
Multiscale Modeling and Dynamic Mutational Profiling of Binding Energetics and Immune Escape for Class I Antibodies with SARS-CoV-2 Spike Protein: Dissecting Mechanisms of High Resistance to Viral Escape Against Emerging Variants



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
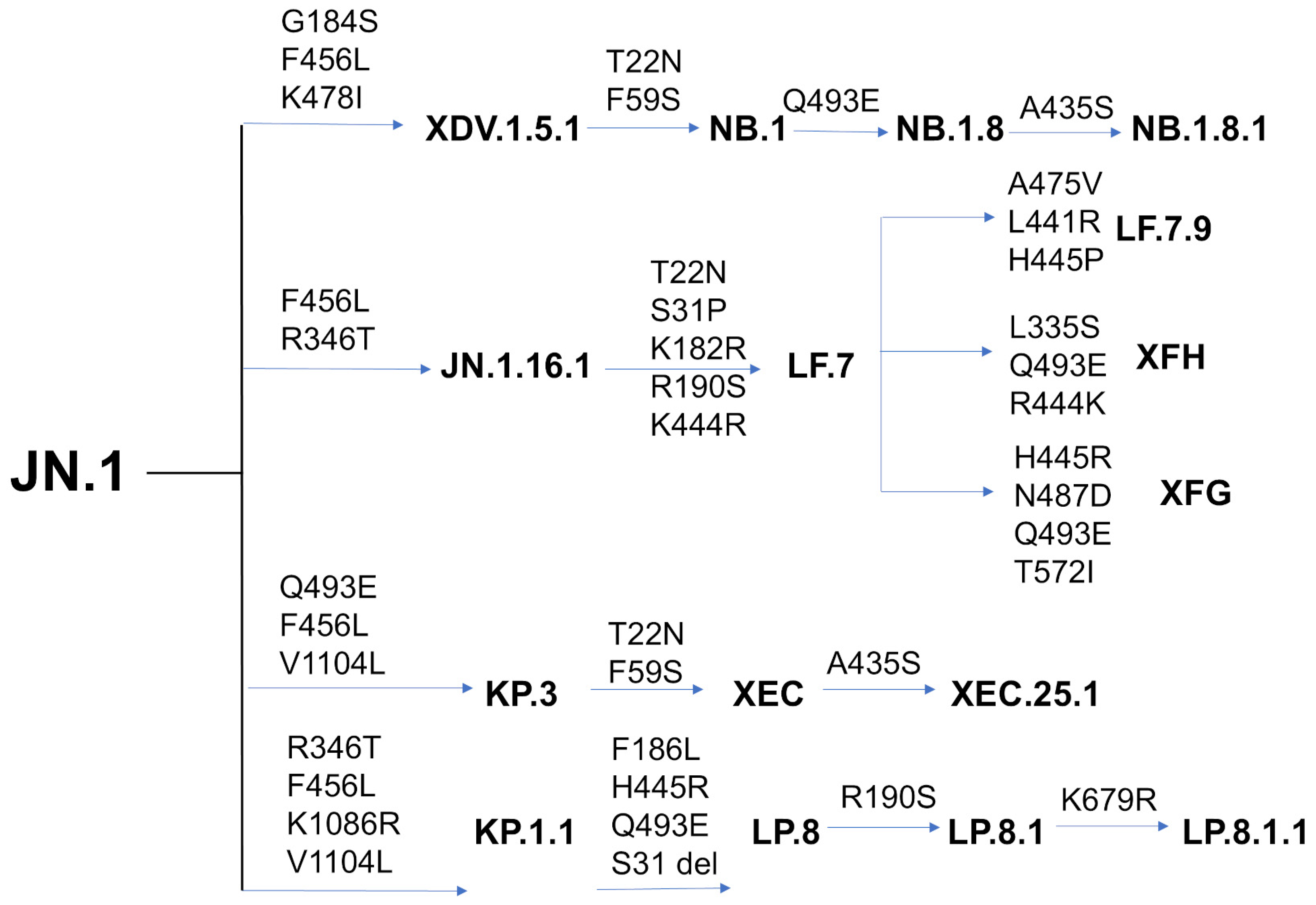
1. Introduction
2. Materials and Methods
2.1. Coarse-Grained Molecular Simulations and Atomistic Reconstruction of Equilibrium Ensembles
2.2. Binding Free Energy Computations: Mutational Scanning Profiling and Analysis
2.3. Binding Free Energy Computations
3. Results
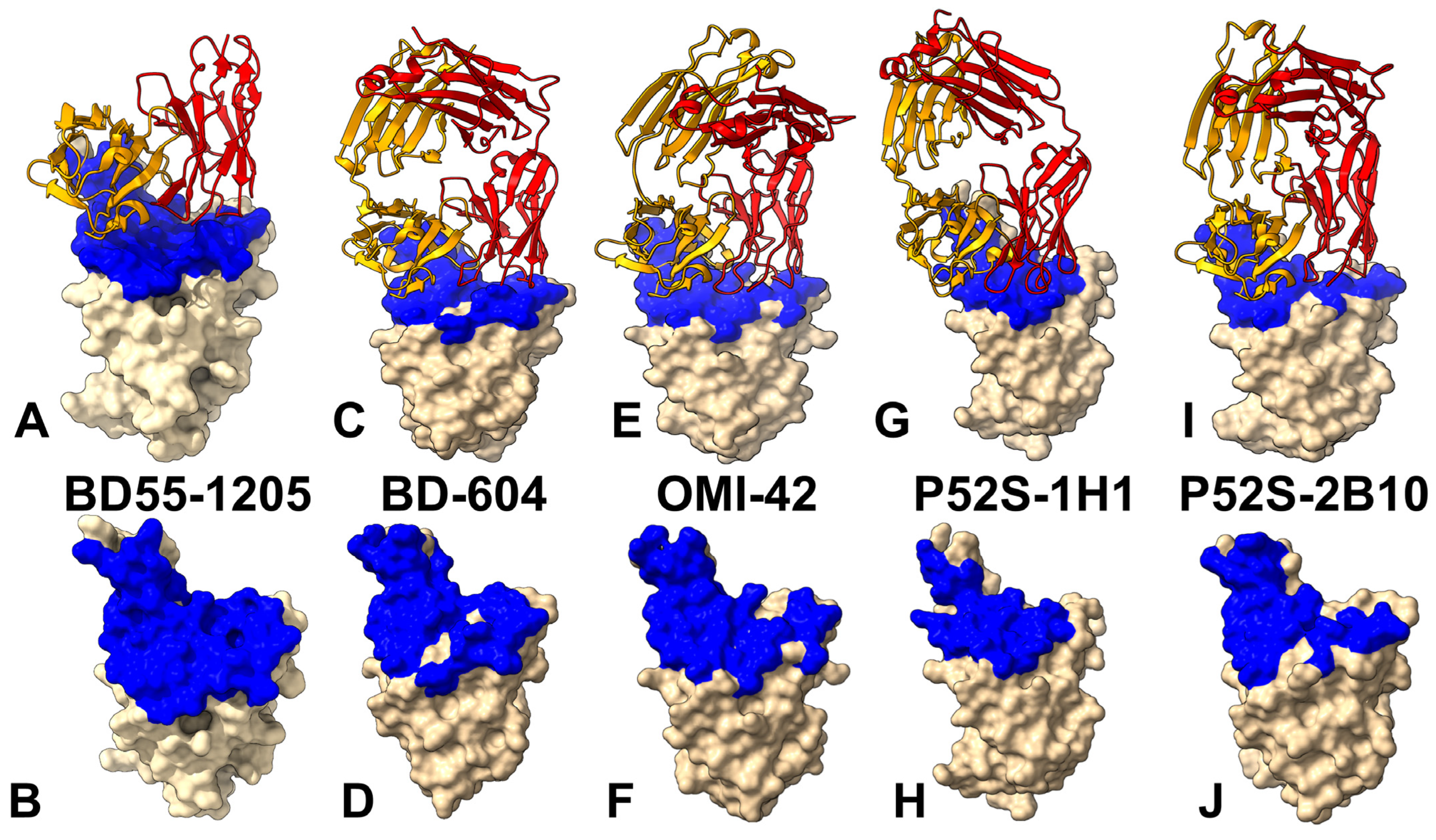
3.1. Structural Analysis of the RBD Complexes
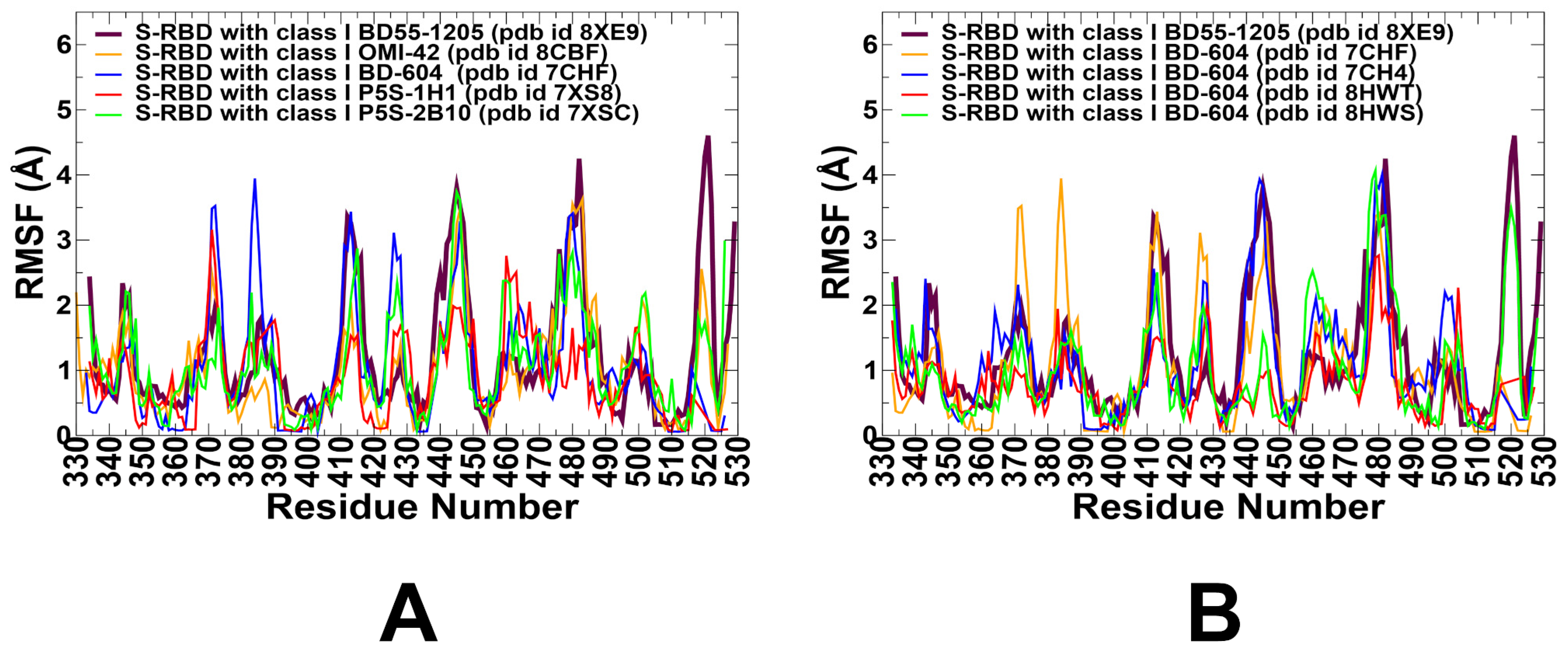
3.2. Coarse-Grained Simulations and Atomistic Reconstruction of the Conformational Ensembles for RBD Complexes with Class I Antibodies
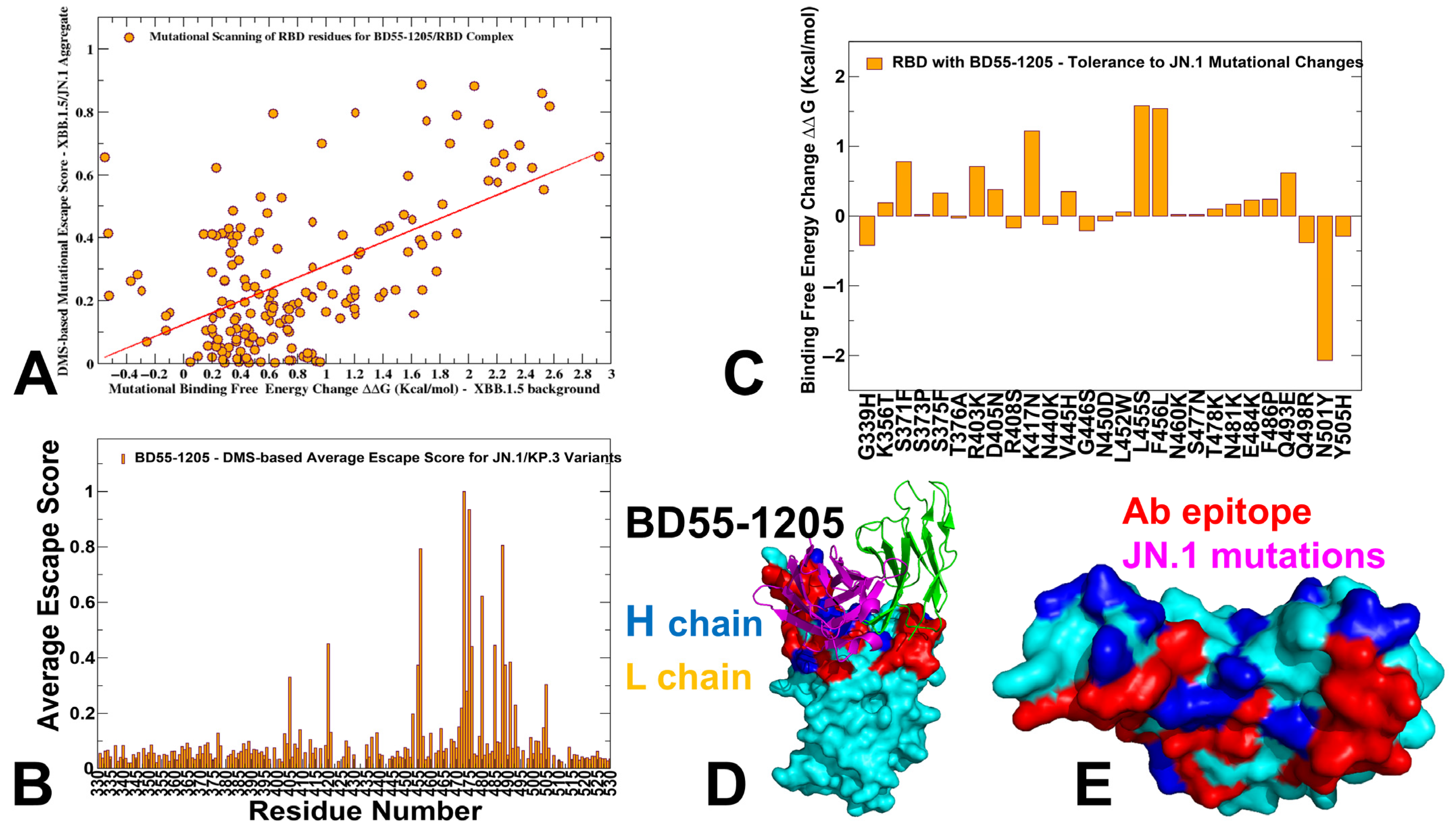
3.3. Mutational Profiling of Antibody-RBD Binding Interactions Interfaces Reveals Molecular Determinants of Immune Sensitivity and Emergence of Convergent Escape Hotspots
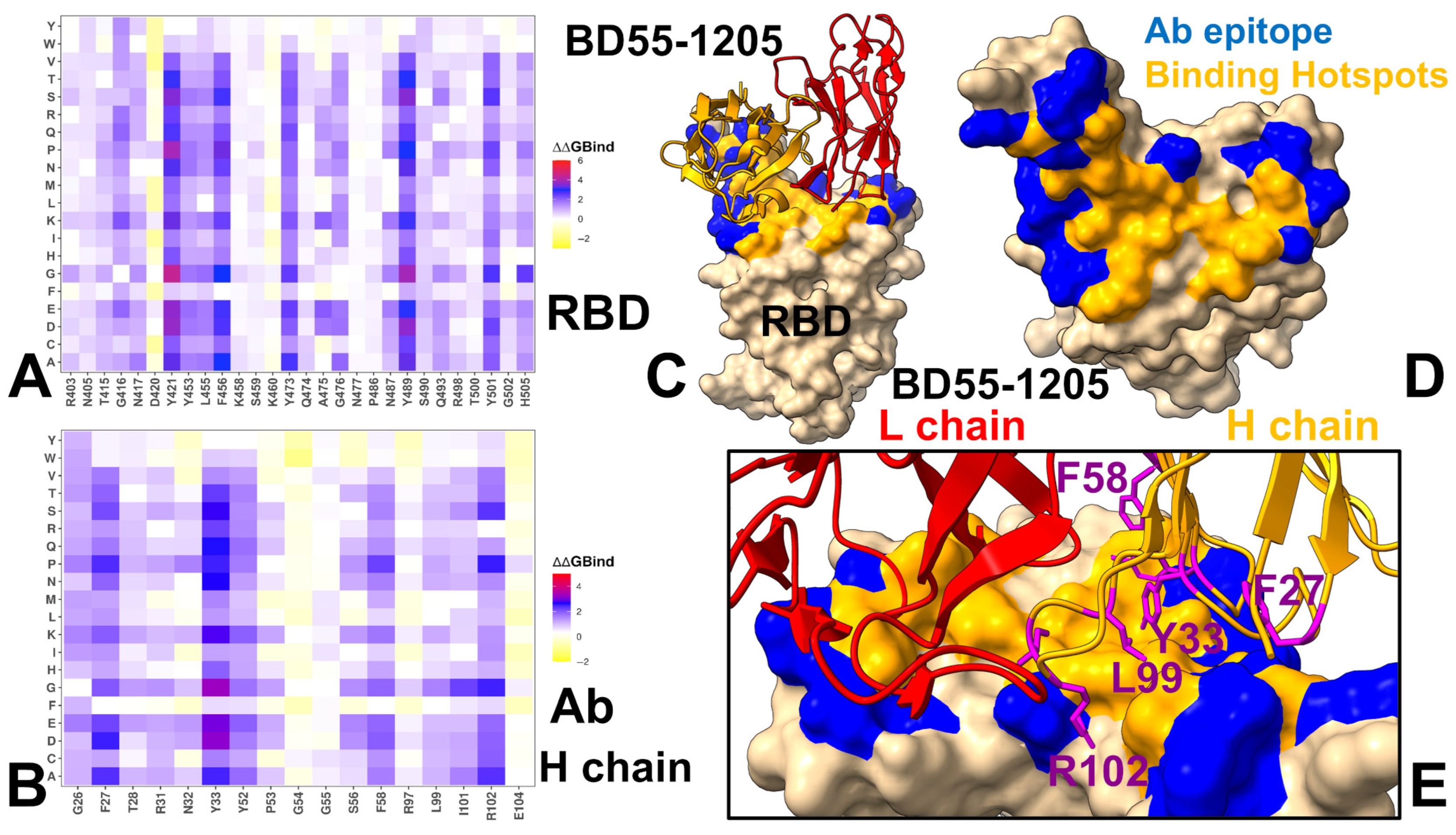
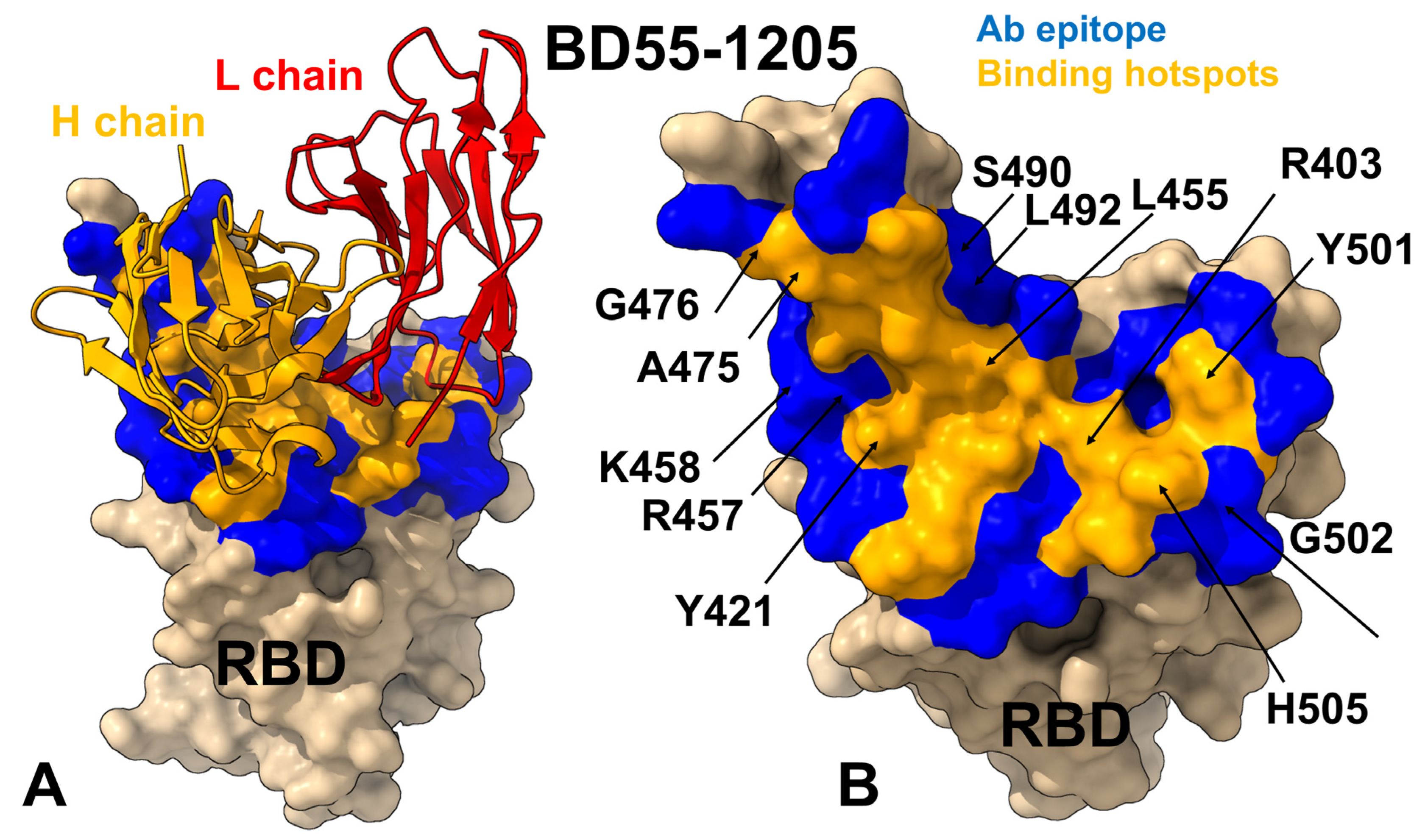
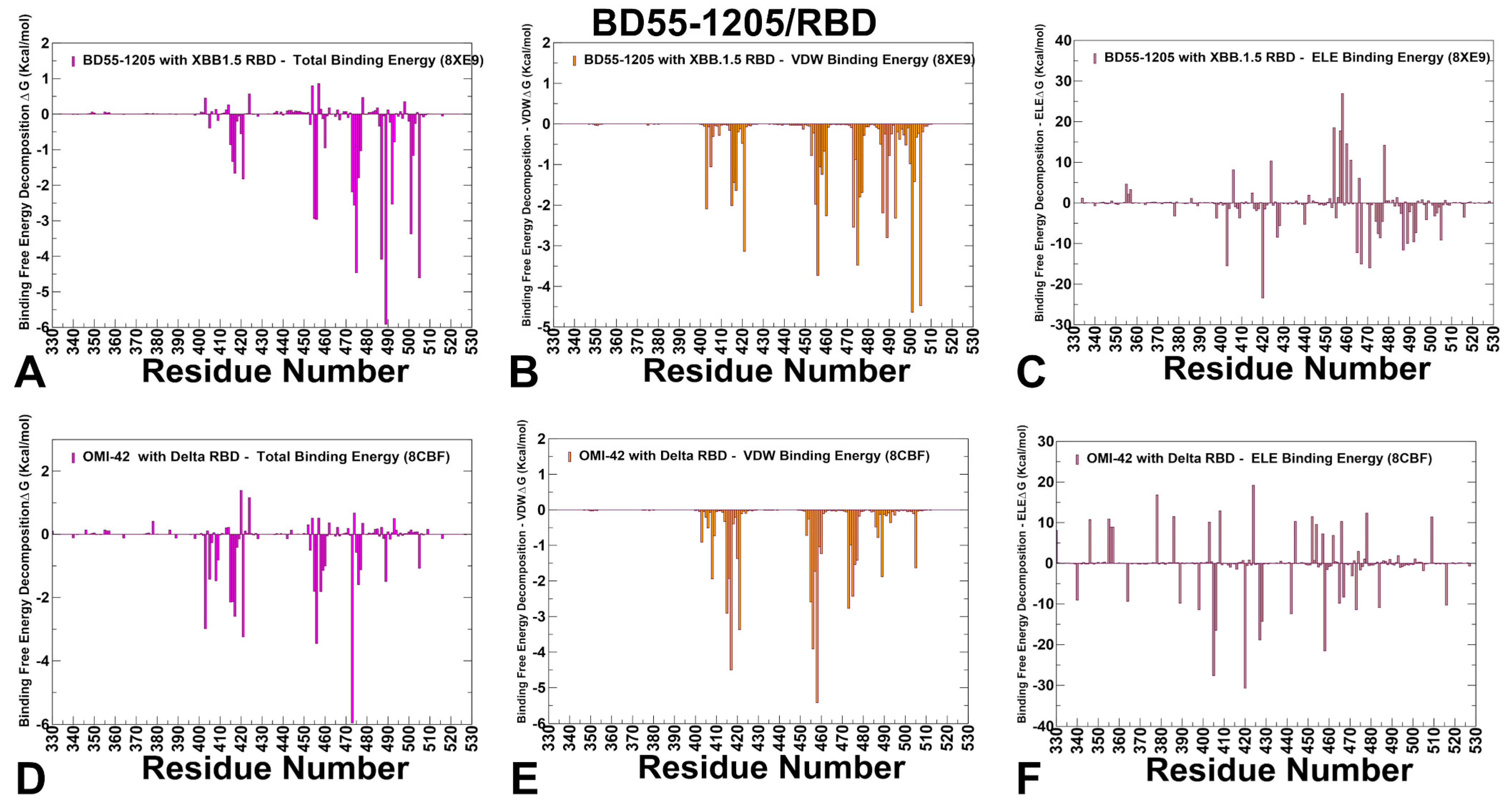
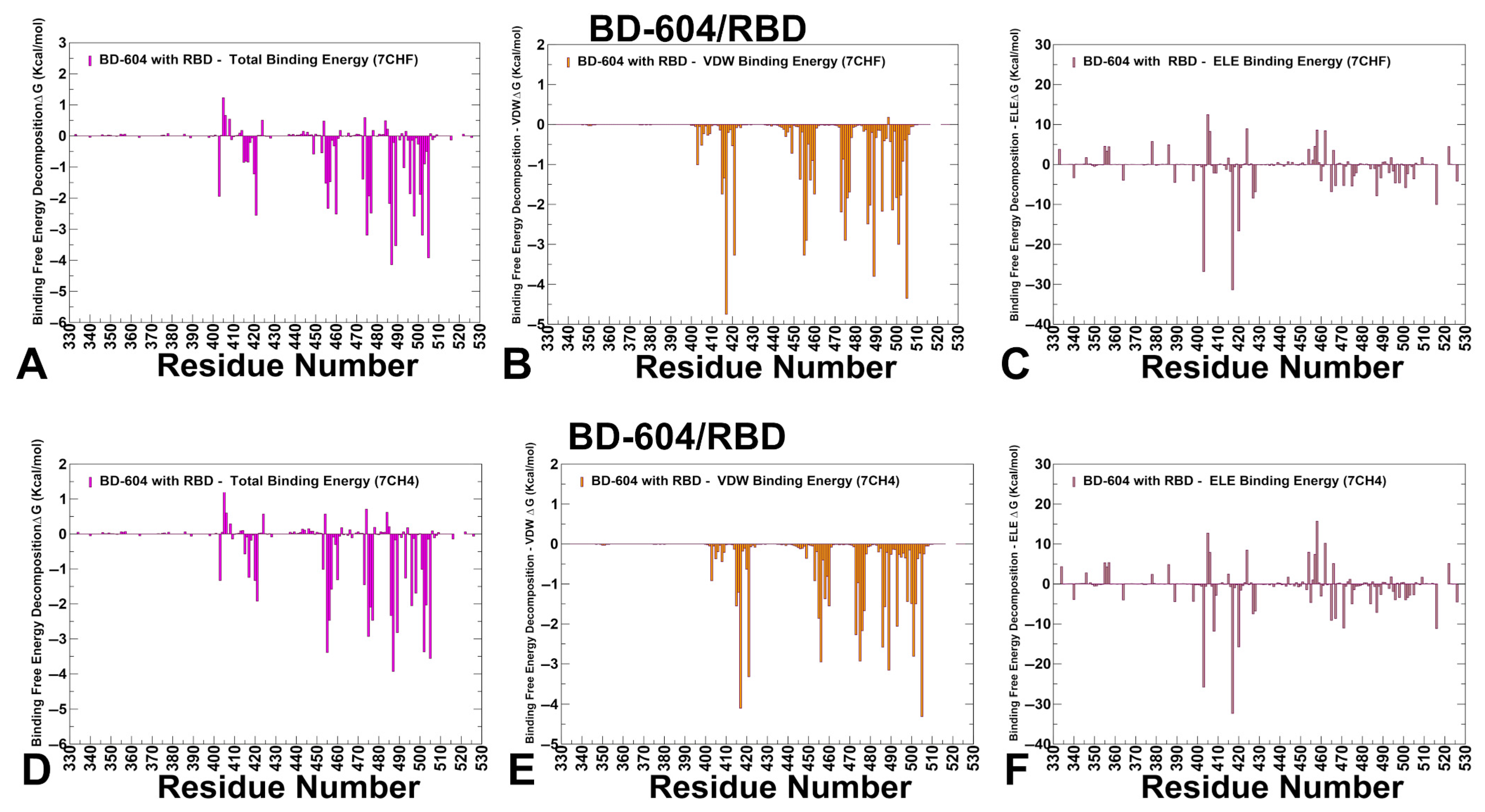
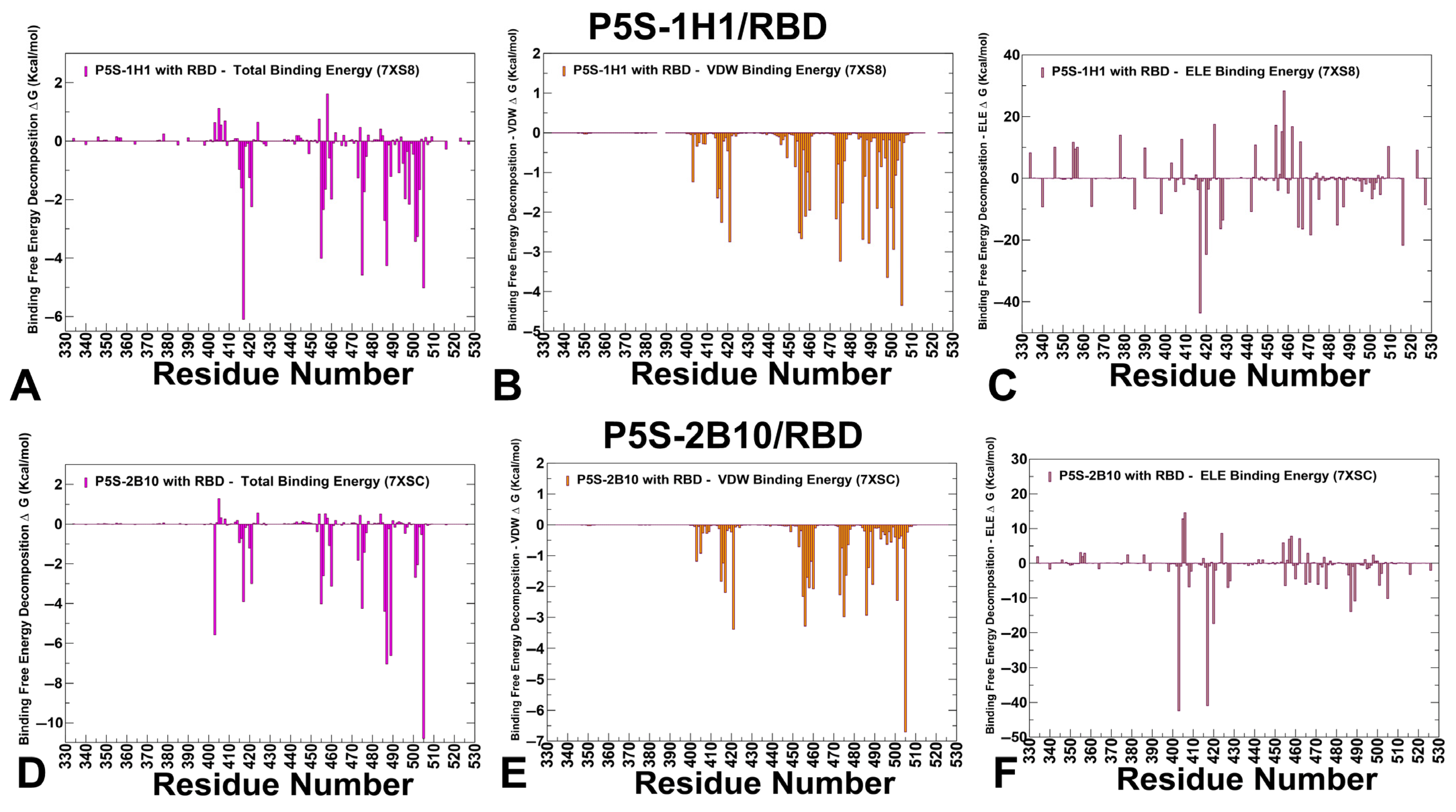
3.4. MM-GBSA Computations of the Binding Energetics and Residue-Based Decomposition Analysis for Class I Antibody-RBD Complexes: Broadly Distributed Footprint of Multiple Binding Hotspots Determines Unique Neutralization Profile of BD55-1205
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Walls, A.C.; Bowen, J.E.; Corti, D.; Veesler, D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020, 27, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.M.; Shoemaker, S.R.; Hobbs, H.T.; Nguyen, A.W.; Hsieh, C.L.; Maynard, J.A.; McLellan, J.S.; Pak, J.E.; Marqusee, S. The SARS-CoV-2 spike reversibly samples an open-trimer conformation exposing novel epitopes. Nat. Struct. Mol. Biol. 2022, 27, 229–238. [Google Scholar] [CrossRef] [PubMed]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The emerging plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, D.; Han, Y.; Lu, M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses 2022, 14, 1255. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Turoňová, B.; Sikora, M.; Schuerman, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Han, Y.; Ding, S.; Shi, W.; Zhou, T.; Finzi, A.; Kwong, P.D.; Mothes, W.; Lu, M. SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy. mBio 2022, 13, e0322721. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. Elife 2022, 11, e75433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural Basis for SARS-CoV-2 Delta Variant Recognition of ACE2 Receptor and Broadly Neutralizing Antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef] [PubMed]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron Variant: Ab Evasion and Cryo-EM Structure of Spike Protein–ACE2 Complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular Basis of Receptor Binding and Ab Neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural Basis of SARS-CoV-2 Omicron Immune Evasion and Receptor Engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike Trimer with ACE2 and an Anti-Omicron Ab. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.-C.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural Diversity of the SARS-CoV-2 Omicron Spike. Mol. Cell 2022, 82, 2050–2068.e6. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and Functional Characterizations of Infectivity and Immune Evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: The XBB.1.5 (‘Kraken’) Subvariant of Omicron SARS-CoV-2 and its Rapid Global Spread. Med. Sci. Monit. 2023, 29, e939580. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming Ab Evasion Properties of Rising SARS-CoV-2 BQ and XBB Subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Arora, P.; Nehlmeier, I.; Kempf, A.; Cossmann, A.; Schulz, S.R.; Morillas Ramos, G.; Manthey, L.A.; Jäck, H.-M.; Behrens, G.M.N.; et al. Profound Neutralization Evasion and Augmented Host Cell Entry Are Hallmarks of the Fast-Spreading SARS-CoV-2 Lineage XBB.1.5. Cell. Mol. Immunol. 2023, 20, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Yamasoba, D.; Uriu, K.; Plianchaisuk, A.; Kosugi, Y.; Pan, L.; Zahradnik, J.; Ito, J.; Sato, K. Virological Characteristics of the SARS-CoV-2 Omicron XBB.1.16 Variant. Lancet Infect. Dis. 2023, 23, 655–656. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, S.; Deguchi, S.; Nomai, T.; Padilla-Blanco, M.; Plianchaisuk, A.; Wang, L.; Begum, M.M.; Uriu, K.; Mizuma, K.; Nao, N.; et al. Virological Characteristics of the SARS-CoV-2 Omicron EG.5.1 Variant. Microbiol Immunol. 2024, 68, 305–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Zhang, R.M.; Ho, J.; Mohri, H.; Valdez, R.; Manthei, D.M.; Gordon, A.; Liu, L.; Ho, D.D. Ab Neutralization of Emerging SARS-CoV-2 Subvariants: EG.5.1 and XBC.1.6. Lancet Infect. Dis. 2023, 23, e397–e398. [Google Scholar] [CrossRef] [PubMed]
- Faraone, J.N.; Qu, P.; Goodarzi, N.; Zheng, Y.-M.; Carlin, C.; Saif, L.J.; Oltz, E.M.; Xu, K.; Jones, D.; Gumina, R.J.; et al. Immune Evasion and Membrane Fusion of SARS-CoV-2 XBB Subvariants EG.5.1 and XBB.2.3. Emerg. Microbes Infect. 2023, 12, 2270069. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, Y.; Plianchaisuk, A.; Putri, O.; Uriu, K.; Kaku, Y.; Hinay, A.A., Jr.; Chen, L.; Kuramochi, J.; Sadamasu, K.; Yoshimura, K.; et al. Characteristics of the SARS-CoV-2 Omicron HK.3 Variant Harbouring the FLip Substitution. Lancet Microbe 2024, 5, e313. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Liu, L.; Schwanz, L.T.; Li, Z.; Nair, M.S.; Ho, J.; Zhang, R.M.; Iketani, S.; Yu, J.; et al. Antigenicity and Receptor Affinity of SARS-CoV-2 BA.2.86 Spike. Nature 2023, 624, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Chen, X.; Xu, Y.; Wang, P.; Wang, J.; Yu, L.; et al. Antigenicity and Infectivity Characterization of SARS-CoV-2 BA.2.86. Lancet Infect. Dis. 2023, 23, e457–e459. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Mizuma, K.; Nasser, H.; Deguchi, S.; Padilla-Blanco, M.; Oda, Y.; Uriu, K.; Tolentino, J.E.M.; Tsujino, S.; Suzuki, R.; et al. Virological Characteristics of the SARS-CoV-2 BA.2.86 Variant. Cell Host Microbe 2024, 32, 170–180.e12. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, D.; Dijokaite-Guraliuc, A.; Supasa, P.; Duyvesteyn, H.M.E.; Ginn, H.M.; Selvaraj, M.; Mentzer, A.J.; Das, R.; de Silva, T.I.; et al. A Structure-Function Analysis SARS-CoV-2 BA.2.86 Balances Ab Escape and ACE2 Affinity. Cell Rep. Med. 2024, 5, 101553. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Lustig, G.; Römer, C.; Reedoy, K.; Jule, Z.; Karim, F.; Ganga, Y.; Bernstein, M.; Baig, Z.; Jackson, L.; et al. Evolution and Neutralization Escape of the SARS-CoV-2 BA.2.86 Subvariant. Nat. Commun. 2023, 14, 8078. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yu, Y.; Xu, Y.; Jian, F.; Song, W.; Yisimayi, A.; Wang, P.; Wang, J.; Liu, J.; Yu, L.; et al. Fast Evolution of SARS-CoV-2 BA.2.86 to JN.1 under Heavy Immune Pressure. Lancet Infect. Dis. 2024, 24, e70–e72. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Okumura, K.; Padilla-Blanco, M.; Kosugi, Y.; Uriu, K.; Hinay, A.A., Jr.; Chen, L.; Plianchaisuk, A.; Kobiyama, K.; Ishii, K.J.; et al. Virological Characteristics of the SARS-CoV-2 JN.1 Variant. Lancet Infect. Dis. 2024, 24, e82. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Faraone, J.N.; Hsu, C.C.; Chamblee, M.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Horowitz, J.C.; Mallampalli, R.K.; Saif, L.J.; et al. Neutralization Escape, Infectivity, and Membrane Fusion of JN.1-Derived SARS-CoV-2 SLip, FLiRT, and KP.2 Variants. Cell Rep. 2024, 43, 114520. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.; Faraone, J.N.; Evans, J.P.; Zheng, Y.-M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Panchal, A.R.; et al. Enhanced Evasion of Neutralizing Ab Response by Omicron XBB.1.5, CH.1.1, and CA.3.1 Variants. Cell Rep. 2023, 42, 112443. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Uriu, K.; Kosugi, Y.; Okumura, K.; Yamasoba, D.; Uwamino, Y.; Kuramochi, J.; Sadamasu, K.; Yoshimura, K.; Asakura, H.; et al. Virological Characteristics of the SARS-CoV-2 KP.2 Variant. Lancet Infect. Dis. 2024, 24, e416. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Yo, M.S.; Tolentino, J.E.; Uriu, K.; Okumura, K.; Ito, J.; Sato, K. Virological Characteristics of the SARS-CoV-2 KP.3, LB.1, and KP.2.3 Variants. Lancet Infect. Dis. 2024, 24, e482–e483. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mellis, I.A.; Ho, J.; Bowen, A.; Kowalski-Dobson, T.; Valdez, R.; Katsamba, P.S.; Wu, M.; Lee, C.; Shapiro, L.; et al. Recurrent SARS-CoV-2 Spike Mutations Confer Growth Advantages to Select JN.1 Sublineages. Emerg. Microbes Infect. 2024, 13, 2402880. [Google Scholar] [CrossRef] [PubMed]
- Jian, F.; Wang, J.; Yisimayi, A.; Song, W.; Xu, Y.; Chen, X.; Niu, X.; Yang, S.; Yu, Y.; Wang, P.; et al. Evolving Antibody Response to SARS-CoV-2 Antigenic Shift from XBB to JN.1. Nature 2025, 637, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.L.; Starr, T.N. Deep Mutational Scanning of SARS-CoV-2 Omicron BA.2.86 and Epistatic Emergence of the KP.3 Variant. Virus Evol. 2024, 10, veae067. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Sun, Z.; Zhang, Y.; Jian, F.; Yang, S.; Xia, K.; Yu, L.; Wang, J.; Shao, F.; Wang, X.; et al. Structural and Molecular Basis of the Epistasis Effect in Enhanced Affinity between SARS-CoV-2 KP.3 and ACE2. Cell Discov. 2024, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, Y.; Jian, F.; Yang, S.; Song, W.; Wang, P.; Yu, L.; Shao, F.; Cao, Y. Enhanced Immune Evasion of SARS-CoV-2 Variants KP.3.1.1 and XEC through N-Terminal Domain Mutations. Lancet Infect. Dis. 2025, 25, e6–e7. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Uriu, K.; Okumura, K.; Ito, J.; Sato, K. Virological Characteristics of the SARS-CoV-2 KP.3.1.1 Variant. Lancet Infect. Dis. 2024, 24, e609. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Okumura, K.; Kawakubo, S.; Uriu, K.; Chen, L.; Kosugi, Y.; Uwamino, Y.; Begum, M.M.; Leong, S.; Ikeda, T.; et al. Virological Characteristics of the SARS-CoV-2 XEC Variant. Lancet Infect. Dis. 2024, 24, e736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Mellis, I.A.; Wu, M.; Mohri, H.; Gherasim, C.; Valdez, R.; Purpura, L.J.; Yin, M.T.; Gordon, A.; et al. Antibody Evasiveness of SARS-CoV-2 Subvariants KP.3.1.1 and XEC. Cell Rep. 2025, 44, 115543. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Huang, J.; Baboo, S.; Diedrich, J.K.; Bangaru, S.; Paulson, J.C.; Yates, J.R., III.; Yuan, M.; Wilson, I.A.; Ward, A.B. Structural and Functional Insights into the Evolution of SARS-CoV-2 KP.3.1.1 Spike Protein. Cell Rep. 2025, 44, 115941. [Google Scholar] [CrossRef] [PubMed]
- Branda, F.; Ciccozzi, M.; Scarpa, F. On the new SARS-CoV-2 variant KP.3.1.1: Focus on its genetic potential. Infect. Dis. 2024, 56, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, Y.; Yang, S.; Jian, F.; Song, W.; Yu, L.; Shao, F.; Cao, Y. Virological and Antigenic Characteristics of SARS-CoV-2 Variants LF.7.2.1, NP.1, and LP.8.1. Lancet Infect. Dis. 2025, 25, e128–e130. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yu, Y.; Liu, J.; Jian, F.; Yang, S.; Song, W.; Yu, L.; Shao, F.; Cao, Y. Antigenic and Virological Characteristics of SARS-CoV-2 Variants BA.3.2, XFG, and NB.1.8.1. Lancet Infect. Dis. 2025, 25, e374–e377. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 Humoral Immunity Induces Convergent Omicron RBD Evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jian, F.; Zhang, Z.; Yisimayi, A.; Hao, X.; Bao, L.; Yuan, F.; Yu, Y.; Du, S.; Wang, J.; et al. Rational Identification of Potent and Broad Sarbecovirus-Neutralizing Antibody Cocktails from SARS Convalescents. Cell Rep. 2022, 41, 111845. [Google Scholar] [CrossRef] [PubMed]
- Yisimayi, A.; Song, W.; Wang, J.; Jian, F.; Yu, Y.; Chen, X.; Xu, Y.; Yang, S.; Niu, X.; Xiao, T.; et al. Repeated Omicron Exposures Override Ancestral SARS-CoV-2 Immune Imprinting. Nature 2024, 625, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Jian, F.; Wec, A.Z.; Feng, L.; Yu, Y.; Wang, L.; Wang, P.; Yu, L.; Wang, J.; Hou, J.; Berrueta, D.M.; et al. Viral Evolution Prediction Identifies Broadly Neutralizing Antibodies to Existing and Prospective SARS-CoV-2 Variants. Nat. Microbiol. 2025, 95, 102560. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.E.; Tortorici, M.A.; De Marco, A.; Pinto, D.; Foreman, W.B.; Taylor, A.L.; Park, Y.-J.; Bohan, D.; Rietz, T.; Errico, J.M.; et al. A Potent Pan-Sarbecovirus Neutralizing Antibody Resilient to Epitope Diversification. Cell 2024, 187, 7196–7213.e26. [Google Scholar] [CrossRef] [PubMed]
- Sztain, T.; Ahn, S.H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls the opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.T.; Acharya, A.; Lynch, D.L.; Pavlova, A.; Gumbart, J.C. SARS-CoV-2 Spike Opening Dynamics and Energetics Reveal the Individual Roles of Glycans and Their Collective Impact. Commun. Biol. 2022, 5, 1170. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Jung, J.; Kobayashi, C.; Dokainish, H.M.; Re, S.; Sugita, Y. Elucidation of interactions regulating conformational stability and dynamics of SARS-CoV-2 S-protein. Biophys. J. 2021, 120, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Dokainish, H.M.; Re, S.; Mori, T.; Kobayashi, C.; Jung, J.; Sugita, Y. The inherent flexibility of receptor binding domains in SARS-CoV-2 spike protein. Elife 2022, 11, e75720. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.H.; Zinno, J.; Piano, F.; Gunsalus, K.C. Omicron Spike Protein Has a Positive Electrostatic Surface That Promotes ACE2 Recognition and Antibody Escape. Front. Virol. 2022, 2, 894531. [Google Scholar] [CrossRef]
- Xiao, S.; Alshahrani, M.; Gupta, G.; Tao, P.; Verkhivker, G. Markov State Models and Perturbation-Based Approaches Reveal Distinct Dynamic Signatures and Hidden Allosteric Pockets in the Emerging SARS-Cov-2 Spike Omicron Variant Complexes with the Host Receptor: The Interplay of Dynamics and Convergent Evolution Modulates Allostery and Functional Mechanisms. J. Chem. Inf. Model. 2023, 63, 5272–5296. [Google Scholar] [CrossRef] [PubMed]
- Raisinghani, N.; Alshahrani, M.; Gupta, G.; Xiao, S.; Tao, P.; Verkhivker, G. AlphaFold2 Predictions of Conformational Ensembles and Atomistic Simulations of the SARS-CoV-2 Spike XBB Lineages Reveal Epistatic Couplings between Convergent Mutational Hotspots That Control ACE2 Affinity. J. Phys. Chem. B. 2024, 128, 4696–4715. [Google Scholar] [CrossRef] [PubMed]
- Alshahrani, M.; Parikh, V.; Foley, B.; Verkhivker, G. Integrative Computational Modeling of Distinct Binding Mechanisms for Broadly Neutralizing Antibodies Targeting SARS-CoV-2 Spike Omicron Variants: Balance of Evolutionary and Dynamic Adaptability in Shaping Molecular Determinants of Immune Escape. Viruses 2025, 17, 741. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Guo, H.; Wang, A.; Cao, L.; Fan, Q.; Jiang, J.; Wang, M.; Lin, L.; Ge, X.; Wang, H.; et al. Structural Basis for the Evolution and Antibody Evasion of SARS-CoV-2 BA.2.86 and JN.1 Subvariants. Nat Commun. 2024, 15, 7715. [Google Scholar] [CrossRef] [PubMed]
- Yajima, H.; Nomai, T.; Okumura, K.; Maenaka, K.; Ito, J.; Hashiguchi, T.; Sato, K.; Matsuno, K.; Nao, N.; Sawa, H.; et al. Molecular and Structural Insights into SARS-CoV-2 Evolution: From BA.2 to XBB Subvariants. mBio 2024, 15, e0322023. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Kolinski, A. Characterization of protein-folding pathways by reduced-space modeling. Proc. Natl. Acad. Sci. USA 2007, 104, 12330–12335. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Kouza, M.; Badaczewska-Dawid, A.E.; Kloczkowski, A.; Kolinski, A. Modeling of protein structural flexibility and large-scale dynamics: Coarse-grained simulations and elastic network models. Int. J. Mol. Sci. 2018, 19, 3496. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.P.; Badaczewska-Dawid, A.E.; Pikuzinska, M.; Kolinski, A.; Kmiecik, S. Modeling of disordered protein structures using monte carlo simulations and knowledge-based statistical force fields. Int. J. Mol. Sci. 2019, 20, 606. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Oleniecki, T.; Ciemny, M.P.; Kuriata, A.; Kolinski, A.; Kmiecik, S. CABS-flex standalone: A simulation environment for fast modeling of protein flexibility. Bioinformatics 2019, 35, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Badaczewska-Dawid, A.E.; Kolinski, A.; Kmiecik, S. Protocols for fast simulations of protein structure flexibility using CABS-Flex and SURPASS. Methods Mol. Biol. 2020, 2165, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Rotkiewicz, P.; Skolnick, J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008, 29, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.E.; Marti, M.A.; Capece, L. CG2AA: Backmapping protein coarse-grained structures. Bioinformatics 2016, 32, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Gilis, D.; Rooman, M. A new generation of statistical potentials for proteins. Biophys. J. 2006, 90, 4010–4017. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks:PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate−DNA Helices. J. Amer. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A Web Server to Predict and Analyze the Protein–Protein Complex Based on Computational Docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef] [PubMed]
- Mongan, J.; Simmerling, C.; McCammon, J.A.; Case, D.A.; Onufriev, A. Generalized Born Model with a Simple, Robust Molecular Volume Correction. J. Chem. Theory Comput. 2007, 3, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Zhan, C.-G. Generalized Methodology for the Quick Prediction of Variant SARS-CoV-2 Spike Protein Binding Affinities with Human Angiotensin-Converting Enzyme II. J. Phys. Chem. B 2022, 126, 2353–2360. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 7. Entropy Effects on the Performance of End-Point Binding Free Energy Calculation Approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III.; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A. PRODIGY: A Contact-Based Predictor of Binding Affinity in Protein-Protein Complexes. Bio Protoc. 2017, 7, e2124. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Cao, Y.; Zhu, Q.; Yu, P.; Qi, F.; Wang, G.; Du, X.; Bao, L.; Deng, W.; Zhu, H.; et al. Structurally Resolved SARS-CoV-2 Antibody Shows High Efficacy in Severely Infected Hamsters and Provides a Potent Cocktail Pairing Strategy. Cell 2020, 183, 1013–1023.e13. [Google Scholar] [CrossRef] [PubMed]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.E.; Dijokaite-Guraliuc, A.; et al. Potent Cross-Reactive Antibodies Following Omicron Breakthrough in Vaccinees. Cell 2022, 185, 2116–2131.e18. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Das, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Mentzer, A.J.; Supasa, P.; Selvaraj, M.; Duyvesteyn, H.M.E.; Ritter, T.G.; Temperton, N.; et al. Emerging Variants Develop Total Escape from Potent Monoclonal Antibodies Induced by BA.4/5 Infection. Nat. Commun. 2024, 15, 3284. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Zhang, Q.; Wang, Z.; Aw, Z.Q.; Chen, P.; Zhou, B.; Wang, R.; Ge, X.; Lv, Q.; Cheng, L.; et al. Infection with Wild-Type SARS-CoV-2 Elicits Broadly Neutralizing and Protective Antibodies against Omicron Subvariants. Nat. Immunol. 2023, 24, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Bloom, J.D. An Antibody-Escape Estimator for Mutations to the SARS-CoV-2 Receptor-Binding Domain. Virus Evol. 2022, 8, veac021. [Google Scholar] [CrossRef] [PubMed]
- Dadonaite, B.; Crawford, K.H.D.; Radford, C.E.; Farrell, A.G.; Yu, T.C.; Hannon, W.W.; Zhou, P.; Andrabi, R.; Burton, D.R.; Liu, L.; et al. A Pseudovirus System Enables Deep Mutational Scanning of the Full SARS-CoV-2 Spike. Cell 2023, 186, 1263–1278.e20. [Google Scholar] [CrossRef] [PubMed]
- Dadonaite, B.; Brown, J.; McMahon, T.E.; Farrell, A.G.; Figgins, M.D.; Asarnow, D.; Stewart, C.; Lee, J.; Logue, J.; Bedford, T.; et al. Spike Deep Mutational Scanning Helps Predict Success of SARS-CoV-2 Clades. Nature 2024, 631, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Jian, F.; Feng, L.; Yang, S.; Yu, Y.; Wang, L.; Song, W.; Yisimayi, A.; Chen, X.; Xu, Y.; Wang, P.; et al. Convergent Evolution of SARS-CoV-2 XBB Lineages on Receptor-Binding Domain 455–456 Synergistically Enhances Antibody Evasion and ACE2 Binding. PLoS Pathog. 2023, 19, e1011868. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Schwanz, L.T.; Mellis, I.A.; Sun, Y.; Qu, Y.; Urtecho, G.; Valdez, R.; Stoneman, E.; Gordon, A.; et al. SARS-CoV-2 omicron BA.2.87.1 exhibits higher susceptibility to serum neutralization than EG.5.1 and JN.1. Emerg. Microbes Infect. 2024, 13, 2359004. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, S.; Yu, Y.; Jian, F.; Yisimayi, A.; Song, W.; Liu, J.; Wang, P.; Xu, Y.; Wang, J.; Niu, X.; et al. Antigenicity Assessment of SARS-CoV-2 Saltation Variant BA.2.87.1. Emerg. Microbes Infect. 2024, 13, 2343909. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshahrani, M.; Parikh, V.; Foley, B.; Verkhivker, G. Multiscale Modeling and Dynamic Mutational Profiling of Binding Energetics and Immune Escape for Class I Antibodies with SARS-CoV-2 Spike Protein: Dissecting Mechanisms of High Resistance to Viral Escape Against Emerging Variants. Viruses 2025, 17, 1029. https://doi.org/10.3390/v17081029
Alshahrani M, Parikh V, Foley B, Verkhivker G. Multiscale Modeling and Dynamic Mutational Profiling of Binding Energetics and Immune Escape for Class I Antibodies with SARS-CoV-2 Spike Protein: Dissecting Mechanisms of High Resistance to Viral Escape Against Emerging Variants. Viruses. 2025; 17(8):1029. https://doi.org/10.3390/v17081029
Chicago/Turabian StyleAlshahrani, Mohammed, Vedant Parikh, Brandon Foley, and Gennady Verkhivker. 2025. "Multiscale Modeling and Dynamic Mutational Profiling of Binding Energetics and Immune Escape for Class I Antibodies with SARS-CoV-2 Spike Protein: Dissecting Mechanisms of High Resistance to Viral Escape Against Emerging Variants" Viruses 17, no. 8: 1029. https://doi.org/10.3390/v17081029
APA StyleAlshahrani, M., Parikh, V., Foley, B., & Verkhivker, G. (2025). Multiscale Modeling and Dynamic Mutational Profiling of Binding Energetics and Immune Escape for Class I Antibodies with SARS-CoV-2 Spike Protein: Dissecting Mechanisms of High Resistance to Viral Escape Against Emerging Variants. Viruses, 17(8), 1029. https://doi.org/10.3390/v17081029