Dual Role of p53 in Innate Antiviral Immunity

Abstract
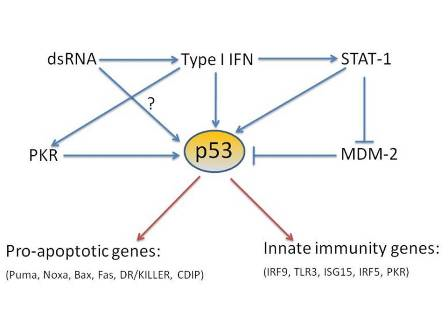
:
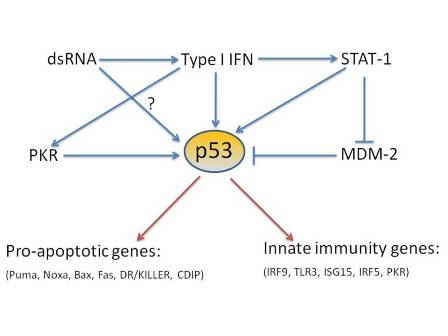{kind=link}
{kind=link}
1. Introduction
2. Regulation of p53 functions
4. p53 direct target genes in the type I interferon pathway
4.1. IRF9
4.2. TLR3
4.3. IRF5
4.4. ISG15
6. Summary
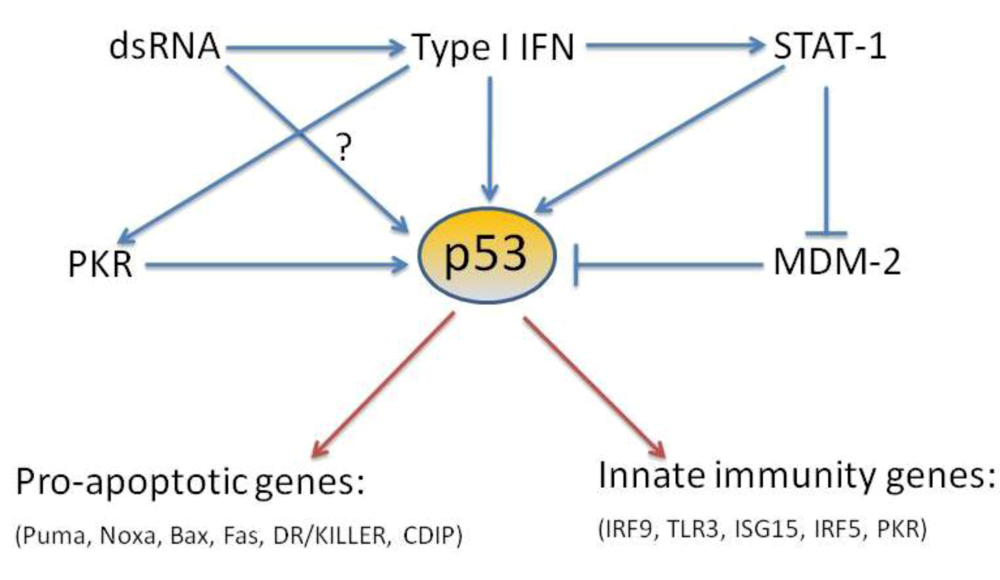
Acknowledgments
References
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- García-Sastre, A.; Biron, C.A. Type 1 interferons and the virus-host relationship: a lesson in détente. Science 2006, 312, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C.; Fish, E.N. Signaling pathways activated by interferons. Exp. Hematol. 1999, 27, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; Taniguchi, T. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Garcia, M.A.; Garcia-Cao, I.; Collado, M.; Arroyo, J.; Esteban, M.; Serrano, M.; Rivas, C. Resistance to viral infection of super p53 mice. Oncogene 2005, 24, 3059–3062. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Macip, S.; Martínez-Sobrido, L.; Brown, L.; Ashour, J.; García-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Turpin, E.; Luke, K.; Jones, J.; Tumpey, T.; Konan, K.; Schultz-Cherry, S. Influenza virus infection increases p53 activity: role of p53 in cell death and viral replication. J. Virol. 2005, 79, 8802–8811. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Kraft, R.M.; Aubert, M.; Goodwin, E.; DiMaio, D.; Blaho, J.A. p53 and hTERT determine sensitivity to viral apoptosis. J. Virol. 2007, 81, 12985–12995. [Google Scholar] [CrossRef] [PubMed]
- Pampin, M.; Simonin, Y.; Blondel, B.; Percherancier, Y.; Chelbi-Alix, M.K. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J. Virol. 2006, 80, 8582–8592. [Google Scholar] [CrossRef] [PubMed]
- Maki, C.G.; Huibregtse, J.M.; Howley, P.M. In vivo ubiquitination and proteasome-mediated degradation of p53(1). Cancer Res. 1996, 56, 2649–2654. [Google Scholar] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–269. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Wu, H.H.; Dasgupta, G. MDM2-master regulator of the p53 tumor suppressor protein. Gene 2000, 242, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Shimokado, K.; Asahara, T.; Dohi, K.; Niwa, O. Analysis of the c-myc, K-ras and p53 genes in methylcholanthrene-induced mouse sarcomas. Jpn. J. Cancer Res. 1999, 90, 40–47. [Google Scholar] [PubMed]
- Kurki, S.; Peltonen, K.; Latonen, L.; Kiviharju, T.M.; Ojala, P.M.; Meek, D.; Laiho, M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 2004, 5, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, R.; Xue, J.; Pilkinton, M.; Salvi, D.; Kiyokawa, H.; Colamonici, O.R. Different requirements for the cytostatic and apoptotic effects of type I interferons. Induction of apoptosis requires ARF but not p53 in osteosarcoma cell lines. J. Biol. Chem. 2004, 279, 32275–32280. [Google Scholar] [CrossRef] [PubMed]
- Townsend, P.A.; Scarabelli, T.M.; Davidson, S.M.; Knight Jr., R.A.; Latchman, D.S.; Stephanou, A. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J. Biol. Chem. 2004, 279, 5811–5820. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Mallette, F.A.; Mukhopadhyay, U.K.; Moores, A.; Ferbeyre, G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol. Biol. Cell 2006, 17, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Cuddihy, A.R.; Wong, A.H.; Tam, N.W.; Li, S.; Koromilas, A.E. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene 1999, 18, 2690–2702. [Google Scholar] [CrossRef] [PubMed]
- Cuddihy, A.R.; Li, S.; Tam, N.W.; Wong, A.H.; Taya, Y.; Abraham, N.; Bell, J.C.; Koromilas, A.E. Double-stranded-RNA-activated protein kinase PKR enhances transcriptional activation by tumor suppressor p53. Mol. Cell Biol. 1999, 19, 2475–2484. [Google Scholar] [PubMed]
- Ullrich, S.J.; Sakaguchi, K.; Lees-Miller, S.P.; Fiscella, M.; Mercer, W.E.; Anderson, C.W.; Appella, E. Phosphorylation at Ser-15 and Ser-392 in mutant p53 molecules from human tumors is altered compared to wild-type p53. Proc. Natl. Acad. Sci. USA 1993, 90, 5954–5958. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Sakamoto, H.; Lewis, M.S.; Anderson, C.W.; Erickson, J.W.; Appella, E.; Xie, D. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 1997, 36, 10117–10124. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.M.; Zeng, X.; Wang, Y.; Zhang, Q.H.; Kapoor, M.; Shu, H.; Goodman, R.; Lozano, G.; Zhao, Y.; Lu, H. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol. Cell 2001, 7, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.C.; Lau, A.S. Tumor suppressor p53 as a component of the tumor necrosis factor-induced, protein kinase PKR-mediated apoptotic pathway in human promonocytic U937 cells. J. Biol. Chem. 1998, 273, 25198–25202. [Google Scholar] [CrossRef] [PubMed]
- Son K.N., Pugazhenthi S. Activation of tumor suppressor protein p53 is required for Theiler's murine encephalomyelitis virus-induced apoptosis in M1-D macrophages. J. Virol. 2009, 83, 10770–10777. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Lee, E.S.; Lim, D.S.; Bae, Y.S. PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53. Proc. Natl. Acad. Sci. USA 2009, 106, 7852–7857. [Google Scholar] [CrossRef]
- Brown, L.; Ongusaha, P.P.; Kim, H.G.; Nuti, S.; Mandinova, A.; Lee, J.W.; Khosravi-Far, R.; Aaronson, S.A.; Lee, S.W. CDIP, a novel pro-apoptotic gene, regulates TNFalpha-mediated apoptosis in a p53-dependent manner. EMBO J. 2007, 26, 3410–3422. [Google Scholar] [CrossRef] [PubMed]
- Julkunen, I.; Melén, K.; Nyqvist, M.; Pirhonen, J.; Sareneva, T.; Matikainen, S. Inflammatory responses in influenza A virus infection. Vaccine 2000, 19, S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Roux-Lombard, P.; Modoux, C.; Cruchaud, A.; Dayer, J.M. Purified blood monocytes from HIV 1-infected patients produce high levels of TNF alpha and IL-1. Clin. Immunol. Immunopathol. 1989, 50, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Caux, C.; Massacrier, C.; Vanbervliet, B.; Dubois, B.; Van Kooten, C.; Durand, I.; Banchereau, J. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 1994, 180, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; Mohamed, M.R.; McFadden, G. Tumor necrosis factor and interferon: cytokines in harmony. Curr. Opin. Microbiol. 2008, 11, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Belardelli, F. Role of interferons and other cytokines in the regulation of the immune response. APMIS 1995, 103, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Mandron, M.; Martin, H.; Bonjean, B.; Lulé, J.; Tartour, E.; Davrinche, C. Dendritic cell-induced apoptosis of human cytomegalovirus-infected fibroblasts promotes cross-presentation of pp65 to CD8+ T cells. J. Gen. Virol. 2008, 89, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Chattergoon, M.A.; Muthumani, K.; Tamura, Y.; Ramanathan, M.; Shames, J.P.; Saulino, V.; Robinson, T.M.; Montaner, L.J.; Weiner, D.B. DR5 activation of caspase-8 induces DC maturation and immune enhancement in vivo. Mol. Ther. 2008, 16, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; Krammer, PH. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Burns, T.F.; Bernhard, E.J.; El-Deiry, W.S. Tissue specific expression of p53 target genes suggests a key role for KILLER/DR5 in p53-dependent apoptosis in vivo. Oncogene 2001, 20, 4601–4612. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Qu, L.K.; Koromilas, A.E. Induction of p53-dependent apoptosis in HCT116 tumor cells by RNA viruses and possible implications in virus-mediated oncolysis. Cell Cycle 2004, 3, 1043–1045. [Google Scholar] [PubMed]
- Marques, J.T.; Rebouillat, D.; Ramana, C.V.; Murakami, J.; Hill, J.E.; Gudkov, A.; Silverman, R.H.; Stark, G.R.; Williams, B.R. Down-regulation of p53 by double-stranded RNA modulates the antiviral response. J. Virol. 2005, 79, 11105–11114. [Google Scholar] [CrossRef] [PubMed]
- Casavant, N.C.; Luo, M.H.; Rosenke, K.; Winegardner, T.; Zurawska, A.; Fortunato, E.A. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J. Virol. 2006, 80, 8390–83401. [Google Scholar] [CrossRef] [PubMed]
- Groskreutz, D.J.; Monick, M.M.; Powers, L.S.; Quelle, D.E.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J. Immunol. 2007, 179, 2741–2747. [Google Scholar] [PubMed]
- Royds, J.A.; Hibma, M.; Dix, B.R.; Hananeia, L.; Russell, I.A.; Wiles, A.; Wynford-Thomas, D.; Braithwaite, A.W. p53 promotes adenoviral replication and increases late viral gene expression. Oncogene 2006, 25, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.G.; Branton, P.E. Regulation of apoptosis by viral gene products. J. Virol. 1997, 7, 3620–3627. [Google Scholar]
- Bluyssen, A.R.; Durbin, J.E.; Levy, D.E. ISGF3 gamma p48, a specificity switch for interferon activated transcription factors. Cytokine Growth Factor Rev. 1996, 7, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kadokawa, Y.; Harada, H.; Matsumoto, M.; Sato, M.; Kashiwazaki, Y.; Tarutani, M.; Tan, R.S.; Takasugi, T.; Matsuyama, T.; Mak, T.W.; Noguchi, S.; Taniguchi, T. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells 1996, 1, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Ouellet, M.; Tremblay, M.J. Microarray study reveals that HIV-1 induces rapid type-I interferon-dependent p53 mRNA up-regulation in human primary CD4+ T cells. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, X.; Guo, L.; Qiu, Y.; Li, X.; Yu, H.; Xiang, H.; Ma, Z. Influenza A virus induces p53 accumulation in a biphasic pattern. Biochem. Biophys. Res. Commun. 2009, 382, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Dharel, N.; Kato, N.; Muroyama, R.; Taniguchi, H.; Otsuka, M.; Wang, Y.; Jazag, A.; Shao, R.X.; Chang, J.H.; Adler, M.K.; Kawabe, T.; Omata, M. Potential contribution of tumor suppressor p53 in the host defense against hepatitis C virus. Hepatology 2008, 47, 1136–1149. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Liu, W.L.; Cheng, C.W.; Chou, Y.B.; Hung, K.H.; Huang, W.H.; Wu, C.L.; Li, Y.T.; Shiau, A.L.; Lai, M.Y. Ribavirin enhances interferon signaling via stimulation of mTOR and p53 activities. FEBS Lett. 2009, 583, 2793–2798. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.S.; Hudson, K.L.; Lin, T.Y.; Anderson, K.V. Dominant and recessive mutations define functional domains of Toll, a transmembrane protein required for dorsal-ventral polarity in the Drosophila embryo. Genes Dev. 1991, 5, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Keith, F.J. Drosophila Toll and IL-1 receptor. Nature 1991, 351, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Antiviral signaling through pattern recognition receptors. J. Biochem. 2007, 14, 137–145. [Google Scholar]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and innate immunity. J. Mol. Med. 2006, 84, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Bosisio, D.; Polentarutti, N.; Stoppacciaro, A.; Mancinelli, R.; van't Veer, C.; Penton-Rol, G.; Ruco, L.P.; Allavena, P.; Mantovani, A. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J. Immunol. 2000, 164, 5998–6004. [Google Scholar] [PubMed]
- Kaisho, T.; Akira, S. Regulation of dendritic cell function through Toll-like receptors. Curr. Mol. Med. 2003, 3, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Taura, M.; Eguma, A.; Suico, M.A.; Shuto, T.; Koga, T.; Komune, T.; Sato, T.; Saya, H.; Li, J.D.; Kai, H. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol. Cell. Biol. 2008, 28, 6557–6567. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Sakaguchi, S.; Nakajima, C.; Watanabe, A.; Yanai, H.; Matsumoto, M.; Ohteki, T.; Kaisho, T.; Takaoka, A.; Akira, S.; Seya, T.; Taniguchi, T. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 10872–10877. [Google Scholar] [CrossRef]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef]
- Honda, K.; Taniguchi, T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Ishihara, M.; Lamphier, M.S.; Nozawa, H.; Matsuyama, T.; Mak, T.W.; Aizawa, S.; Tokino, T.; Oren, M.; Taniguchi, T. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature 1996, 382, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.; Lubyova, B.; Pitha, P.M. On the role of IRF in host defense. J. Interferon Cytokine Res. 2002, 22, 59–71. [Google Scholar] [PubMed]
- Panne, D.; Maniatis, T.; Harrison, S.C. An atomic model of the interferon-beta enhanceosome. Cell 2007, 129, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Yanai, H.; Kondo, S.; Duncan, G.; Negishi, H.; Mizutani, T.; Kano, S.; Honda, K.; Ohba, Y.; Mak, T.W.; Taniguchi, T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 2005, 434, 243–249. [Google Scholar] [CrossRef] [PubMed]
- López, C.B.; Moltedo, B.; Alexopoulou, L.; Bonifaz, L.; Flavell, R.A.; Moran; T.M. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J. Immunol. 2004, 173, 6882–6889. [Google Scholar] [PubMed]
- Barnes, B.J.; Moore, P.A.; Pitha, P.M. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J. Biol. Chem. 2001, 276, 23382–23390. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Chen, H.M.; Inuzuka, T.; Kondo, S.; Mak, T.W.; Takaoka, A.; Honda, K.; Taniguchi, T. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. USA 2007, 104, 3402–3407. [Google Scholar] [CrossRef]
- Mori, T.; Anazawa, Y.; Iiizumi, M.; Fukuda, S.; Nakamura, Y.; Arakawa, H. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene 2002, 21, 2914–2918. [Google Scholar] [CrossRef] [PubMed]
- Pitha-Rowe, I.F.; Pitha, P.M. Viral defense, carcinogenesis and ISG15: novel roles for an old ISG. Cytokine Growth Factor Rev. 2007, 18, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J.; Broeze, R.J.; Lengyel, P. Accumulation of an mRNA and protein in interferon-treated Ehrlich ascites tumour cells. Nature 1979, 279, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar] [CrossRef]
- Lu, G.; Reinert, J.T.; Pitha-Rowe, I.; Okumura, A.; Kellum, M.; Knobeloch, K. P.; Hassel, B.; Pitha, P.M. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell Mol. Biol. (Noisy-le-grand) 2006, 52, 29–41. [Google Scholar] [PubMed]
- Blomstrom, D.C.; Fahey, D.; Kutny, R.; Korant, B.D.; Knight Jr., E. Molecular characterization of the interferon-induced 15-kDa protein. Molecular cloning and nucleotide and amino acid sequence. J. Biol. Chem. 1986, 261, 8811–8816. [Google Scholar] [PubMed]
- Okumura, A.; Pitha, P.M.; Harty, R.N. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3974–3979. [Google Scholar] [CrossRef]
- Okumura, A.; Lu, G.; Pitha-Rowe, I.; Pitha, P.M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. USA 2006, 103, 1440–1445. [Google Scholar] [CrossRef]
- Yuan, W.; Krug, R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Hummer, B.T.; Li, X.L.; Hassel, B.A. Role for p53 in gene induction by double-stranded RNA. J. Virol. 2001, 75, 7774–7777. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Klein, G. Perspectives in studies of human tumor viruses. Front Biosci. 2002, 7, 268–274. [Google Scholar] [CrossRef]
- Pan, H.; Yin, C.; Van Dyke, T. Apoptosis and cancer mechanisms. Cancer Surv. 1997, 29, 305–327. [Google Scholar] [PubMed]
- Martin, M.E.; Berk, A.J. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 1998, 72, 3146–3154. [Google Scholar] [PubMed]
- Zhang, Q.; Gutsch, D.; Kenney, S. Functional and physical interaction between p53 and BZLF1: implications for Epstein-Barr virus latency. Mol. Cell Biol. 1994, 14, 1929–1938. [Google Scholar] [PubMed]
- Parker, G.A.; Crook, T.; Bain, M.; Sara, E.A.; Farrell, P.J.; Allday, M.J. Epstein-Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene 1996, 13, 2541–2549. [Google Scholar] [PubMed]
- Friborg Jr., J.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [PubMed]
- Rivas, C.; Thlick, A.E.; Parravicini, C.; Moore, P.S.; Chang, Y. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 2001, 75, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Damania, B.; Jeong, J.H.; Bowser, B.S.; DeWire, S.M.; Staudt, M.R.; Dittmer, D.P. Comparison of the Rta/Orf50 transactivator proteins of gamma-2-herpesviruses. J. Virol. 2004, 78, 5491–5499. [Google Scholar] [CrossRef] [PubMed]
- Kashanchi, F.; Araujo, J.; Doniger, J.; Muralidhar, S.; Hoch, R.; Khleif, S.; Mendelson, E.; Thompson, J.; Azumi, N.; Brady, J.N.; Luppi, M.; Torelli, G.; Rosenthal, L.J. Human herpesvirus 6 (HHV-6) ORF-1 transactivating gene exhibits malignant transforming activity and its protein binds to p53. Oncogene 1997, 14, 359–367. [Google Scholar] [PubMed]
- Zantema, A.; Fransen, J.A.; Davis-Olivier, A.; Ramaekers, F.C.; Vooijs, G.P.; DeLeys, B.; Van der Eb, A.J. Localization of the E1B proteins of adenovirus 5 in transformed cells, as revealed by interaction with monoclonal antibodies. Virology 1985, 142, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Dobner, T.; Horikoshi, N.; Rubenwolf, S.; Shenk, T. Blockage by adenovirus E4orf6 of transcriptional activation by the p53 tumor suppressor. Science 1996, 272, 1470–1473. [Google Scholar] [PubMed]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [PubMed]
- Szekely, L.; Selivanova, G.; Magnusson, K.P.; Klein, G.; Wiman, K.G. EBNA-5, an Epstein-Barr virus-encoded nuclear antigen, binds to the retinoblastoma and p53 proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 5455–5459. [Google Scholar] [CrossRef]
- Lee, H.R.; Toth, Z.; Shin, Y.C.; Lee, J.S.; Chang, H.; Gu, W.; Oh, T.K.; Kim, M.H.; Jung, J.U. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J. Virol. 2009, 83, 6739–6747. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Vega, F.M.; Blanco, S.; Barcia, R.; Lazo, P.A. The vaccinia virus B1R kinase induces p53 downregulation by an Mdm2-dependent mechanism. Virology 2004, 328, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Banks, L. Repression of p53 transcriptional activity by the HPV E7 proteins. Virology 1997, 227, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Seavey, S.E.; Holubar, M.; Saucedo, L.J.; Perry, M.E. The E7 oncoprotein of human papillomavirus type 16 stabilizes p53 through a mechanism independent of p19(ARF). J. Virol. 1999, 73, 7590–7598. [Google Scholar] [PubMed]
- Li, Z; Day, C.P.; Yang, J.Y.; Tsai, W.B.; Lozano, G.; Shih, H.M.; Hung, M.C. Adenoviral E1A targets Mdm4 to stabilize tumor suppressor p53. Cancer Res. 2004, 64, 9080–9085. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W; Ruley, H.E. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993, 7, 535–545. [Google Scholar] [CrossRef] [PubMed]
- de Stanchina, E; McCurrach, M.E.; Zindy, F.; Shieh, S.Y.; Ferbeyre, G.; Samuelson, A.V.; Prives, C.; Roussel, M.F.; Sherr, C.J.; Lowe, S.W. E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev. 1998, 12, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Seo, T.; Park, J.; Lee, D.; Hwang, S.G.; Choe, J. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J. Virol. 2001, 75, 6193–6198. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Nakamura, H.; Liang, X.; Feng, P.; Chang, H.; Kowalik, T.F.; Jung, J.U. Inhibition of the ATM/p53 signal transduction pathway by Kaposi's sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006, 80, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Kashuba, E.; Mattsson, K.; Pokrovskaja, K.; Kiss, C.; Protopopova, M.; Ehlin-Henriksson, B.; Klein, G.; Szekely, L. EBV-encoded EBNA-5 associates with P14ARF in extranucleolar inclusions and prolongs the survival of P14ARF-expressing cells. Int. J. Cancer 2003, 105, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Moule, M.G.; Collins, C.H.; McCormick, F.; Fried, M. Role for PP2A in ARF signaling to p53. Proc. Natl. Acad. Sci. USA 2004, 101, 14063–14066. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Zhu, M.; Duan, L.X.; London, W.T. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene 1993, 8, 1109–1117. [Google Scholar] [PubMed]
- Li, L.; Zhou, S.; Chen, X.; Guo, L.; Li, Z.; Hu, D.; Luo, X.; Ma, X.; Tang, M.; Yi, W.; Tsao, S.W.; Cao, Y. The activation of p53 mediated by Epstein-Barr virus latent membrane protein 1 in SV40 large T-antigen transformed cells. FEBS Lett. 2008, 582, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Marker, P.H.; Belcher, J.D.; Wilcken, D.E.; Burns, L.J.; Vercellotti, G.M.; Wang, X.L. Human cytomegalovirus immediate early proteins upregulate endothelial p53 function. FEBS Lett. 2000, 474, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Eckardt-Michel, J.; Lorek, M.; Baxmann, D.; Grunwald, T.; Kei, G.M.; Zimmer, G. The fusion protein of respiratory syncytial virus triggers p53-dependent apoptosis. J. Virol. 2008, 82, 3236–3249. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Sheeter, D.; Rought, S.; Zaunders, J.J.; Susin, S.A.; Kroemer, G.; Richman, D.D.; Carson, D.A.; Corbeil, J.; Leoni, L.M. HIV induces lymphocyte apoptosis by a p53-initiated, mitochondrial-mediated mechanism. FASEB J. 2001, 15, 5–6. [Google Scholar] [PubMed]
- Verschuren, E.W.; Klefstrom, J.; Evan, G.I.; Jones, N. The oncogenic potential of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2002, 2, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Besaratinia, A. Mutational spectra of human cancer. Hum. Genet. 2009, 125, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 58, 15–16. [Google Scholar] [CrossRef]
- Sutcliffe, J.E.; Brehm, A. Of flies and men; p53, a tumour suppressor. FEBS Lett. 2004, 567, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Abrams, J.M. Lessons from p53 in non-mammalian models. Cell Death Differ. 2006, 13, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Graeber, T.G.; Peterson, J.F.; Tsai, M.; Monica, K.; Fornace Jr., A.J.; Giaccia, A.J. Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol. Cell Biol. 1994, 14, 6264–6277. [Google Scholar] [PubMed]
- Yu, X.; Riley, T.; Levine, A.J. The regulation of the endosomal compartment by p53 the tumor suppressor gene. FEBS J. 2009, 276, 2201–2212. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Rivas, C.; Aaronson, S.A.; Munoz-Fontela, C. Dual Role of p53 in Innate Antiviral Immunity. Viruses 2010, 2, 298-313. https://doi.org/10.3390/v2010298
Rivas C, Aaronson SA, Munoz-Fontela C. Dual Role of p53 in Innate Antiviral Immunity. Viruses. 2010; 2(1):298-313. https://doi.org/10.3390/v2010298
Chicago/Turabian StyleRivas, Carmen, Stuart A. Aaronson, and Cesar Munoz-Fontela. 2010. "Dual Role of p53 in Innate Antiviral Immunity" Viruses 2, no. 1: 298-313. https://doi.org/10.3390/v2010298
APA StyleRivas, C., Aaronson, S. A., & Munoz-Fontela, C. (2010). Dual Role of p53 in Innate Antiviral Immunity. Viruses, 2(1), 298-313. https://doi.org/10.3390/v2010298