010, 2, 1110-1145. https://doi.org/10.3390/v2051110
Studamire B, Goff SP. Interactions of Host Proteins with the Murine Leukemia Virus Integrase. Viruses. 2010; 2(5):1110-1145. https://doi.org/10.3390/v2051110
Chicago/Turabian StyleStudamire, Barbara, and Stephen P. Goff. 2010. "Interactions of Host Proteins with the Murine Leukemia Virus Integrase" Viruses 2, no. 5: 1110-1145. https://doi.org/10.3390/v2051110
APA StyleStudamire, B., & Goff, S. P. (2010). Interactions of Host Proteins with the Murine Leukemia Virus Integrase. Viruses, 2(5), 1110-1145. https://doi.org/10.3390/v2051110