Herpesviruses and Autophagy: Catch Me If You Can!

Abstract
:1. Overview of Autophagy
1.1. Autophagosome Formation and Expansion
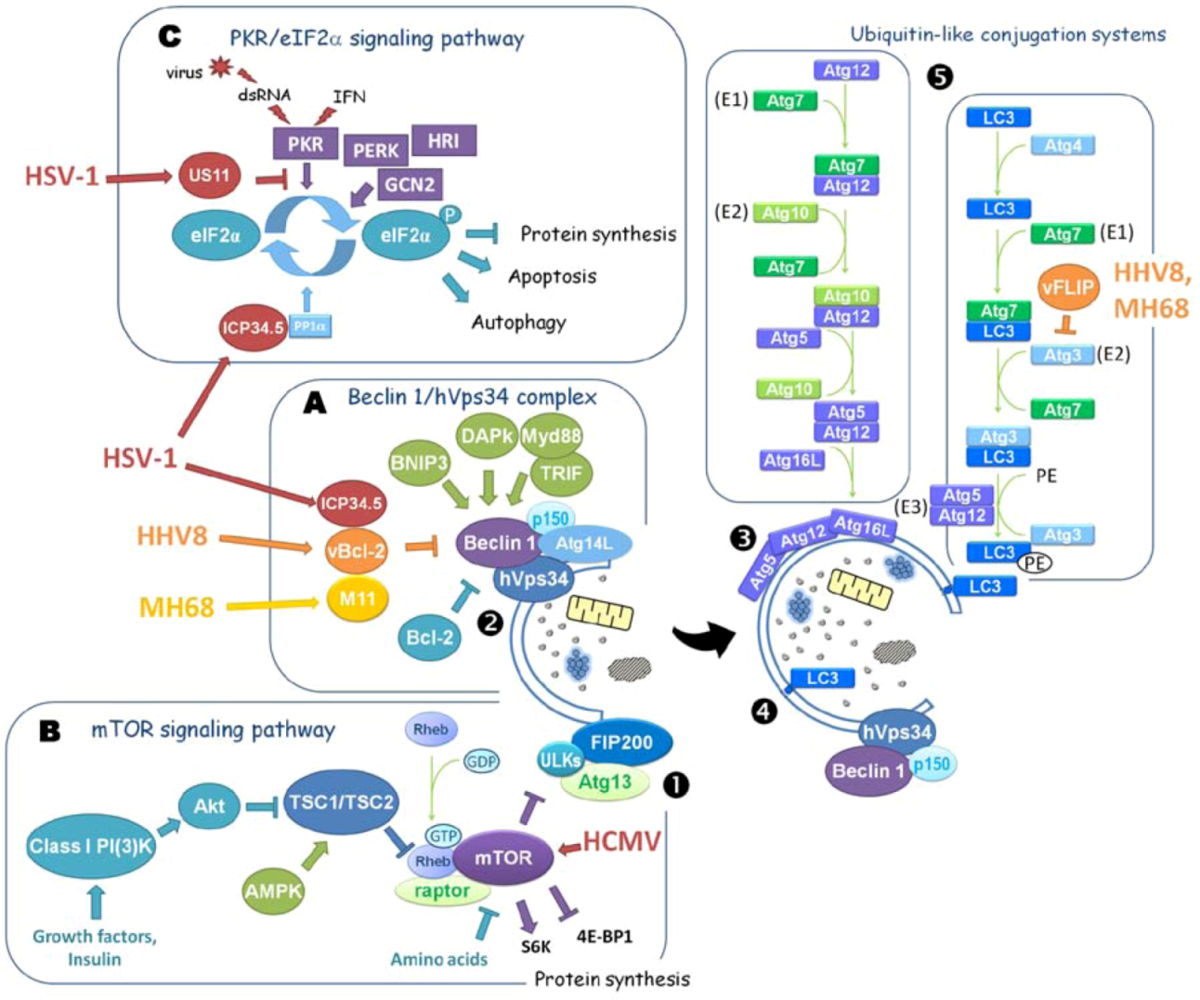
1.2. Regulation of Autophagy
1.3. Autophagy and Viruses
2. Autophagy is Modulated by Herpesviruses
2.1. Alphaherpesvirinae
2.1.1. Herpes Simplex Virus Type 1 (HSV-1)
Impact of Autophagy on HSV-1 Viral Replication
2.1.2. Bovine Herpes Simplex Virus Type 1 (BHV-1)
2.1.3. Varicella-zoster Virus (VZV)
2.2. Betaherpesvirinae
2.2.1. Human Cytomegalovirus (HCMV)
2.2.2. Human Herpesvirus (HHV-6)
2.3. Gammaherpesvirinae
2.3.1. Epstein-Barr Virus (EBV)
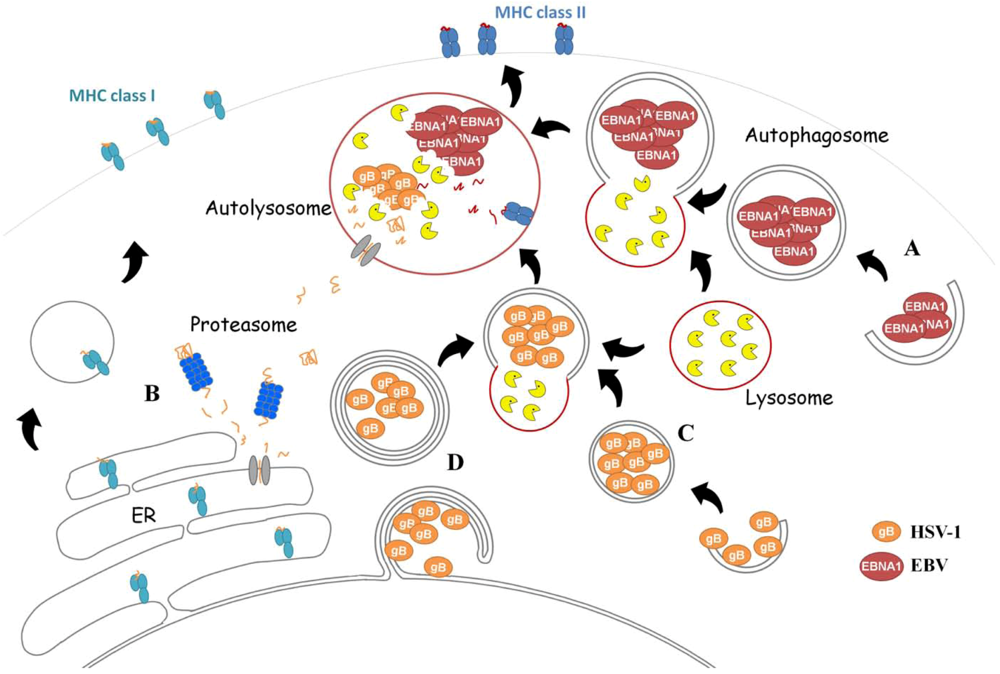
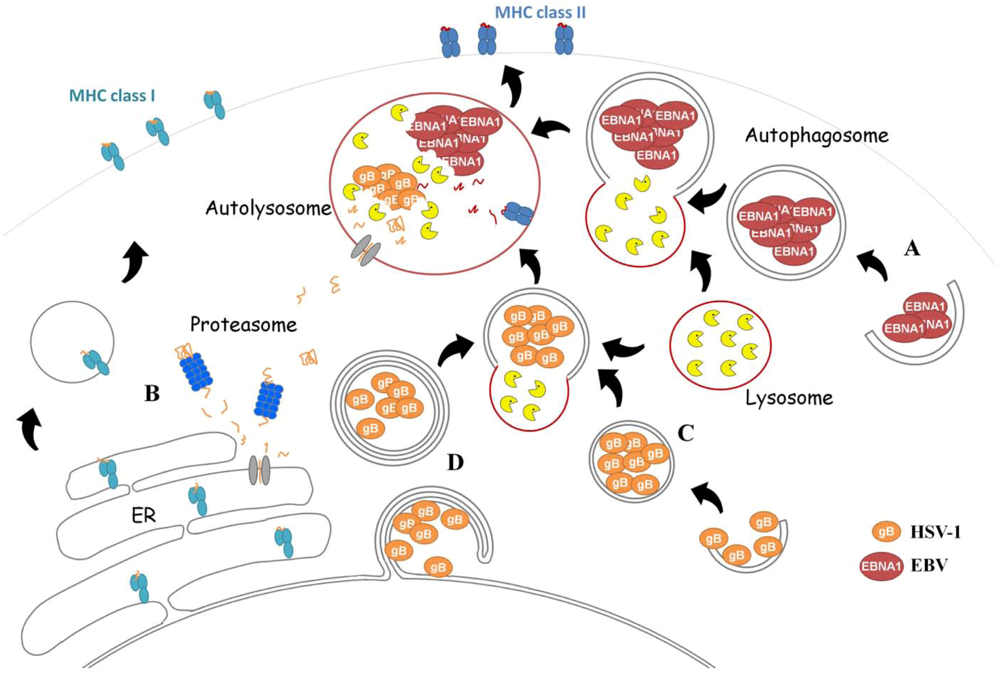
EBNA1 Is Loaded onto MHC Class II after Macroautophagy
Latency-associated LMP1 Induces Autophagy
2.3.2. Viral Bcl-2 Homologs of γHerpesviruses Are both Antiapoptotic and Antiautophagic
3. Conclusions

{kind=link}
{kind=link}
| Herpesvirus | Viral evasion of autophagy | Autophagy promotion | Host-virus interactions and host antiviral defense | Viral proteins/mechanisms | Ref. | |
|---|---|---|---|---|---|---|
| Alphaherpesvirinae | ||||||
| Herpes simplex virus type 1 (HSV-1) | HSV-1 infection inhibits autophagy in neurons and in fibroblasts | HSV-1 infection induces autophagy in macrophages | Viral control of autophagy is involved in neurovirulenceAutophagy contributes to MHC class I presentation of gB to CD8+ cells | ICP34.5 blocks autophagy by inhibiting PKR signaling and by direct interaction with Beclin 1gB | [38][36][61][47] | |
| Bovine herpesvirus type 1 (BHV-1) | BHV-1 WT may inhibit autophagy whereas the bICP0 null mutant may induce autophagy in MDBK cells | bICP0 may block autophagy | [70] | |||
| Varicella-zoster Virus (VZV) | VZV stimulates autophagy in fibroblasts and in vivo in human zoster vesicles | Unknown | [57] | |||
| Betaherpesvirinae | ||||||
| Human cytomegalovirus (HCMV) | HCMV inhibits autophagy in human fibroblasts | May activate mTOR pathway? | [71] | |||
| Gammaherpesvirinae | ||||||
| Epstein-Barr virus (EBV) | A latency-associated protein (LMP1) induces autophagy | Autophagy contributes to MHC class II presentation of EBNA1 to CD4+ cells | LMP1Proposed role in regulating its own expression levelLatency associated protein EBNA1 | [80][48] | ||
| Human Herpesvirus 8(HHV8),murine -HV68 | HHV8 may inhibit autophagy | Unknown | Viral homologs of Bcl-2(vBcl-2, M11) inhibit autophagy by its interaction with Beclin 1Viral homologs of FLIP (vFLIP) inhibit autophagy by interaction with Atg3 | [30][86][89][88] | ||
Acknowledgments
References
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W.; Levine, B. Autophagy genes in immunity. Nat. Immunol. 2009, 10, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; Ohsumi, Y. A unified nomenclature for yeast autophagy-related genes. Dev. Cell. 2003, 5, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Sasaki, T.; Iemura, S.I.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Mercer, C.A.; Kaliappan, A.; Dennis, P.B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009, 5, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Kamada, Y.; Kabeya, Y.; Sekito, T.; Ohsumi, Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol. Biol. Cell 2008, 19, 2039–2050. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Yorimitsu, T.; Reggiori, F.; Legakis, J.E.; Wang, C.W.; Klionsky, D.J. Atg17 regulates the magnitude of the autophagic response. Mol. Biol. Cell 2005, 16, 3438–3453. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Nair, U.; Geng, J.; Klionsky, D.J. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol. Biol. Cell 2008, 19, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Fan, W.; Chen, K.; Ding, X.; Chen, S.; Zhong, Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl Acad. Sci. USA 2008, 105, 19211–19216. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. Embo J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. Embo J. 1999, 18, 5234–5241. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Tanida-Miyake, E.; Komatsu, M.; Ueno, T.; Kominami, E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP; MAP-LC3; facilitates the conjugation of hApg12p to hApg5p. J. Biol. Chem. 2002, 277, 13739–13744. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; Noda, T.; Ohsumi, Y. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–92. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; Akira, S.; Noda, T.; Yoshimori, T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Signalling and autophagy regulation in health, aging and disease. Mol. Aspects Med. 2006, 27, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Tooze, S.A. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009, 16, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; Goldberg, A.L.; Schiaffino, S.; Sandri, M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Li, B.; Chen, Y.; Ren, X.; Liu, C.G.; Yang, J.M. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009, 5, 816–823. [Google Scholar] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Bauvy, C.; Carpentier, S.; Levade, T.; Levine, B.; Codogno, P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009, 284, 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. Embo J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Esclatine, A.; Chaumorcel, M.; Codogno, P. Macroautophagy signaling and regulation. Curr. Top. Microbiol. Immunol. 2009, 335, 33–70. [Google Scholar] [PubMed]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.t.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef]
- Kirkegaard, K.; Taylor, M.P.; Jackson, W.T. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2004, 2, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; Shiosaka, S.; Hammarback, J.A.; Urano, F.; Imaizumi, K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity; microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Levine, B. Autophagy, antiviral immunity; viral countermeasures. Biochim. Biophys. Acta 2009, 1793, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Virgin, H.W.t.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [PubMed]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [PubMed]
- Liu, Y.; Schiff, M.; Czymmek, K.; Talloczy, Z.; Levine, B.; Dinesh-Kumar, S.P. Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005, 121, 567–577. [Google Scholar] [CrossRef] [PubMed]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; Desjardins, M. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Munz, C. Macroautophagy in immunity and tolerance. Traffic 2009, 10, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS One 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; Federico, M.; Panganiban, A.; Vergne, I.; Deretic, V. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, K. Subversion of the cellular autophagy pathway by viruses. Curr. Top. Microbiol. Immunol. 2009, 335, 323–333. [Google Scholar] [PubMed]
- Jackson, W.T.; Giddings, T.H.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Burgon, T.B.; Kirkegaard, K.; Jackson, W.T. Role of microtubules in extracellular release of poliovirus. J. Virol. 2009, 83, 6599–6609. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.N.; Jackson, W.; Laird, D.T.; Culp, T.D.; Grose, C.; Haynes 2nd, J.I.; Benetti, L. Varicella-zoster virus infection induces autophagy in both cultured cells and human skin vesicles. J. Virol. 2009, 83, 5466–5476. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [PubMed]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [PubMed]
- Cheng, G.; Brett, M.E.; He, B. Val193 and Phe195 of the gamma 1 34.5 protein of herpes simplex virus 1 are required for viral resistance to interferon-alpha/beta. Virology 2001, 290, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.E.; Ward, S.L.; Mizushima, N.; Levine, B.; Leib, D.A. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 2007, 81, 12128–12134. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Roizman, B. Herpes simplex virus 1 gamma(1)34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc. Natl. Acad. Sci. USA 1994, 91, 5247–5251. [Google Scholar] [CrossRef]
- Leib, D.A.; Machalek, M.A.; Williams, B.R.; Silverman, R.H.; Virgin, H.W. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6097–6101. [Google Scholar] [CrossRef]
- Cassady, K.A.; Gross, M. The herpes simplex virus type 1 U(S)11 protein interacts with protein kinase R in infected cells and requires a 30-amino-acid sequence adjacent to a kinase substrate domain. J. Virol. 2002, 76, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Khoo, D.; Mohr, I.; Sen, G.C. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J. Virol. 2002, 76, 11054–11064. [Google Scholar] [CrossRef] [PubMed]
- Cassady, K.A.; Gross, M.; Roizman, B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J. Virol. 1998, 72, 7005–7011. [Google Scholar] [PubMed]
- Leib, D.A.; Alexander, D.E.; Cox, D.; Yin, J.; Ferguson, T.A. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T cell responses. J. Virol. 2009. [Google Scholar]
- English, L.; Chemali, M.; Desjardins, M. Nuclear membrane-derived autophagy, a novel process that participates in the presentation of endogenous viral antigens during HSV-1 infection. Autophagy 2009, 5, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Geiser, V.; Rose, S.; Jones, C. Bovine herpesvirus type 1 induces cell death by a cell-type-dependent fashion. Microb. Pathog. 2008, 44, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Chaumorcel, M.; Souquere, S.; Pierron, G.; Codogno, P.; Esclatine, A. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 2008, 4, 46–53. [Google Scholar] [PubMed]
- Kudchodkar, S.B.; Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc. Natl. Acad. Sci. USA 2006, 103, 14182–14187. [Google Scholar] [CrossRef]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, F.; Granata, R.; Meijer, A.J.; Codogno, P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009, 16, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- McCormick, A.L. Control of apoptosis by human cytomegalovirus. Curr. Top. Microbiol. Immunol. 2008, 325, 281–295. [Google Scholar] [PubMed]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y. Recent topics related to human herpesvirus 6 cell tropism. Cell Microbiol. 2009, 11, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Meiffren, G.; Gregoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; Codogno, P.; Rabourdin-Combe, C.; Faure, M. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Munz, C. Innate and adaptive immunity through autophagy. Immunity 2007, 27, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.S.; Long, H.M.; Haigh, T.A.; Larsen, M.; Brooks, J.; Rickinson, A.B. A role for intercellular antigen transfer in the recognition of EBV-transformed B cell lines by EBV nuclear antigen-specific CD4+ T cells. J. Immunol. 2006, 177, 3746–3756. [Google Scholar] [PubMed]
- Gannage, M.; Munz, C. Monitoring macroautophagy by major histocompatibility complex class II presentation of targeted antigens. Methods Enzymol. 2009, 452, 403–421. [Google Scholar] [PubMed]
- Lam, N.; Sugden, B. CD40 and its viral mimic, LMP1: similar means to different ends. Cell Signal. 2003, 15, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 2008, 111, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Colbert, C.L.; Becker, N.; Wei, Y.; Levine, B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 2008, 4, 989–997. [Google Scholar] [PubMed]
- Ku, B.; Woo, J.S.; Liang, C.; Lee, K.H.; Hong, H.S.; E, X.; Kim, K.S.; Jung, J.U.; Oh, B.H. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog. 2008, 4, e25. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; Jung, J.U. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; E, X.; Jung, J.U. Downregulation of autophagy by herpesvirus Bcl-2 homologs. Autophagy 2008, 4, 268–272. [Google Scholar] [PubMed]
- Lee, M.A.; Yates, J.L. BHRF1 of Epstein-Barr virus, which is homologous to human proto-oncogene bcl2, is not essential for transformation of B cells or for virus replication in vitro. J. Virol. 1992, 66, 1899–1906. [Google Scholar] [PubMed]
- Cheng, E.H.; Nicholas, J.; Bellows, D.S.; Hayward, G.S.; Guo, H.G.; Reitz, M.S.; Hardwick, J.M. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. USA 1997, 94, 690–694. [Google Scholar] [CrossRef]
- Virgin, H.W.t.; Latreille, P.; Wamsley, P.; Hallsworth, K.; Weck, K.E.; Dal Canto, A.J.; Speck, S.H. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 1997, 71, 5894–5904. [Google Scholar] [PubMed]
- Gangappa, S.; van Dyk, L.F.; Jewett, T.J.; Speck, S.H.; Virgin, H.W.t. Identification of the in vivo role of a viral bcl-2. J. Exp. Med. 2002, 195, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Shi, Y. FLIP and the death effector domain family. Oncogene 2008, 27, 6216–6227. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.S.; Camara, N.O.; Nogueira, E.; Pereira, M.G.; Granato, C.; Melaragno, C.; Camargo, L.F.; Pacheco-Silva, A. The use of sirolimus in ganciclovir-resistant cytomegalovirus infections in renal transplant recipients. Clin. Transplant. 2007, 21, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Trotter, J.F.; Wallack, A.; Steinberg, T. Low incidence of cytomegalovirus disease in liver transplant recipients receiving sirolimus primary immunosuppression with 3-day corticosteroid taper. Transpl. Infect. Dis. 2003, 5, 174–180. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Cavignac, Y.; Esclatine, A. Herpesviruses and Autophagy: Catch Me If You Can! Viruses 2010, 2, 314-333. https://doi.org/10.3390/v2010314
Cavignac Y, Esclatine A. Herpesviruses and Autophagy: Catch Me If You Can! Viruses. 2010; 2(1):314-333. https://doi.org/10.3390/v2010314
Chicago/Turabian StyleCavignac, Yolaine, and Audrey Esclatine. 2010. "Herpesviruses and Autophagy: Catch Me If You Can!" Viruses 2, no. 1: 314-333. https://doi.org/10.3390/v2010314
APA StyleCavignac, Y., & Esclatine, A. (2010). Herpesviruses and Autophagy: Catch Me If You Can! Viruses, 2(1), 314-333. https://doi.org/10.3390/v2010314