
Intra-Host Evolution During Relapsing Parvovirus B19 Infection in Immunocompromised Patients
 , , , ,
, , , ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
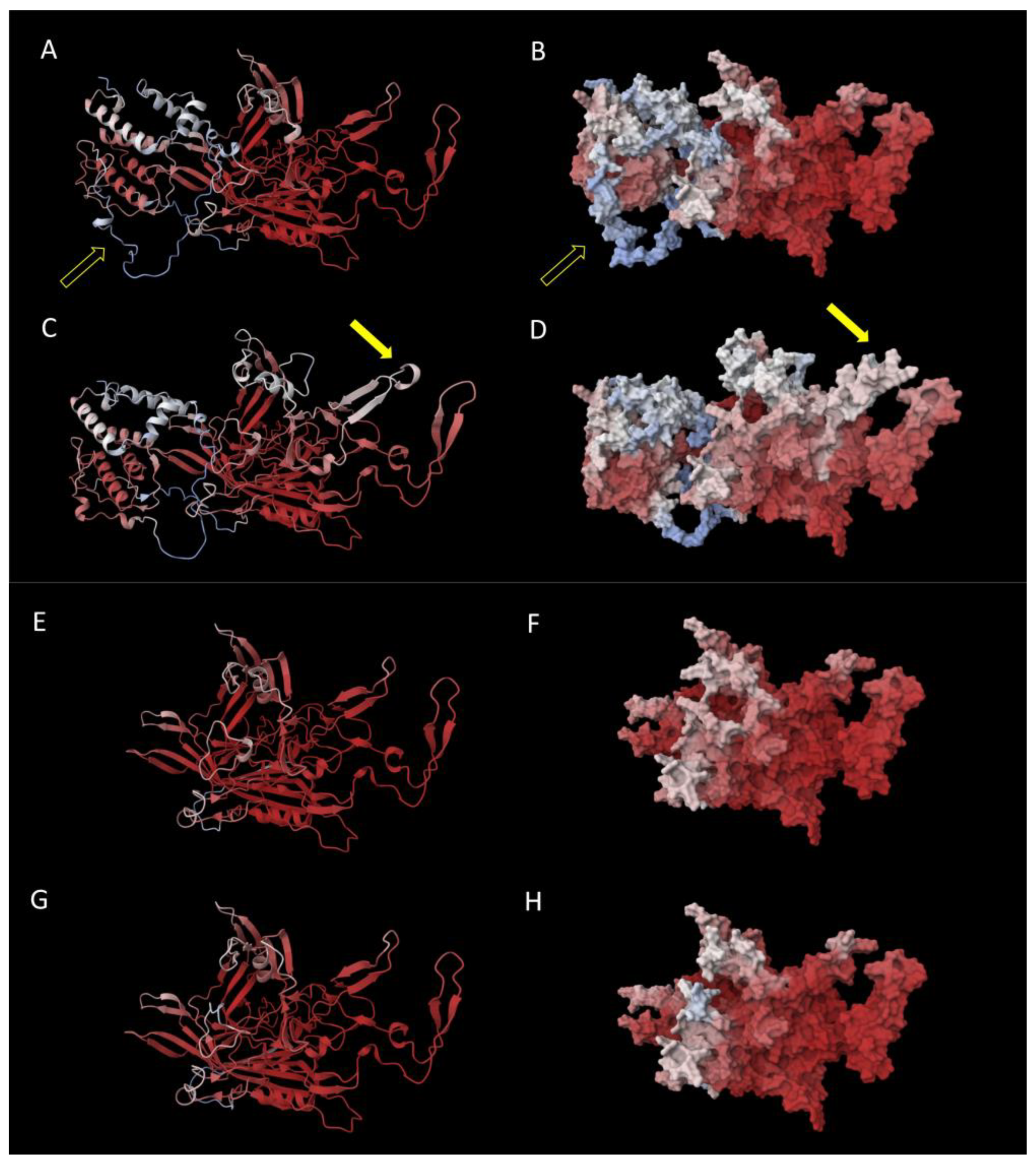
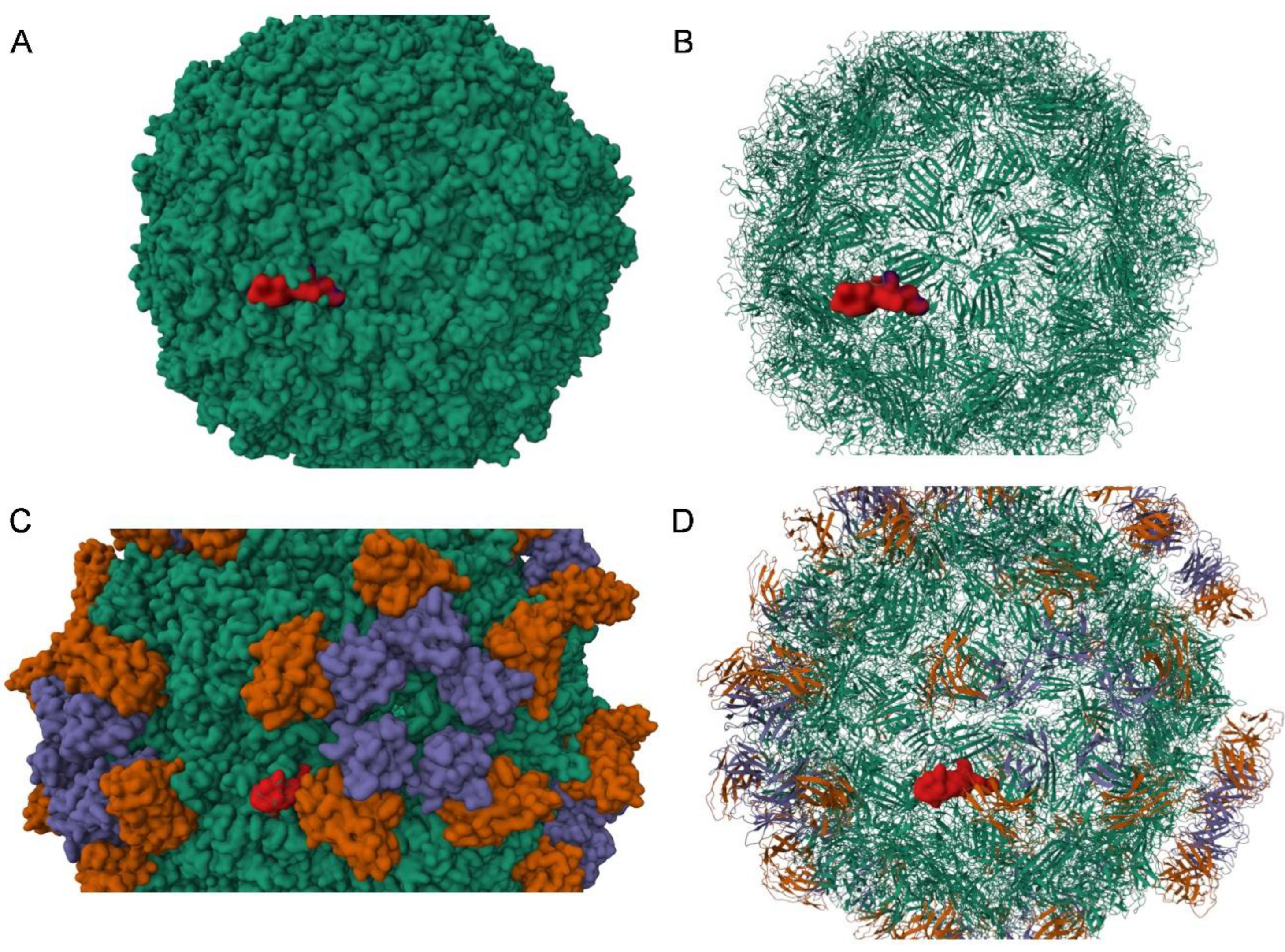
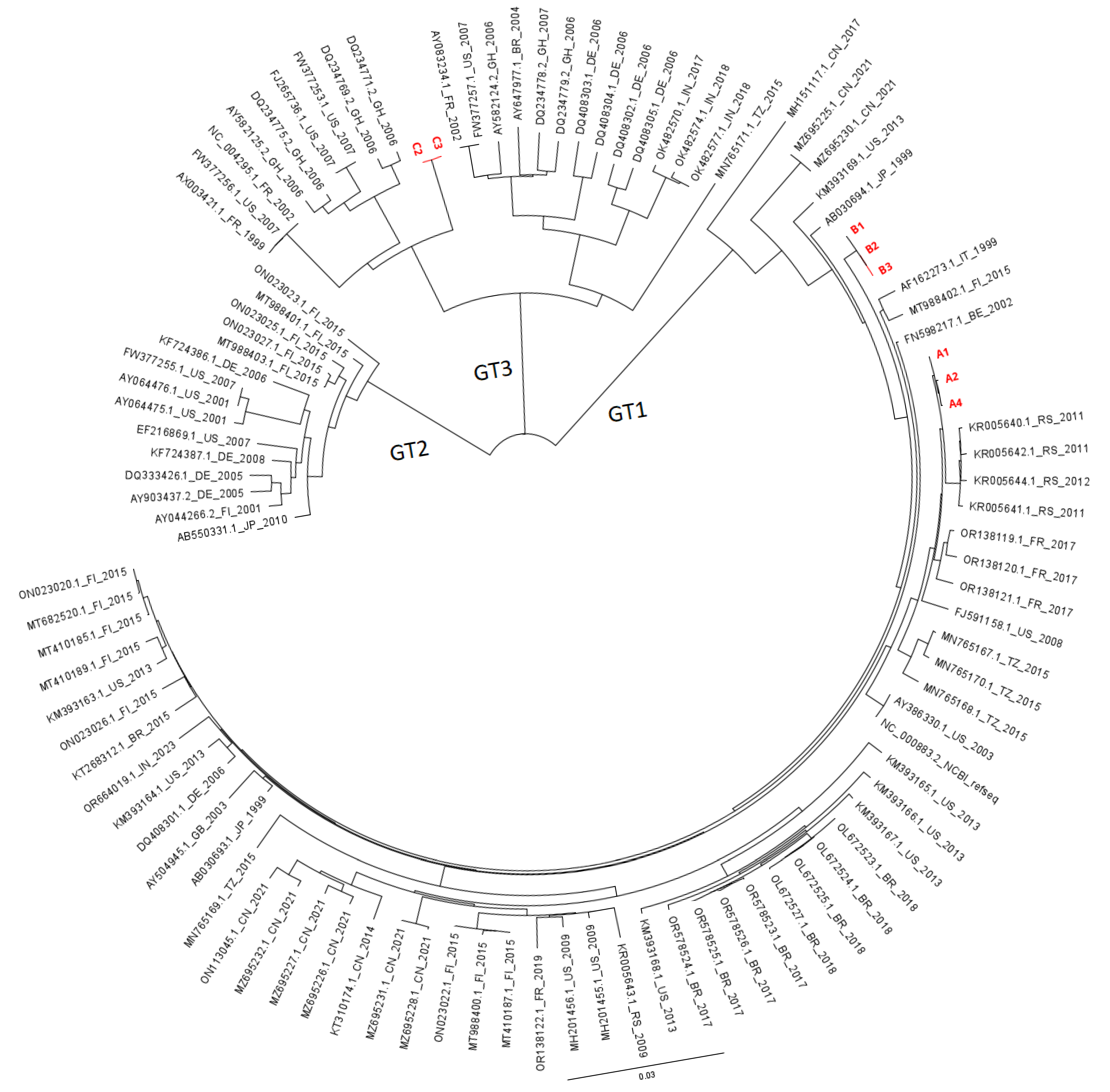
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ganaie, S.S.; Qiu, J. Recent Advances in Replication and Infection of Human Parvovirus B19. Front. Cell Infect. Microbiol. 2018, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.J.; Ardura, M.I. Human parvovirus B19 in solid organ transplantation: Guidelines from the American society of transplantation infectious diseases community of practice. Clin. Transplant. 2019, 33, e13535. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Canto, R.; Carrillo-Pérez, D.L.; Jiménez, J.V.; Cuellar-Rodríguez, J.; Parra-Avila, I.; Alberú, J.; Morales-Buenrostro, L.E. Treatment strategies and outcome of parvovirus B19 infection in kidney transplant recipients: A case series and literature review of 128 patients. Rev. Investig. Clin. 2019, 71, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Worp, N.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Haagmans, B.; Fouchier, R.A.M.; Koopmans, M. The next phase of SARS-CoV-2 surveillance: Real-time molecular epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Gooskens, J.; Jonges, M.; Claas, E.C.; Meijer, A.; Kroes, A.C. Prolonged influenza virus infection during lymphocytopenia and frequent detection of drug-resistant viruses. J. Infect. Dis. 2009, 199, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.B.; Ribeiro, J.; Boutolleau, D.; Sousa, H. Human cytomegalovirus antiviral drug resistance in hematopoietic stem cell transplantation: Current state of the art. Rev. Med. Virol. 2016, 26, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Nijhuis, R.H.T.; Worp, N.; Boter, M.; Weller, B.; Verstrepen, B.E.; Geurtsvankessel, C.; Corsten, M.F.; Russcher, A.; Koopmans, M. Highly Divergent SARS-CoV-2 Alpha Variant in Chronically Infected Immunocompromised Person. Emerg. Infect. Dis. 2022, 28, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Gallinella, G.; Venturoli, S.; Gentilomi, G.; Musiani, M.; Zerbini, M. Extent of sequence variability in a genomic region coding for capsid proteins of B19 parvovirus. Arch. Virol. 1995, 40, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.C.; Sheng, W.H.; Lee, K.L.; Yang, S.J.; Chen, M.Y. Genetic drift of parvovirus B19 is found in AIDS patients with persistent B19 infection. J. Med. Virol. 2006, 78, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Ito, Y.; Shimada, A.; Saito, M.; Muramatsu, H.; Hama, A.; Takahashi, Y.; Kimura, H.; Kojima, S. Long-term parvovirus B19 infections with genetic drift after cord blood transplantation complicated by persistent CD4+ lymphocytopenia. J. Pediatr. Hematol. Oncol. 2014, 36, e65–e68. [Google Scholar] [CrossRef] [PubMed]
- Plentz, A.; Hahn, J.; Holler, E.; Jilg, W.; Modrow, S. Long-term parvovirus B19 viraemia associated with pure red cell aplasia after allogeneic bone marrow transplantation. J. Clin. Virol. 2004, 31, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Lu, I.N.; Muller, C.P.; He, F.Q. Applying next-generation sequencing to unravel the mutational landscape in viral quasispecies. Virus Res. 2020, 283, 197963. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Hung, S.J.; Wang, J.R. Application of deep sequencing methods for inferring viral population diversity. J. Virol. Methods. 2019, 266, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Knoester, M.; von dem Borne, P.A.; Vossen, A.C.; Kroes, A.C.; Claas, E.C. Human parvovirus B19 genotype 3 associated with chronic anemia after stem cell transplantation, missed by routine PCR testing. J. Clin. Virol. 2012, 54, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Carbo, E.C.; Russcher, A.; Kraakman, M.E.M.; de Brouwer, C.S.; Sidorov, I.A.; Feltkamp, M.C.W.; Kroes, A.C.M.; Claas, E.C.; de Vries, J.J.C. Longitudinal Monitoring of DNA Viral Loads in Transplant Patients Using Quantitative Metagenomic Next-Generation Sequencing. Pathogens 2022, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Jaroslav, K.; Rose, R.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, w431–w437. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.B.; Huntington, B.; Perminov, A.; Smith, K.; Hastings, N.; Dennis, P.; Kelley-Loughnane, N.; Berry, R. AlphaFold2 modeling and molecular dynamics simulations of an intrinsically disordered protein. PLoS ONE 2024, 19, e0301866. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.; Simpson, A.; Rossmann, M. The structure of human parvovirus B19. Proc. Natl. Acad. Sci. USA 2004, 101, 11628–11633. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Klose, T.; Liu, Y.; Modrow, S.; Rossmann, M. Structure of Parvovirus B19 decorated by Fabs from a human antibody. J. Virol. 2019, 93, e01732-18. [Google Scholar] [CrossRef] [PubMed]
- Saikawa, T.; Anderson, S.; Momoeda, M.; Kajigaya, S.; Young, N.S. Neutralizing linear epitopes of B19 parvovirus cluster in the VP1 unique and VP1-VP2 junction regions. J. Virol. 1993, 67, 3004–3009. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Momoeda, M.; Kawase, M.; Kajigaya, S.; Young, N.S. Peptides derived from the unique region of B19 parvovirus minor capsid protein elicit neutralizing antibodies in rabbits. Virology 1995, 206, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Ros, C.; Bieri, J.; Leisi, R. The VP1u of Human Parvovirus B19: A Multifunctional Capsid Protein with Biotechnological Applications. Viruses 2020, 12, 1463. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, K.; Rosenfeld, S.; Frickhofen, N.; Kennedy, D.; Hills, R.; Kajigaya, S.; Young, N.S. A second neutralizing epitope of B19 parvovirus implicates the spike region in the immune response. J. Virol. 1991, 65, 7056–7060. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Jain, P.; Kumar, A.; Prakash, S.; Khan, D.N.; Kant, R. Incidence and progression of Parvovirus B19 infection and molecular changes in circulating B19V strains in children with haematological malignancy: A follow up study. Infect. Genet. Evol. 2018, 57, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Holmes, E.C. Phylogenetic evidence for the rapid evolution of human B19 erythrovirus. J. Virol. 2006, 80, 3666–3669. [Google Scholar] [CrossRef] [PubMed]
- Norja, P.; Hokynar, K.; Aaltonen, L.-M.; Chen, R.; Ranki, A.; Partio, E.K.; Kiviluoto, O.; Davidkin, I.; Leivo, T.; Eis-Hübinger, A.M.; et al. Bioportfolio: Lifelong persistence of variant and prototypic erythrovirus DNA genomes in human tissue. Proc. Natl. Acad. Sci. USA 2006, 103, 7450–7453. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Yoto, Y.; Ishikawa, A.; Tsutsumi, H. Analysis of nucleotide sequences of human parvovirus B19 genome reveals two different modes of evolution, a gradual alteration and a sudden replacement: A retrospective study in Sapporo, Japan, from 1980 to 2008. J. Virol. 2009, 83, 10975–10980. [Google Scholar] [CrossRef] [PubMed]
- Toppinen, M.; Perdomo, M.F.; Palo, J.U.; Simmonds, P.; Lycett, S.J.; Soderlund-Venermo, M.; Sajantila, A.; Hedman, K. Bones hold the key to DNA virus history and epidemiology. Sci. Rep. 2015, 5, 17226. [Google Scholar] [CrossRef] [PubMed]
- Stamenković, G.G.; Ćirković, V.S.; Šiljić, M.M.; Blagojević, J.V.; Knežević, A.M.; Joksić, I.D.; Stanojević, M.P. Substitution rate and natural selection in parvovirus B19. Sci. Rep. 2016, 6, 35759. [Google Scholar] [CrossRef] [PubMed]
- Mühlemann, B.; Margaryan, A.; Damgaard, P.d.B.; Allentoft, M.E.; Vinner, L.; Hansen, A.J.; Weber, A.; Bazaliiskii, V.I.; Molak, M.; Arneborg, J.; et al. Ancient human parvovirus B19 in Eurasia reveals its long-term association with humans. Proc. Natl. Acad. Sci. USA 2018, 115, 7557–7562. [Google Scholar] [CrossRef] [PubMed]
- A Guzmán-Solís, A.; Villa-Islas, V.; Bravo-López, M.J.; Sandoval-Velasco, M.; Wesp, J.K.; A Gómez-Valdés, J.; Moreno-Cabrera, M.d.l.L.; Meraz, A.; Solís-Pichardo, G.; Schaaf, P.; et al. Ancient viral genomes reveal introduction of human pathogenic viruses into Mexico during the transatlantic slave trade. Elife 2021, 10, e68612. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, M.; Donoghue, P.C.; Yang, Z. Bayesian molecular clock dating of species divergences in the genomics era. Nat. Rev. Genet. 2016, 17, 71–80. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Presence C3742G (% Reads) | Presence A3892T (% Reads) | AA Substitution | AA Substitution | AA Substitution | ||
|---|---|---|---|---|---|---|
| VP1 | VP2 | NS | ||||
| Patient A | 0 weeks | |||||
| 10 weeks | 20% | 2% | - | - | - | |
| 14 weeks | 48% | 16% | T372S | T145S | na 1 | |
| 16 weeks | 51% | 38% | T372S | T145S | na | |
| 20 weeks | 27% | 71% | Q422L | Q195L | na |
| First Author | Year | Study Population | B19V Treatment | Sequencing Method | Nucleotide Region Sequenced | Follow-Up Period | Results |
|---|---|---|---|---|---|---|---|
| Gallinella | 1996 | 1 patient; chronic anemia | Not mentioned | Sanger | 2400–3400 | 16 months | No changes in viral genome |
| Plentz | 2004 | 1 patient; bone marrow transplantation | IVIG | Not mentioned | Not mentioned | 8 months | 3 lasting changes after temporary variations: T3463C C4852G T4867C |
| Hung | 2006 | 3 patients; AIDS | IVIG (n = 3); HAART (n = 2) | Sanger | 436–2431; 3125–4283 | 11 months | Pt 1: A3271C Pt 2: 18 SNP (14 N 1) Pt 3: 15 SNP (9 N) |
| Suzuki | 2014 | 1 pediatric patient; cord blood transplantation | IVIG | Sanger | 602–5014 | 29 months | 6 SNP T/C941T T/C1037T A1048A/G (N) T1112C T1118T/C A1266A/G (N) |
| Jain | 2018 | 13 pediatric hematological malignancy patients followed up; 3 patients with at least one mutation detected | Not mentioned | Sanger | 1747–2691 | 6 months | Pt 1: G579A Pt 2: C577G Pt 3: C672G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russcher, A.; Mohammed, Y.; Kraakman, M.E.M.; Chow, X.; Kok, S.T.; Claas, E.C.J.; Wuhrer, M.; Vossen, A.C.T.M.; Kroes, A.C.M.; de Vries, J.J.C. Intra-Host Evolution During Relapsing Parvovirus B19 Infection in Immunocompromised Patients. Viruses 2025, 17, 1034. https://doi.org/10.3390/v17081034
Russcher A, Mohammed Y, Kraakman MEM, Chow X, Kok ST, Claas ECJ, Wuhrer M, Vossen ACTM, Kroes ACM, de Vries JJC. Intra-Host Evolution During Relapsing Parvovirus B19 Infection in Immunocompromised Patients. Viruses. 2025; 17(8):1034. https://doi.org/10.3390/v17081034
Chicago/Turabian StyleRusscher, Anne, Yassene Mohammed, Margriet E. M. Kraakman, Xavier Chow, Stijn T. Kok, Eric C. J. Claas, Manfred Wuhrer, Ann C. T. M. Vossen, Aloys C. M. Kroes, and Jutte J. C. de Vries. 2025. "Intra-Host Evolution During Relapsing Parvovirus B19 Infection in Immunocompromised Patients" Viruses 17, no. 8: 1034. https://doi.org/10.3390/v17081034
APA StyleRusscher, A., Mohammed, Y., Kraakman, M. E. M., Chow, X., Kok, S. T., Claas, E. C. J., Wuhrer, M., Vossen, A. C. T. M., Kroes, A. C. M., & de Vries, J. J. C. (2025). Intra-Host Evolution During Relapsing Parvovirus B19 Infection in Immunocompromised Patients. Viruses, 17(8), 1034. https://doi.org/10.3390/v17081034