
Complete Genome and Recombination Analysis of a Novel Porcine Reproductive and Respiratory Syndrome Virus 2 (Variant 1H.18) Identified in the Midwestern USA

 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequencing and Genome Assemble
2.2. Sequence Comparison and Classification
2.3. Datasets and Pre-Processing
2.4. Recombination Analysis
2.5. Phylogenetic Analisys
3. Results
3.1. Sequence Comparisons Reveal a Close Relationship Between PRRSV-2 Variant 1H.18 and Variants 1H.31 and 1C.3
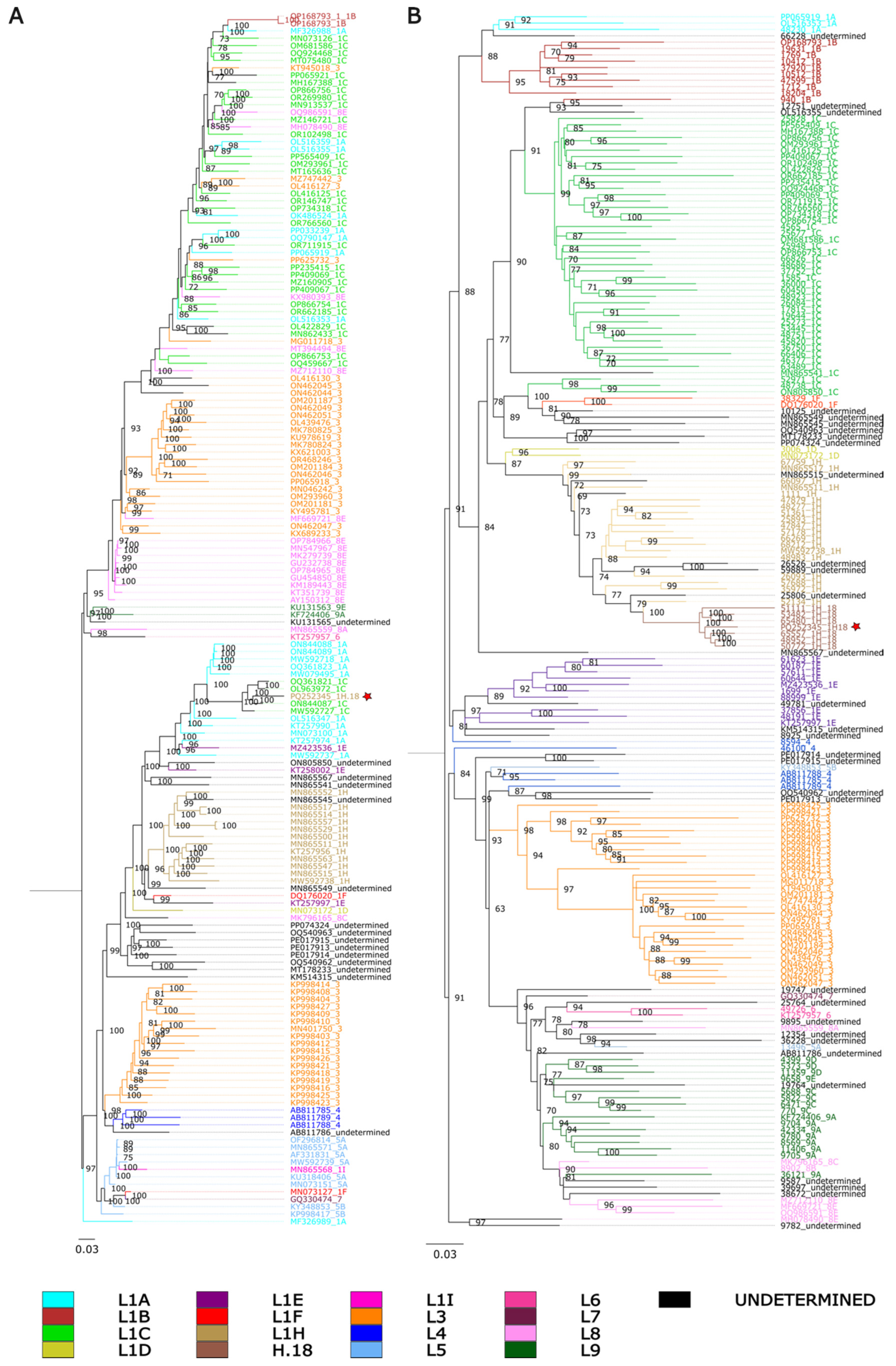
3.2. Genealogical Discordances Between Whole-Genome and ORF5 Phylogenies Suggest Evidence of Recombination in the PRRSV-2 Variant 1H.18
3.3. Evidence of Recombination in Both Whole-Genome and ORF5 Sequences of PRRSV-2 Variant 1H.18 Suggests an Evolutionary Origin Involving Genetic Exchange
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruedas-Torres, I.; Sánchez-Carvajal, J.M.; Salguero, F.J.; Pallarés, F.J.; Carrasco, L.; Mateu, E.; Gómez-Laguna, J.; Rodríguez-Gómez, I.M. The scene of lung pathology during PRRSV-1 infection. Front. Vet. Sci. 2024, 11, 1330990. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, D.; Kliebenstein, J.; Neumann, E.; Zimmerman, J.; Rotto, H.; Yoder, T.; Wang, C.; Yeske, P.; Mowrer, C.; Haley, C. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J. Swine Health Prod. 2013, 21, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Roepke, D. Growing Losses from PRRS Cost Pork Producers $1.2 Billion Per Year, New Study Shows. Iowa State University News Service. 2024. Available online: https://research.iastate.edu/2024/07/30/growing-losses-from-prrs-cost-pork-producers-1-2-billion-per-year-new-study-shows/ (accessed on 15 April 2025).
- Brinton, M.A.; Gulyaeva, A.A.; Balasuriya, U.B.R.; Dunowska, M.; Faaberg, K.S.; Goldberg, T.; Leung, F.C.C.; Nauwynck, H.J.; Snijder, E.J.; Stadejek, T.; et al. ICTV Virus Taxonomy Profile: Arteriviridae 2021. J. Gen. Virol. 2021, 102, 001632. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lam, T.T.-Y.; Hon, C.-C.; Hui, R.K.-H.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Leung, F.C.-C. Molecular epidemiology of PRRSV: A phylogenetic perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef]
- VanderWaal, K.; Pamornchainavakul, N.; Kikuti, M.; Zhang, J.; Zeller, M.; Trevisan, G.; Rossow, S.; Schwartz, M.; Linhares, D.C.L.; Holtkamp, D.J.; et al. PRRSV-2 variant classification: A dynamic nomenclature for enhanced monitoring and surveillance. mSphere 2025, 10, e0070924. [Google Scholar] [CrossRef]
- VanderWaal, K.; Pamornchainavakul, N.; Kikuti, M.; Linhares, D.C.L.; Trevisan, G.; Zhang, J.; Anderson, T.K.; Zeller, M.; Rossow, S.; Holtkamp, D.J.; et al. Phylogenetic-based methods for fine-scale classification of PRRSV-2 ORF5 sequences: A comparison of their robustness and reproducibility. Front. Virol. 2024, 4, 1433931. [Google Scholar] [CrossRef]
- Yim-Im, W.; Anderson, T.K.; Paploski, I.A.D.; VanderWaal, K.; Gauger, P.; Krueger, K.; Shi, M.; Main, R.; Zhang, J.; Chao, D.-Y. Refining PRRSV-2 genetic classification based on global ORF5 sequences and investigation of their geographic distributions and temporal changes. Microbiol. Spectr. 2023, 11, e0291623. [Google Scholar] [CrossRef]
- Paploski, I.A.D.; Corzo, C.; Rovira, A.; Murtaugh, M.P.; Sanhueza, J.M.; Vilalta, C.; Schroeder, D.C.; VanderWaal, K. Temporal Dynamics of Co-circulating Lineages of Porcine Reproductive and Respiratory Syndrome Virus. Front. Microbiol. 2019, 10, 2486. [Google Scholar] [CrossRef]
- Shi, M.; Lam, T.T.; Hon, C.C.; Murtaugh, M.P.; Davies, P.R.; Hui, R.K.; Li, J.; Wong, L.T.; Yip, C.W.; Jiang, J.W.; et al. Phylogeny-Based Evolutionary, Demographical, and Geographical Dissection of North American Type 2 Porcine Reproductive and Respiratory Syndrome Viruses. J. Virol. 2010, 84, 8700–8711. [Google Scholar] [CrossRef]
- Hanada, K.; Suzuki, Y.; Nakane, T.; Hirose, O.; Gojobori, T. The Origin and Evolution of Porcine Reproductive and Respiratory Syndrome Viruses. Mol. Biol. Evol. 2005, 22, 1024–1031. [Google Scholar] [CrossRef]
- Pamornchainavakul, N.; Kikuti, M.; Paploski, I.A.D.; Makau, D.N.; Rovira, A.; Corzo, C.A.; VanderWaal, K. Measuring How Recombination Re-shapes the Evolutionary History of PRRSV-2: A Genome-Based Phylodynamic Analysis of the Emergence of a Novel PRRSV-2 Variant. Front. Vet. Sci. 2022, 9, 846904. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.-Y.; Xia, D.-S.; Luo, L.-Z.; An, T.-Q. Recombination of Porcine Reproductive and Respiratory Syndrome Virus: Features, Possible Mechanisms, and Future Directions. Viruses 2024, 16, 929. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xia, D.; Huang, X.; Sun, Y.; Shi, M.; Zhang, J.; Li, G.; Yang, Y.; Wang, H.; Cai, X.; et al. Analysis of Recombinant Characteristics Based on 949 PRRSV-2 Genomic Sequences Obtained from 1991 to 2021 Shows That Viral Multiplication Ability Contributes to Dominant Recombination. Microbiol. Spectr. 2022, 10, e0293422. [Google Scholar] [CrossRef] [PubMed]
- Kikuti, M.; Paploski, I.; Pamornchainavakul, N.; Melini, M.; Yue, X.; Vadnais, S.; Baker, J.; da Silva, J.P.H.; Wagner, M.; Kurt, J.; et al. Monitoring the Detection of PRRSV Variant 1H.18. 2024. Available online: https://mshmp.umn.edu/sites/mnshmp.umn.edu/files/2024-05/SHMP%202023l24.43%20%5BMonitoring%20PRRS%20Variant%5D2.pdf (accessed on 15 April 2025).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 April 2025).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet—Next generation sequence assembly visualization. Bioinformatics 2009, 26, 401–402. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.M.; Linhares, D.C.L.; Arruda, A.G.; VanderWaal, K.; Machado, G.; Vilalta, C.; Sanhueza, J.M.; Torrison, J.; Torremorell, M.; Corzo, C.A. Individual or Common Good? Voluntary Data Sharing to Inform Disease Surveillance Systems in Food Animals. Front. Vet. Sci. 2019, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, Z.; Teng, K.; Lai, J.; Zhang, Y.; Huang, Y.; Li, Y.; Liang, L.; Wang, Y.; Chu, C.; et al. Up-regulation ofLSB1/GDU3affects geminivirus infection by activating the salicylic acid pathway. Plant J. 2009, 62, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible Emergence of New Geminiviruses by Frequent Recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef]
- Martin, D.; Posada, D.; Crandall, K.; Williamson, C. A Modified Bootscan Algorithm for Automated Identification of Recombinant Sequences and Recombination Breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Posada, D. Evaluation of Methods for Detecting Recombination from DNA Sequences: Empirical Data. Mol. Biol. Evol. 2002, 19, 708–717. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol. Biol. Evol. 2017, 35, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2020, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Boukadida, K.; Cachot, J.; Morin, B.; Clerandeau, C.; Banni, M. Moderate temperature elevation increases susceptibility of early-life stage of the Mediterranean mussel, Mytilus galloprovincialis, to metal-induced genotoxicity. Sci. Total Environ. 2019, 663, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Eclercy, J.; Renson, P.; Lebret, A.; Hirchaud, E.; Normand, V.; Andraud, M.; Paboeuf, F.; Blanchard, Y.; Rose, N.; Bourry, O. A Field Recombinant Strain Derived from Two Type 1 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV-1) Modified Live Vaccines Shows Increased Viremia and Transmission in SPF Pigs. Viruses 2019, 11, 296. [Google Scholar] [CrossRef]
- Wang, A.; Chen, Q.; Wang, L.; Madson, D.; Harmon, K.; Gauger, P.; Zhang, J.; Li, G. Recombination between Vaccine and Field Strains of Porcine Reproductive and Respiratory Syndrome Virus. Emerg. Infect. Dis. 2019, 25, 2335–2337. [Google Scholar] [CrossRef]
- Pamornchainavakul, N.; Paploski, I.A.D.; Makau, D.N.; Kikuti, M.; Rovira, A.; Lycett, S.; Corzo, C.A.; VanderWaal, K. Mapping the Dynamics of Contemporary PRRSV-2 Evolution and Its Emergence and Spreading Hotspots in the U.S. Using Phylogeography. Pathogens 2023, 12, 740. [Google Scholar] [CrossRef]

{kind=link}
| A | Breakpoint Positions in Whole Genome | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| In Alignment | In Recombinant Sequence | ||||||||
| Event | Begin | End | Begin | End | Recombinant Sequence(s) | Minor Parental Sequence(s) | Major Parental Sequence(s) | Detection Methods # | p-Value * |
| 1 | 164 | 476 | 164 | 473 | PQ252345 | MN865563 (1H) | Unknown | RBMCS | 9.97 × 10−11 |
| 2 | 569 | 3224 | 569 | 2740 | PQ252345 | Unknown | ON805850 (Undetermined) | RGBMCS3 | 4.33 × 10−25 |
| 3 | 4246 | 5834 | 3557 | 5140 | PQ252345 | OQ924468 (L1C) | MW592737 (L1A) | RGBMCS3 | 8.41 × 10−50 |
| 4 | 5835 | 6952 | 5141 | 6258 | PQ252345 | MW592739 (L1A) | MW592737 (L1A) | RGBMCS3 | 6.02 × 10−49 |
| 5 | 6967 | 11,841 | 6363 | 11,222 | PQ252345 | KT257974 (L1A) | Unknown | MCS3 | 8.82 × 10−34 |
| 6 | 12,198 | 15,387 | 11,576 | 14,695 | PQ252345 | MN865552 (L1H) | OP168793 (L1B) | RBCS3 | 4.05 × 10−12 |
| B | Breakpoint Positions in ORF-5 | ||||||||
| In Alignment | In Recombinant Sequence | ||||||||
| Event | Begin | End | Begin | End | Recombinant Sequence(s) | Minor Parental Sequence(s) | Major Parental Sequence(s) | Detection Methods # | p-Value * |
| 1 | 112 | 341 | 112 | 341 | PQ252345 | Shmp_62265 (1H.31) | Shmp65283 (1C.3) | RBMCS3 | 4.32 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera da Silva, J.P.; Rossow, S.; Paploski, I.A.D.; Kikuti, M.; Corzo, C.A.; VanderWaal, K. Complete Genome and Recombination Analysis of a Novel Porcine Reproductive and Respiratory Syndrome Virus 2 (Variant 1H.18) Identified in the Midwestern USA. Viruses 2025, 17, 863. https://doi.org/10.3390/v17060863
Herrera da Silva JP, Rossow S, Paploski IAD, Kikuti M, Corzo CA, VanderWaal K. Complete Genome and Recombination Analysis of a Novel Porcine Reproductive and Respiratory Syndrome Virus 2 (Variant 1H.18) Identified in the Midwestern USA. Viruses. 2025; 17(6):863. https://doi.org/10.3390/v17060863
Chicago/Turabian StyleHerrera da Silva, Joao P., Stephanie Rossow, Igor A. D. Paploski, Mariana Kikuti, Cesar A. Corzo, and Kimberly VanderWaal. 2025. "Complete Genome and Recombination Analysis of a Novel Porcine Reproductive and Respiratory Syndrome Virus 2 (Variant 1H.18) Identified in the Midwestern USA" Viruses 17, no. 6: 863. https://doi.org/10.3390/v17060863
APA StyleHerrera da Silva, J. P., Rossow, S., Paploski, I. A. D., Kikuti, M., Corzo, C. A., & VanderWaal, K. (2025). Complete Genome and Recombination Analysis of a Novel Porcine Reproductive and Respiratory Syndrome Virus 2 (Variant 1H.18) Identified in the Midwestern USA. Viruses, 17(6), 863. https://doi.org/10.3390/v17060863