

Primary Sequence-Intrinsic Immune Evasion by Viral Proteins Guides CTL-Based Vaccine Strategies


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Trans and Cis-Acting MHC Class I-Mediated CTL Immune Evasion
3. Viral Sequence-Dependent Intrinsic CTL Immune Evasion
3.1. G-Quadruplexes Regulate Viral Protein Expression and Antigen Presentation
3.2. Resistance to Proteasomal Degradation
3.3. Rare Codon Usage
3.4. Intrinsic Immune Evasion in RNA Viruses
4. Enhanced CTL Vaccines by Reversing Intrinsic Immune Evasion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the γ-herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Are There More Human Cancer Viruses Left to Be Found? Annu. Rev. Virol. 2024, 11, 239–259. [Google Scholar] [CrossRef]
- Redpath, S.; Angulo, A.; Gascoigne, N.R.; Ghazal, P. Immune checkpoints in viral latency. Annu. Rev. Microbiol. 2001, 55, 531–560. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, W.; Gao, S.J.; Lu, C. KSHV microRNAs: Tricks of the Devil. Trends Microbiol. 2017, 25, 648–661. [Google Scholar] [CrossRef]
- Toptan, T.; Abere, B.; Nalesnik, M.A.; Swerdlow, S.H.; Ranganathan, S.; Lee, N.; Shair, K.H.; Moore, P.S.; Chang, Y. Circular DNA tumor viruses make circular RNAs. Proc. Natl. Acad. Sci. USA 2018, 115, E8737–E8745. [Google Scholar] [CrossRef]
- Wan, L.; Xie, B.; Shuda, M.; Delgoffe, G.; Chang, Y.; Moore, P.S. Engineered protein destabilization reverses intrinsic immune evasion for candidate vaccine pan-strain KSHV and SARS-CoV-2 antigens. bioRxiv 2024. 2024.2010.2022.619692. [Google Scholar]
- Williams, A.; Peh, C.A.; Elliott, T. The cell biology of MHC class I antigen presentation. Tissue Antigens 2002, 59, 3–17. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Antón, L.C.; Bennink, J.R. Defective ribosomal products (DRiPs): A major source of antigenic peptides for MHC class I molecules? J. Immunol. 1996, 157, 1823–1826. [Google Scholar] [CrossRef]
- Khan, S.; de Giuli, R.; Schmidtke, G.; Bruns, M.; Buchmeier, M.; van den Broek, M.; Groettrup, M. Cutting edge: Neosynthesis is required for the presentation of a T cell epitope from a long-lived viral protein. J. Immunol. 2001, 167, 4801–4804. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, E.W. The MHC class I antigen presentation pathway: Strategies for viral immune evasion. Immunology 2003, 110, 163–169. [Google Scholar] [CrossRef]
- Zhou, F. Molecular mechanisms of IFN-γ to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.D.; Verweij, M.C.; Wiertz, E.J. Herpesviruses and immunity: The art of evasion. Vet. Microbiol. 2010, 143, 89–100. [Google Scholar] [CrossRef] [PubMed]
- van de Weijer, M.L.; Luteijn, R.D.; Wiertz, E.J. Viral immune evasion: Lessons in MHC class I antigen presentation. Semin Immunol. 2015, 24, 125–137. [Google Scholar] [CrossRef]
- Jasinski-Bergner, S.; Mandelboim, O.; Seliger, B. Molecular mechanisms of human herpes viruses inferring with host immune surveillance. J. Immunother. Cancer 2020, 8, e000841. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus immunoevasion and tumorigenesis: Two sides of the same coin? Annu. Rev. Microbiol. 2003, 57, 609–639. [Google Scholar] [CrossRef]
- Song, J.; Perreault, J.P.; Topisirovic, I.; Richard, S. RNA G-quadruplexes and their potential regulatory roles in translation. Translation 2016, 4, e1244031. [Google Scholar] [CrossRef] [PubMed]
- Lech, C.J.; Heddi, B.; Phan, A.T.n. Guanine base stacking in G-quadruplex nucleic acids. Nucleic Acids Res. 2013, 41, 2034–2046. [Google Scholar] [CrossRef]
- Millevoi, S.; Moine, H.; Vagner, S. G-quadruplexes in RNA biology. Wiley Interdiscip. Rev. RNA 2012, 3, 495–507. [Google Scholar] [CrossRef]
- Ruggiero, E.; Zanin, I.; Terreri, M.; Richter, S.N. G-quadruplex targeting in the fight against viruses: An update. Int. J. Mol. Sci. 2021, 22, 10984. [Google Scholar] [CrossRef]
- Lavezzo, E.; Berselli, M.; Frasson, I.; Perrone, R.; Palù, G.; Brazzale, A.R.; Richter, S.N.; Toppo, S. G-quadruplex forming sequences in the genome of all known human viruses: A comprehensive guide. PLoS Comput. Biol. 2018, 14, e1006675. [Google Scholar] [CrossRef] [PubMed]
- Abiri, A.; Lavigne, M.; Rezaei, M.; Nikzad, S.; Zare, P.; Mergny, J.-L.; Rahimi, H.-R. Unlocking G-quadruplexes as antiviral targets. Pharmacol. Rev. 2021, 73, 897–923. [Google Scholar] [CrossRef] [PubMed]
- Estep, K.N.; Butler, T.J.; Ding, J.; Brosh, R.M. G4-interacting DNA helicases and polymerases: Potential therapeutic targets. Curr. Med. Chem. 2019, 26, 2881–2897. [Google Scholar] [CrossRef] [PubMed]
- Dinh, V.T.; Loaëc, N.; Quillévéré, A.; Le Sénéchal, R.; Keruzoré, M.; Martins, R.P.; Granzhan, A.; Blondel, M. The hide-and-seek game of the oncogenic Epstein-Barr virus-encoded EBNA1 protein with the immune system: An RNA G-quadruplex tale. Biochimie 2023, 214 Pt A, 57–68. [Google Scholar] [CrossRef]
- Murat, P.; Zhong, J.; Lekieffre, L.; Cowieson, N.P.; Clancy, J.L.; Preiss, T.; Balasubramanian, S.; Khanna, R.; Tellam, J. G-quadruplexes regulate Epstein-Barr virus–encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 2014, 10, 358–364. [Google Scholar] [CrossRef]
- Dabral, P.; Babu, J.; Zareie, A.; Verma, S.C. LANA and hnRNP A1 Regulate the Translation of LANA mRNA through G-Quadruplexes. J. Virol. 2020, 94, e01508-19. [Google Scholar] [CrossRef]
- Kwun, H.J.; Toptan, T.; Ramos da Silva, S.; Atkins, J.F.; Moore, P.S.; Chang, Y. Human DNA tumor viruses generate alternative reading frame proteins through repeat sequence recoding. Proc. Natl. Acad. Sci. USA 2014, 111, E4342–E4349. [Google Scholar] [CrossRef]
- Agarwala, P.; Pandey, S.; Maiti, S. The tale of RNA G-quadruplex. Org. Biomol. Chem. 2015, 13, 5570–5585. [Google Scholar] [CrossRef]
- Bugaut, A.; Balasubramanian, S. 5′-UTR RNA G-quadruplexes: Translation regulation and targeting. Nucleic Acids Res. 2012, 40, 4727–4741. [Google Scholar] [CrossRef]
- Zareie, A.R.; Verma, S.C. Nucleolin Regulates the Expression of Kaposi’s Sarcoma-Associated Herpesvirus’ Latency-Associated Nuclear Antigen through G-Quadruplexes in the mRNA. Viruses 2023, 15, 2438. [Google Scholar] [CrossRef]
- Lista, M.J.; Martins, R.P.; Billant, O.; Contesse, M.A.; Findakly, S.; Pochard, P.; Daskalogianni, C.; Beauvineau, C.; Guetta, C.; Jamin, C.; et al. Nucleolin directly mediates Epstein-Barr virus immune evasion through binding to G-quadruplexes of EBNA1 mRNA. Nat. Commun. 2017, 8, 16043. [Google Scholar] [CrossRef]
- Vossen, M.T.; Westerhout, E.M.; Söderberg-Nauclér, C.; Wiertz, E.J. Viral immune evasion: A masterpiece of evolution. Immunogenetics 2002, 54, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.d.M.; Alves, C.E.d.C.; Pontes, G.S. Epstein-Barr virus: The mastermind of immune chaos. Front. Immunol. 2024, 15, 1297994. [Google Scholar] [CrossRef]
- Ressing, M.E.; Horst, D.; Griffin, B.D.; Tellam, J.; Zuo, J.; Khanna, R.; Rowe, M.; Wiertz, E.J. Epstein-Barr virus evasion of CD8+ and CD4+ T cell immunity via concerted actions of multiple gene products. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 397–408. [Google Scholar]
- Sorel, O.; Dewals, B.G. The critical role of genome maintenance proteins in immune evasion during gammaherpesvirus latency. Front. Microbiol. 2019, 9, 3315. [Google Scholar] [CrossRef] [PubMed]
- Davenport, M.G.; Pagano, J.S. Expression of EBNA-1 mRNA is regulated by cell cycle during Epstein-Barr virus type I latency. J. Virol. 1999, 73, 3154–3161. [Google Scholar] [CrossRef]
- Coppotelli, G.; Mughal, N.; Marescotti, D.; Masucci, M.G. High avidity binding to DNA protects ubiquitylated substrates from proteasomal degradation. J. Biol. Chem. 2011, 286, 19565–19575. [Google Scholar] [CrossRef]
- Singh, G.; Aras, S.; Zea, A.H.; Koochekpour, S.; Aiyar, A. Optimal transactivation by Epstein-Barr nuclear antigen 1 requires the UR1 and ATH1 domains. J. Virol. 2009, 83, 4227–4235. [Google Scholar] [CrossRef] [PubMed]
- Sears, J.; Ujihara, M.; Wong, S.; Ott, C.; Middeldorp, J.; Aiyar, A. The Amino Terminus of Epstein-Barr Virus (EBV) Nuclear Antigen 1 Contains AT Hooks That Facilitate the Replication and Partitioning of Latent EBV Genomes by Tethering Them to Cellular Chromosomes. J. Virol. 2004, 78, 11487–11505. [Google Scholar] [CrossRef]
- Huth, J.R.; Bewley, C.A.; Nissen, M.S.; Evans, J.N.; Reeves, R.; Gronenborn, A.M.; Clore, G.M. The solution structure of an HMG-I (Y)–DNA complex defines a new architectural minor groove binding motif. Nat. Struct. Biol. 1997, 4, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Haigh, T.A.; Mackay, L.K.; Rickinson, A.B.; Taylor, G.S. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. USA 2010, 107, 2165–2170. [Google Scholar] [CrossRef]
- Sausen, D.G.; Poirier, M.C.; Spiers, L.M.; Smith, E.N. Mechanisms of T cell evasion by Epstein-Barr virus and implications for tumor survival. Front. Immunol. 2023, 14, 1289313. [Google Scholar] [CrossRef] [PubMed]
- Münz, C.; Bickham, K.L.; Subklewe, M.; Tsang, M.L.; Chahroudi, A.; Kurilla, M.G.; Zhang, D.; O’Donnell, M.; Steinman, R.M. Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J. Exp. Med. 2000, 191, 1649–1660. [Google Scholar] [CrossRef]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef] [PubMed]
- Voo, K.S.; Fu, T.; Wang, H.Y.; Tellam, J.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Wang, R.F. Evidence for the presentation of major histocompatibility complex class I-restricted Epstein-Barr virus nuclear antigen 1 peptides to CD8+ T lymphocytes. J. Exp. Med. 2004, 199, 459–470. [Google Scholar] [CrossRef]
- Tellam, J.; Connolly, G.; Green, K.J.; Miles, J.J.; Moss, D.J.; Burrows, S.R.; Khanna, R. Endogenous presentation of CD8+ T cell epitopes from Epstein-Barr virus-encoded nuclear antigen 1. J. Exp. Med. 2004, 199, 1421–1431. [Google Scholar] [CrossRef]
- Heessen, S.; Leonchiks, A.; Issaeva, N.; Sharipo, A.; Selivanova, G.; Masucci, M.G.; Dantuma, N.P. Functional p53 chimeras containing the Epstein-Barr virus Gly-Ala repeat are protected from Mdm2- and HPV-E6-induced proteolysis. Proc. Natl. Acad. Sci. USA 2002, 99, 1532–1537. [Google Scholar] [CrossRef]
- Kwun, H.J.; da Silva, S.R.; Shah, I.M.; Blake, N.; Moore, P.S.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 2007, 81, 8225–8235. [Google Scholar] [CrossRef]
- Kelley-Clarke, B.; De Leon-Vazquez, E.; Slain, K.; Barbera, A.J.; Kaye, K.M. Role of Kaposi’s sarcoma-associated herpesvirus C-terminal LANA chromosome binding in episome persistence. J. Virol. 2009, 83, 4326–4337. [Google Scholar] [CrossRef]
- Gao, S.J.; Kingsley, L.; Li, M.; Zheng, W.; Parravicini, C.; Ziegler, J.; Newton, R.; Rinaldo, C.R.; Saah, A.; Phair, J.; et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat. Med. 1996, 2, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Roshan, R.; Labo, N.; Trivett, M.; Miley, W.; Marshall, V.; Coren, L.; Castro, E.M.C.; Perez, H.; Holdridge, B.; Davis, E. T-cell responses to KSHV infection: A systematic approach. Oncotarget 2017, 8, 109402. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Verma, S.C.; Robertson, E.S. ORF73 of herpesvirus saimiri, a viral homolog of Kaposi’s sarcoma-associated herpesvirus, modulates the two cellular tumor suppressor proteins p53 and pRb. J. Virol. 2004, 78, 10336–10347. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Coulson, J.M.; Whitehouse, A.; Blake, N. Reduction in RNA levels rather than retardation of translation is responsible for the inhibition of major histocompatibility complex class I antigen presentation by the glutamic acid-rich repeat of herpesvirus saimiri open reading frame 73. J. Virol. 2009, 83, 273–282. [Google Scholar] [CrossRef]
- Sorel, O.; Chen, T.; Myster, F.; Javaux, J.; Vanderplasschen, A.; Dewals, B.G. Macavirus latency-associated protein evades immune detection through regulation of protein synthesis in cis depending upon its glycin/glutamate-rich domain. PLoS Pathog. 2017, 13, e1006691. [Google Scholar] [CrossRef]
- Jitobaom, K.; Phakaratsakul, S.; Sirihongthong, T.; Chotewutmontri, S.; Suriyaphol, P.; Suptawiwat, O.; Auewarakul, P. Codon usage similarity between viral and some host genes suggests a codon-specific translational regulation. Heliyon 2020, 6, e03915. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Q.; Zhao, F. Synonymous but Not Silent: The Codon Usage Code for Gene Expression and Protein Folding. Annu. Rev. Biochem. 2021, 90, 375–401. [Google Scholar] [CrossRef]
- Lyu, X.; Yang, Q.; Zhao, F.; Liu, Y. Codon usage and protein length-dependent feedback from translation elongation regulates translation initiation and elongation speed. Nucleic Acids Res. 2021, 49, 9404–9423. [Google Scholar] [CrossRef]
- Angov, E. Codon usage: Nature’s roadmap to expression and folding of proteins. Biotechnol. J. 2011, 6, 650–659. [Google Scholar] [CrossRef]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The adaptation of codon usage of +ssRNA viruses to their hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef]
- Kanduc, D. Rare Human Codons and HCMV Translational Regulation. J. Mol. Microbiol. Biotechnol. 2017, 27, 213–216. [Google Scholar] [CrossRef]
- Pintó, R.M.; Pérez-Rodríguez, F.J.; D’Andrea, L.; de Castellarnau, M.; Guix, S.; Bosch, A. Hepatitis A Virus Codon Usage: Implications for Translation Kinetics and Capsid Folding. Cold Spring Harb. Perspect. Med. 2018, 8, a031781. [Google Scholar] [CrossRef]
- Pintó, R.M.; Aragonès, L.; Costafreda, M.I.; Ribes, E.; Bosch, A. Codon usage and replicative strategies of hepatitis A virus. Virus Res. 2007, 127, 158–163. [Google Scholar] [CrossRef]
- Karlin, S.; Blaisdell, B.E.; Schachtel, G.A. Contrasts in codon usage of latent versus productive genes of Epstein-Barr virus: Data and hypotheses. J. Virol. 1990, 64, 4264–4273. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tong, W.; Yuying, Z.; Wei, F. Evasion mechanisms of the type I interferons responses by influenza A virus. Crit. Rev. Microbiol. 2020, 46, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Berkhoff, E.G.; de Wit, E.; Geelhoed-Mieras, M.M.; Boon, A.C.; Symons, J.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Functional constraints of influenza A virus epitopes limit escape from cytotoxic T lymphocytes. J. Virol. 2005, 79, 11239–11246. [Google Scholar] [CrossRef]
- Gotch, F.; McMichael, A.; Smith, G.; Moss, B. Identification of viral molecules recognized by influenza-specific human cytotoxic T lymphocytes. J. Exp. Med. 1987, 165, 408–416. [Google Scholar] [CrossRef] [PubMed]
- van de Sandt, C.E.; Kreijtz, J.H.; Rimmelzwaan, G.F. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses 2012, 4, 1438–1476. [Google Scholar] [CrossRef]
- Zanker, D.J.; Oveissi, S.; Tscharke, D.C.; Duan, M.; Wan, S.; Zhang, X.; Xiao, K.; Mifsud, N.A.; Gibbs, J.; Izzard, L.; et al. Influenza A Virus Infection Induces Viral and Cellular Defective Ribosomal Products Encoded by Alternative Reading Frames. J. Immunol. 2019, 202, 3370–3380. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K. Outbreak. info genomic reports: Scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat. Methods 2023, 20, 512–522. [Google Scholar] [CrossRef]
- Meng, W.; Guo, S.; Cao, S.; Shuda, M.; Robinson-McCarthy, L.R.; McCarthy, K.R.; Shuda, Y.; Paniz Mondolfi, A.E.; Bryce, C.; Grimes, Z.; et al. Development and characterization of a new monoclonal antibody against SARS-CoV-2 NSP12 (RdRp). J. Med. Virol. 2023, 95, e28246. [Google Scholar] [CrossRef] [PubMed]
- Woolard, S.N.; Kumaraguru, U. Viral vaccines and CTL response. BioMed Res. Int. 2010, 2010, 141657. [Google Scholar] [CrossRef] [PubMed]
- Bevan, M.J. Antigen presentation to cytotoxic T lymphocytes in vivo. J. Exp. Med. 1995, 182, 639–641. [Google Scholar] [CrossRef]
- Saunders, K.O.; Lee, E.; Parks, R.; Martinez, D.R.; Li, D.; Chen, H.; Edwards, R.J.; Gobeil, S.; Barr, M.; Mansouri, K. Neutralizing antibody vaccine for pandemic and pre-emergent coronaviruses. Nature 2021, 594, 553–559. [Google Scholar] [CrossRef]
- Karch, C.P.; Matyas, G.R. The current and future role of nanovaccines in HIV-1 vaccine development. Expert. Rev. Vaccines 2021, 20, 935–944. [Google Scholar] [CrossRef]
- Koup, R.A.; Douek, D.C. Vaccine design for CD8 T lymphocyte responses. Cold Spring Harb. Perspect. Med. 2011, 1, a007252. [Google Scholar] [CrossRef]
- Kwun, H.J.; da Silva, S.R.; Qin, H.; Ferris, R.L.; Tan, R.; Chang, Y.; Moore, P.S. The central repeat domain 1 of Kaposi’s sarcoma-associated herpesvirus (KSHV) latency associated-nuclear antigen 1 (LANA1) prevents cis MHC class I peptide presentation. Virology 2011, 412, 357–365. [Google Scholar] [CrossRef]
- Dersh, D.; Yewdell, J.W.; Wei, J. A SIINFEKL-Based System to Measure MHC Class I Antigen Presentation Efficiency and Kinetics. Methods Mol. Biol. 2019, 1988, 109–122. [Google Scholar] [CrossRef]
- Yewdell, J.W. Plumbing the sources of endogenous MHC class I peptide ligands. Curr. Opin. Immunol. 2007, 19, 79–86. [Google Scholar] [CrossRef]
- Otano, I.; Azpilikueta, A.; Glez-Vaz, J.; Alvarez, M.; Medina-Echeverz, J.; Cortés-Domínguez, I.; Ortiz-de-Solorzano, C.; Ellmark, P.; Fritzell, S.; Hernandez-Hoyos, G. CD137 (4-1BB) costimulation of CD8+ T cells is more potent when provided in cis than in trans with respect to CD3-TCR stimulation. Nat. Commun. 2021, 12, 7296. [Google Scholar] [CrossRef] [PubMed]
- Shang, M.; Yang, H.; Yang, R.; Chen, T.; Fu, Y.; Li, Y.; Fang, X.; Zhang, K.; Zhang, J.; Li, H.; et al. The folate cycle enzyme MTHFD2 induces cancer immune evasion through PD-L1 up-regulation. Nat. Commun. 2021, 12, 1940. [Google Scholar] [CrossRef] [PubMed]
- Duttagupta, P.A.; Boesteanu, A.C.; Katsikis, P.D. Costimulation signals for memory CD8+ T cells during viral infections. Crit. Rev. Immunol. 2009, 29, 469–486. [Google Scholar] [CrossRef]
- Dogra, P.; Schiavone, C.; Wang, Z.; Ruiz-Ramírez, J.; Caserta, S.; Staquicini, D.I.; Markosian, C.; Wang, J.; Sostman, H.D.; Pasqualini, R.; et al. A modeling-based approach to optimize COVID-19 vaccine dosing schedules for improved protection. JCI Insight 2023, 8, e169860. [Google Scholar] [CrossRef] [PubMed]
- Berzofsky, J.A.; Ahlers, J.D.; Belyakov, I.M. Strategies for designing and optimizing new generation vaccines. Nat. Rev. Immunol. 2001, 1, 209–219. [Google Scholar] [CrossRef]
- Kanagavelu, S.; Termini, J.M.; Gupta, S.; Raffa, F.N.; Fuller, K.A.; Rivas, Y.; Philip, S.; Kornbluth, R.S.; Stone, G.W. HIV-1 adenoviral vector vaccines expressing multi-trimeric BAFF and 4-1BBL enhance T cell mediated anti-viral immunity. PLoS ONE 2014, 9, e90100. [Google Scholar] [CrossRef]
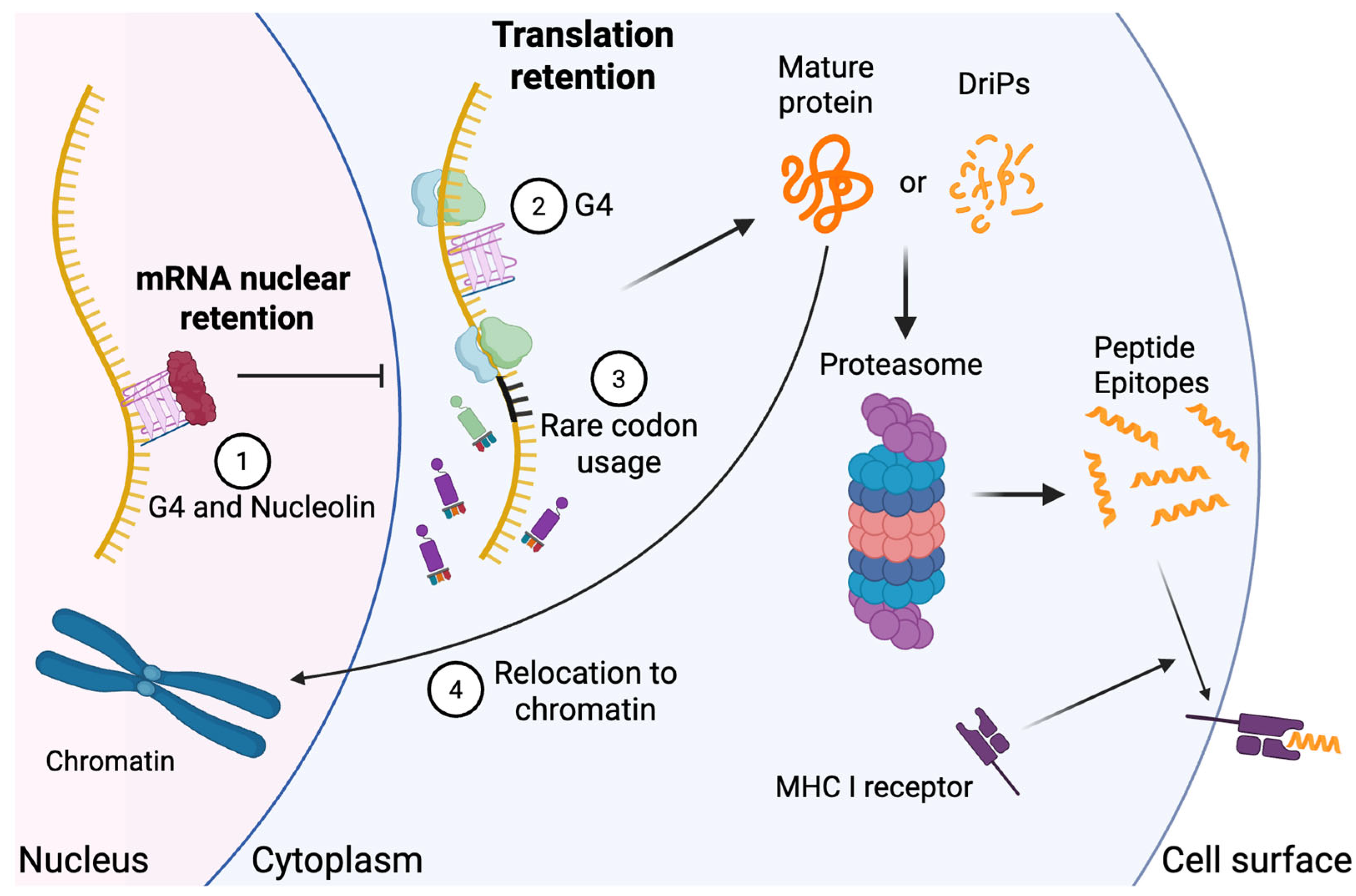
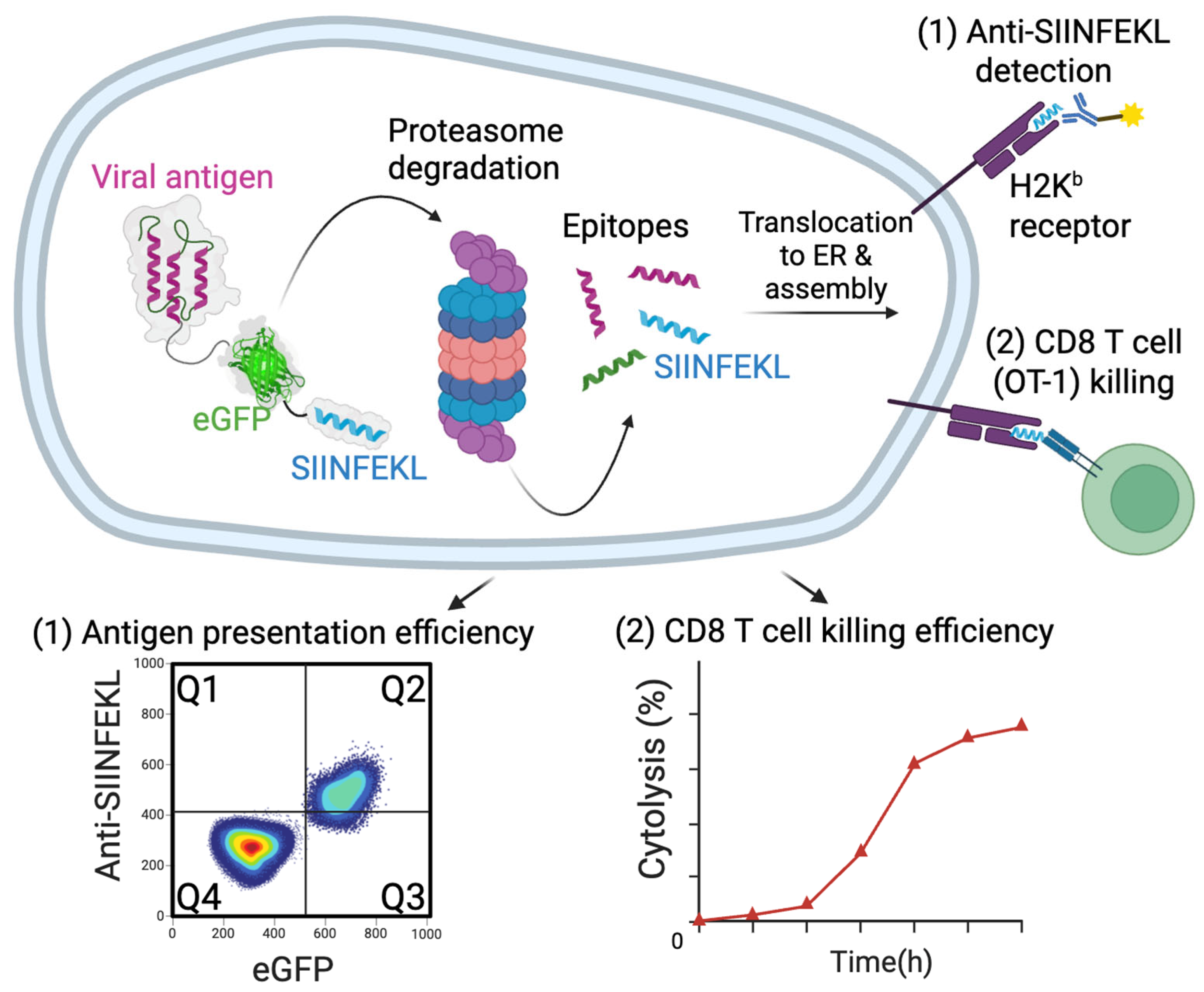
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, L.; Shuda, M.; Chang, Y.; Moore, P.S. Primary Sequence-Intrinsic Immune Evasion by Viral Proteins Guides CTL-Based Vaccine Strategies. Viruses 2025, 17, 1035. https://doi.org/10.3390/v17081035
Wan L, Shuda M, Chang Y, Moore PS. Primary Sequence-Intrinsic Immune Evasion by Viral Proteins Guides CTL-Based Vaccine Strategies. Viruses. 2025; 17(8):1035. https://doi.org/10.3390/v17081035
Chicago/Turabian StyleWan, Li, Masahiro Shuda, Yuan Chang, and Patrick S. Moore. 2025. "Primary Sequence-Intrinsic Immune Evasion by Viral Proteins Guides CTL-Based Vaccine Strategies" Viruses 17, no. 8: 1035. https://doi.org/10.3390/v17081035
APA StyleWan, L., Shuda, M., Chang, Y., & Moore, P. S. (2025). Primary Sequence-Intrinsic Immune Evasion by Viral Proteins Guides CTL-Based Vaccine Strategies. Viruses, 17(8), 1035. https://doi.org/10.3390/v17081035