
Intersegment Recombination During Influenza A Virus Replication Gives Rise to a Novel Class of Defective Viral Genomes

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Propagation of Influenza A PR8
2.2. Egg Infectious Dose 50% Endpoint Assay (EID50)
2.3. RNA Extraction
2.4. Illumina Sequencing and Analysis
2.5. Virus Concentration and Sucrose Gradient Purification
2.6. cDNA Synthesis
2.7. RT-PCR of Multisegment DVGs
2.8. DNA Cloning
2.9. Sanger Sequencing
3. Results
3.1. DVG Recombination Events Detected by ViReMa
3.2. Frequency of Recombination Events Across Different Segments of Influenza A PR8
3.3. Multisegment DVGs Are Encapsidated Within Viral Particles
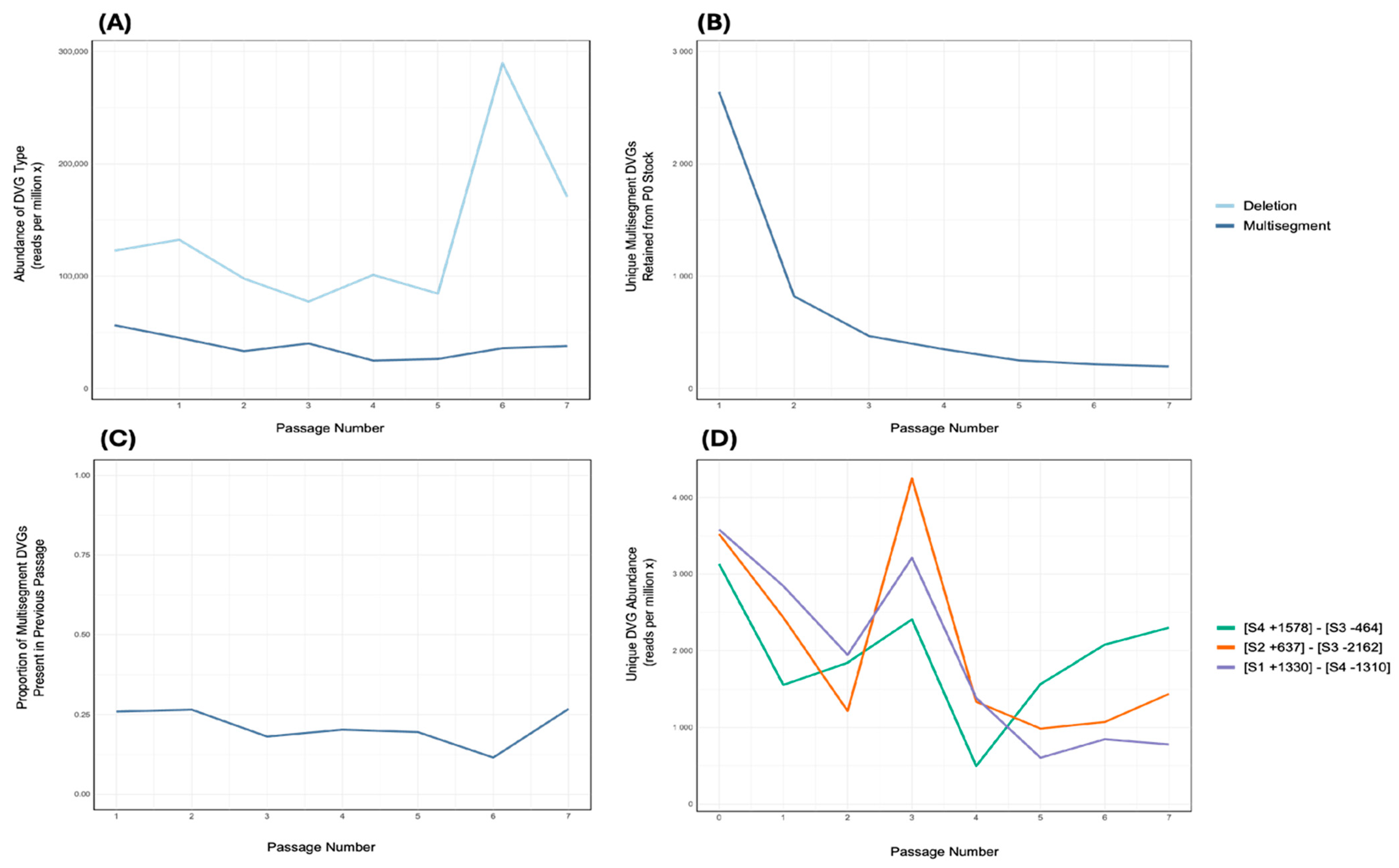
3.4. Persistence of Multisegment DVGs During Serial Passaging in Embryonated Chicken Eggs
3.5. Dominance of a Unique Multisegment DVG and Enrichment During Passaging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IAV | Influenza A Virus |
| DVG | Defective Viral Genome |
| DIP | Defective Interfering Particle |
| HA | Haemagglutinin |
| NA | Neuraminidase |
| NP | Nucleoprotein |
| PB1 | Polymerase Basic 1 |
| PB2 | Polymerase Basic 2 |
| PA | Polymerase Acidic |
| NS | Non-Structural Segment |
| M | Matrix |
| IFN | Interferon |
| RIG-I | Retinoic Acid-Inducible Gene I |
| MDA5 | Melanoma Differentiation-Associated Protein 5 |
| NGS | Next-Generation Sequencing |
| RT-PCR | Reverse Transcription Polymerase Chain Reaction |
| cDNA | Complementary DNA |
| EID50 | Egg Infectious Dose 50% |
| NCBI | National Center for Biotechnology Information |
| Nt | Nucleotide |
| SD | Standard Deviation |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Primer Sequence (5′ to 3′) |
|---|---|
| Segment 1 Forward | GATCATGGTACCCGCAGTCTCGCACCCGCGA |
| Segment 1 Reverse | ATGATCGCGGCCGCGCTGTCTGGCTGTCAGTAAG |
| Segment 2 Forward | GATCATGGTACCGCTATAAGCACAACTTTCCCTTATACC |
| Segment 2 Reverse | ATGATCGCGGCCGCCTAAATTCACTATTTTTGCCGTCTGAG |
| Segment 3 Forward | GATCATGGTACCCCGATGATTGTCGAGCTTGCG |
| Segment 3 Reverse | ATGATCGCGGCCGCGGACAGTATGGATAGCAAATAG |
| Segment 4 Forward | GAGTTCTACCACAAGTGTGACAATG |
| Segment Comparison | p-Value |
|---|---|
| 2-1 | 0.425 |
| 3-1 | 0.714 |
| 4-1 | 0.074 |
| 5-1 | 0.009 (**) |
| 6-1 | 0.003 (**) |
| 7-1 | <0.001 (***) |
| 8-1 | <0.001 (***) |
| 3-2 | 1.000 |
| 4-2 | 0.001 (**) |
| 5-2 | <0.001 (***) |
| 6-2 | <0.001 (***) |
| 7-2 | <0.001 (***) |
| 8-2 | <0.001 (***) |
| 4-3 | 0.003 (**) |
| 5-3 | <0.001 (***) |
| 6-3 | <0.001 (***) |
| 7-3 | <0.001 (***) |
| 8-3 | <0.001 (***) |
| 5-4 | 0.951 |
| 6-4 | 0.724 |
| 7-4 | 0.163 |
| 8-4 | 0.055 |
| 6-5 | 0.999 |
| 7-5 | 0.678 |
| 8-5 | 0.338 |
| 7-6 | 0.931 |
| 8-6 | 0.645 |
| 8-7 | 0.988 |
| Segments of Origin | Predicted Full Length of DVG (nt) | Sanger Sequencing File ID | Sample No. |
|---|---|---|---|
| 1 and 2 | 417 | EIN537/EIN536 | 1 |
| 3 and 2 | 477 | EIN541/EIN540 | |
| 2 and 3 | 602 | EIN539/EIN544 | |
| 3 and 1 | 306 | EIN547/EIN546 | |
| 1 and 2 | 417 | HAH679/HAH678 | 2 |
| 3 and 2 | 684 | HAH695/HAH694 | |
| 3 and 1 | 466 | HAH697/HAH696 | |
| 1 and 2 | 566 | HAH701/HAH700 | |
| 3 and 2 | 910 | HQZ427 | 3 |
| 4 and 2 | 1934 | HNS027/HNS026 | 4 |
References
- Uyeki, T.M.; Hui, D.S.; Zambon, M.; Wentworth, D.E.; Monto, A.S. Influenza. Lancet 2022, 400, 693–706. [Google Scholar] [CrossRef] [PubMed]
- WHO Influenza (Seasonal). Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 5 May 2025).
- Tanner, A.R.; Dorey, R.B.; Brendish, N.J.; Clark, T.W. Influenza Vaccination: Protecting the Most Vulnerable. Eur. Respir. Rev. 2021, 30, 200258. [Google Scholar] [CrossRef] [PubMed]
- Memoli, M.J.; Athota, R.; Reed, S.; Czajkowski, L.; Bristol, T.; Proudfoot, K.; Hagey, R.; Voell, J.; Fiorentino, C.; Ademposi, A.; et al. The Natural History of Influenza Infection in the Severely Immunocompromised vs. Nonimmunocompromised Hosts. Clin. Infect. Dis. 2014, 58, 214–224. [Google Scholar] [CrossRef]
- Chan, L.; Alizadeh, K.; Alizadeh, K.; Fazel, F.; Kakish, J.E.; Karimi, N.; Knapp, J.P.; Mehrani, Y.; Minott, J.A.; Morovati, S.; et al. Review of Influenza Virus Vaccines: The Qualitative Nature of Immune Responses to Infection and Vaccination Is a Critical Consideration. Vaccines 2021, 9, 979. [Google Scholar] [CrossRef]
- Lampejo, T. Influenza and Antiviral Resistance: An Overview. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1201–1208. [Google Scholar] [CrossRef]
- Vignuzzi, M.; López, C.B. Defective Viral Genomes Are Key Drivers of the Virus–Host Interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef]
- von Magnus, P. Incomplete Forms of Influenza Virus. In Advances in Virus Research; Smith, K.M., Lauffer, M.A., Eds.; Academic Press: Cambridge, MA, USA, 1954; Volume 2, pp. 59–79. [Google Scholar]
- Huang, A.S.; Baltimore, D. Defective Viral Particles and Viral Disease Processes. Nature 1970, 226, 325–327. [Google Scholar] [CrossRef]
- Alnaji, F.G.; Reiser, W.K.; Rivera-Cardona, J.; Te Velthuis, A.J.W.; Brooke, C.B. Influenza A Virus Defective Viral Genomes Are Inefficiently Packaged into Virions Relative to Wild-Type Genomic RNAs. mBio 2021, 12, e0295921. [Google Scholar] [CrossRef] [PubMed]
- Vasilijevic, J.; Zamarreño, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gómez, G.; Rodriguez, G.; Pérez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F.; et al. Reduced Accumulation of Defective Viral Genomes Contributes to Severe Outcome in Influenza Virus Infected Patients. PLoS Pathog. 2017, 13, e1006650. [Google Scholar] [CrossRef]
- Dimmock, N.J.; Dove, B.K.; Meng, B.; Scott, P.D.; Taylor, I.; Cheung, L.; Hallis, B.; Marriott, A.C.; Carroll, M.W.; Easton, A.J. Comparison of the Protection of Ferrets against Pandemic 2009 Influenza A Virus (H1N1) by 244 DI Influenza Virus and Oseltamivir. Antivir. Res. 2012, 96, 376–385. [Google Scholar] [CrossRef]
- Manzoni, T.B.; López, C.B. Defective (Interfering) Viral Genomes Re-Explored: Impact on Antiviral Immunity and Virus Persistence. Future Virol. 2018, 13, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Garushyants, S.K.; Rogozin, I.B.; Koonin, E.V. Template Switching and Duplications in SARS-CoV-2 Genomes Give Rise to Insertion Variants That Merit Monitoring. Commun. Biol. 2021, 4, 1343. [Google Scholar] [CrossRef] [PubMed]
- Alnaji, F.G.; Holmes, J.R.; Rendon, G.; Vera, J.C.; Fields, C.J.; Martin, B.E.; Brooke, C.B. Sequencing Framework for the Sensitive Detection and Precise Mapping of Defective Interfering Particle-Associated Deletions across Influenza A and B Viruses. J. Virol. 2019, 93, e00354-19. [Google Scholar] [CrossRef]
- Jaworski, E.; Routh, A. Parallel ClickSeq and Nanopore Sequencing Elucidates the Rapid Evolution of Defective-Interfering RNAs in Flock House Virus. PLoS Pathog. 2017, 13, e1006365. [Google Scholar] [CrossRef]
- Genoyer, E.; López, C.B. The Impact of Defective Viruses on Infection and Immunity. Annu. Rev. Virol. 2019, 6, 547–566. [Google Scholar] [CrossRef]
- Xu, J.; Mercado-López, X.; Grier, J.T.; Kim, W.; Chun, L.F.; Irvine, E.B.; Del Toro Duany, Y.; Kell, A.; Hur, S.; Gale, M.; et al. Identification of a Natural Viral RNA Motif That Optimizes Sensing of Viral RNA by RIG-I. mBio 2015, 6, e01265-15. [Google Scholar] [CrossRef]
- Killip, M.J.; Young, D.F.; Gatherer, D.; Ross, C.S.; Short, J.A.L.; Davison, A.J.; Goodbourn, S.; Randall, R.E. Deep Sequencing Analysis of Defective Genomes of Parainfluenza Virus 5 and Their Role in Interferon Induction. J. Virol. 2013, 87, 4798–4807. [Google Scholar] [CrossRef] [PubMed]
- Gould, P.S.; Easton, A.J.; Dimmock, N.J. Live Attenuated Influenza Vaccine Contains Substantial and Unexpected Amounts of Defective Viral Genomic RNA. Viruses 2017, 9, 269. [Google Scholar] [CrossRef]
- Bdeir, N.; Arora, P.; Gärtner, S.; Hoffmann, M.; Reichl, U.; Pöhlmann, S.; Winkler, M. A System for Production of Defective Interfering Particles in the Absence of Infectious Influenza A Virus. PLoS ONE 2019, 14, e0212757. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA Sequencing at 40: Past, Present and Future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Zhou, T.; Gilliam, N.J.; Li, S.; Spandau, S.; Osborn, R.M.; Connor, S.; Anderson, C.S.; Mariani, T.J.; Thakar, J.; Dewhurst, S.; et al. Generation and Functional Analysis of Defective Viral Genomes during SARS-CoV-2 Infection. mBio 2023, 14, e0025023. [Google Scholar] [CrossRef] [PubMed]
- Routh, A.; Johnson, J.E. Discovery of Functional Genomic Motifs in Viruses with ViReMa–a Virus Recombination Mapper–for Analysis of next-Generation Sequencing Data. Nucleic Acids Res. 2014, 42, e11. [Google Scholar] [CrossRef]
- Beauclair, G.; Mura, M.; Combredet, C.; Tangy, F.; Jouvenet, N.; Komarova, A.V. DI-Tector: Defective Interfering Viral Genomes’ Detector for next-Generation Sequencing Data. RNA 2018, 24, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Bosma, T.J.; Karagiannis, K.; Santana-Quintero, L.; Ilyushina, N.; Zagorodnyaya, T.; Petrovskaya, S.; Laassri, M.; Donnelly, R.P.; Rubin, S.; Simonyan, V.; et al. Identification and Quantification of Defective Virus Genomes in High Throughput Sequencing Data Using DVG-Profiler, a Novel Post-Sequence Alignment Processing Algorithm. PLoS ONE 2019, 14, e0216944. [Google Scholar] [CrossRef]
- Boussier, J.; Munier, S.; Achouri, E.; Meyer, B.; Crescenzo-Chaigne, B.; Behillil, S.; Enouf, V.; Vignuzzi, M.; van der Werf, S.; Naffakh, N. RNA-Seq Accuracy and Reproducibility for the Mapping and Quantification of Influenza Defective Viral Genomes. RNA 2020, 26, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Pelz, L.; Rüdiger, D.; Dogra, T.; Alnaji, F.G.; Genzel, Y.; Brooke, C.B.; Kupke, S.Y.; Reichl, U. Semi-Continuous Propagation of Influenza A Virus and Its Defective Interfering Particles: Analyzing the Dynamic Competition to Select Candidates for Antiviral Therapy. J. Virol. 2021, 95, e0117421. [Google Scholar] [CrossRef]
- Duhaut, S.D.; Dimmock, N.J. Heterologous Protection of Mice from a Lethal Human H1N1 Influenza A Virus Infection by H3N8 Equine Defective Interfering Virus: Comparison of Defective RNA Sequences Isolated from the DI Inoculum and Mouse Lung. Virology 1998, 248, 241–253. [Google Scholar] [CrossRef]
- Klimov, A.; Balish, A.; Veguilla, V.; Sun, H.; Schiffer, J.; Lu, X.; Katz, J.M.; Hancock, K. Influenza Virus Titration, Antigenic Characterization, and Serological Methods for Antibody Detection. In Influenza Virus: Methods and Protocols; Kawaoka, Y., Neumann, G., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 25–51. ISBN 978-1-61779-621-0. [Google Scholar]
- Reed, L.J.; Muench, H. A Simple Method of Estimating Fifty Per Cent Endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Penn, R.; Tregoning, J.S.; Flight, K.E.; Baillon, L.; Frise, R.; Goldhill, D.H.; Johansson, C.; Barclay, W.S. Levels of Influenza A Virus Defective Viral Genomes Determine Pathogenesis in the BALB/c Mouse Model. J. Virol. 2022, 96, e01178-22. [Google Scholar] [CrossRef]
- Fujii, Y.; Goto, H.; Watanabe, T.; Yoshida, T.; Kawaoka, Y. Selective Incorporation of Influenza Virus RNA Segments into Virions. Proc. Natl. Acad. Sci. USA 2003, 100, 2002–2007. [Google Scholar] [CrossRef]
- Liang, Y.; Hong, Y.; Parslow, T.G. Cis-Acting Packaging Signals in the Influenza Virus PB1, PB2, and PA Genomic RNA Segments. J. Virol. 2005, 79, 10348–10355. [Google Scholar] [CrossRef] [PubMed]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The Structure of the Influenza A Virus Genome. Nat. Microbiol. 2019, 4, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Tapia, K.; Kim, W.; Sun, Y.; Mercado-López, X.; Dunay, E.; Wise, M.; Adu, M.; López, C.B. Defective Viral Genomes Arising In Vivo Provide Critical Danger Signals for the Triggering of Lung Antiviral Immunity. PLoS Pathog. 2013, 9, e1003703. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Forst, C.V.; Chou, T.; Geber, A.; Wang, M.; Hamou, W.; Smith, M.; Sebra, R.; Zhang, B.; Zhou, B.; et al. Cell-to-Cell Variation in Defective Virus Expression and Effects on Host Responses During Influenza Virus Infection. mBio 2020, 11, e02880-19. [Google Scholar] [CrossRef]
- Thompson, K.A.S.; Yin, J. Population dynamics of an RNA virus and its defective interfering particles in passage cultures. Virol. J. 2010, 7, 257. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The Coronavirus Proofreading Exoribonuclease Mediates Extensive Viral Recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Alnaji, F.G.; Brooke, C.B. Influenza Virus DI Particles: Defective Interfering or Delightfully Interesting? PLoS Pathog. 2020, 16, e1008436. [Google Scholar] [CrossRef]
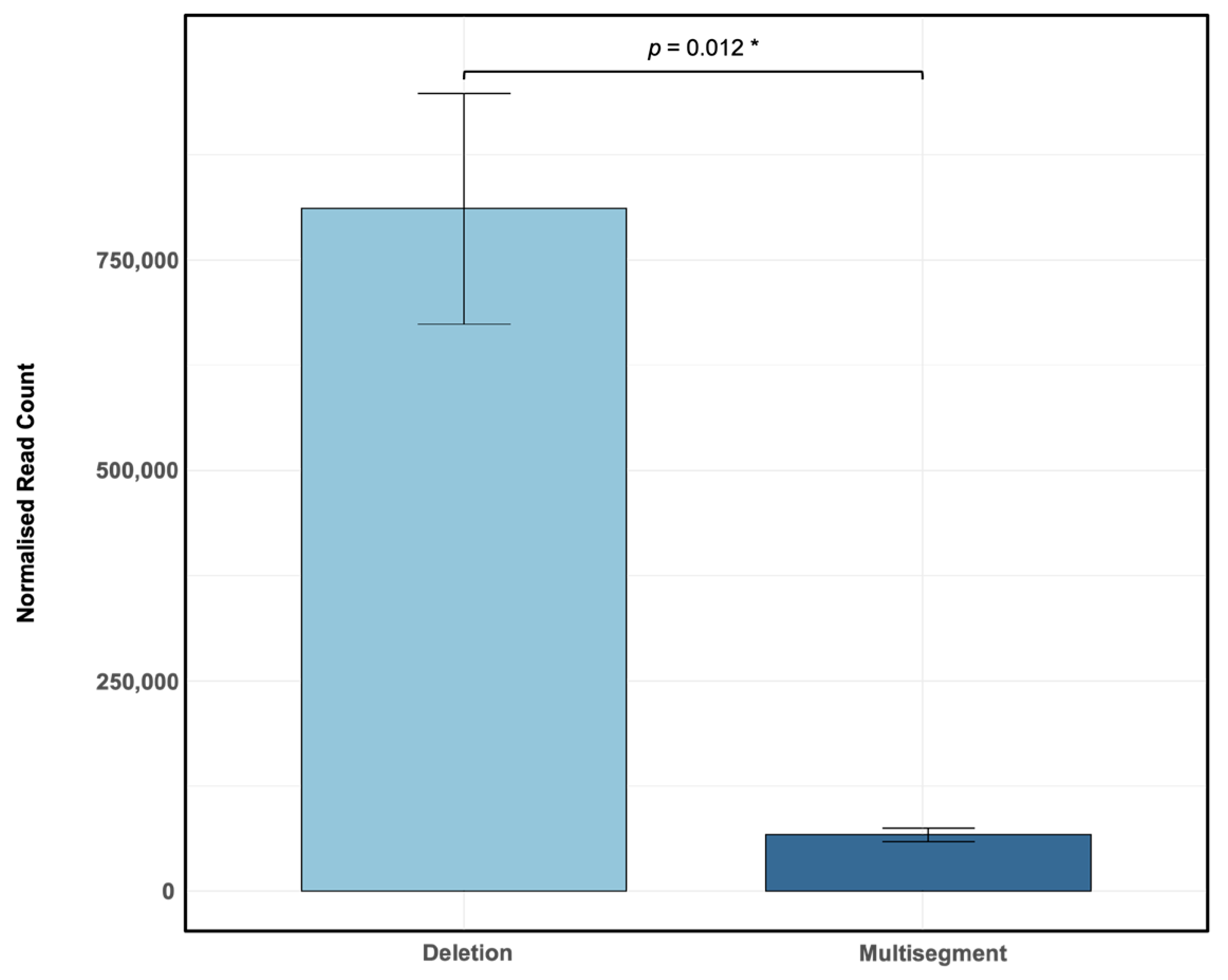

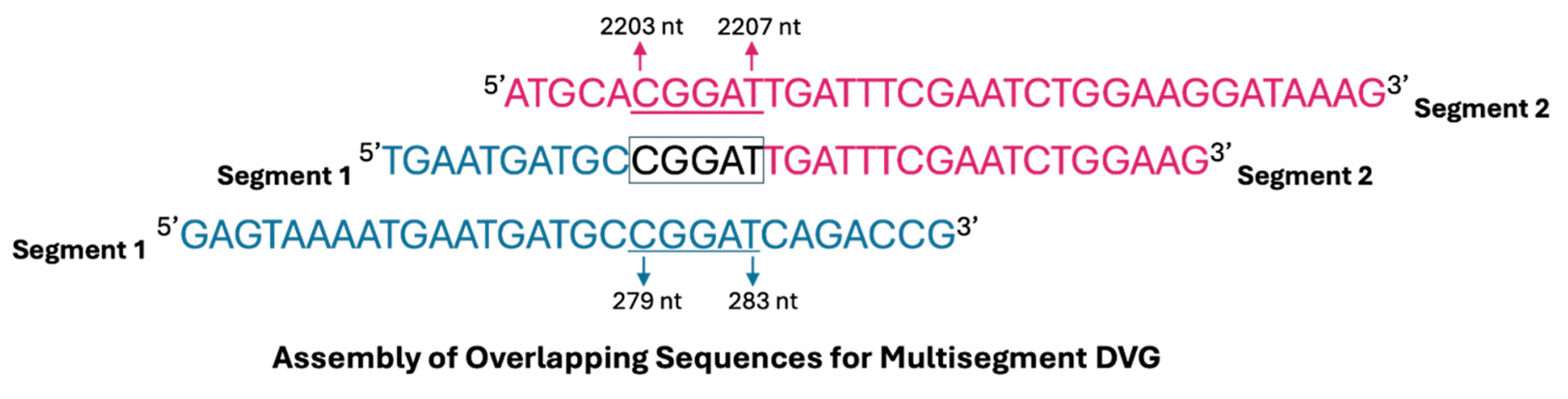

| Segments of Origin | Range of 5′ Breakpoint (nts) | Range of 3′ Breakpoint (nts) | Full Length of DVG (nt) | Sample |
|---|---|---|---|---|
| 1 and 2 | 279–283 | 2203–2207 | 417 | 1 |
| 3 and 2 | 101–108 | 1965–1972 | 477 | |
| 2 and 3 | 111–113 | 1742–1744 | 602 | |
| 3 and 1 | 166–167 | 2201–2202 | 306 | |
| 1 and 2 | 279–283 | 2203–2207 | 417 | 2 |
| 3 and 2 | 161 | 1819 | 684 | |
| 3 and 1 | 97–100 | 1976–1979 | 466 | |
| 1 and 2 | 483–488 | 2258–2263 | 566 | |
| 3 and 2 | 690–699 | 2121–2130 | 910 | 3 |
| Segment of Origin (5′) | Segment of Origin (3′) | 5′ Breakpoint | 3′ Breakpoint | Type of DVG | Normalised Reads |
|---|---|---|---|---|---|
| 4 | 2 | 1700 | 2107 | Multisegment | 54,695.19 |
| 3 | 3 | 137 | 2074 | Deletion | 18,372.05 |
| 4 | 4 | 97 | 1636 | Deletion | 15,295.15 |
| 3 | 3 | 470 | 474 | Deletion | 14,100.86 |
| 2 | 4 | 158 | 1972 | Multisegment | 12,592.70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anisi, S.; Noble, G.; Williams, R.; Hales, J.; Bridgewater, H.E.; Easton, A.; Collier, W.; Gould, P. Intersegment Recombination During Influenza A Virus Replication Gives Rise to a Novel Class of Defective Viral Genomes. Viruses 2025, 17, 856. https://doi.org/10.3390/v17060856
Anisi S, Noble G, Williams R, Hales J, Bridgewater HE, Easton A, Collier W, Gould P. Intersegment Recombination During Influenza A Virus Replication Gives Rise to a Novel Class of Defective Viral Genomes. Viruses. 2025; 17(6):856. https://doi.org/10.3390/v17060856
Chicago/Turabian StyleAnisi, Soraya, George Noble, Rory Williams, Jack Hales, Hannah E. Bridgewater, Andrew Easton, William Collier, and Phillip Gould. 2025. "Intersegment Recombination During Influenza A Virus Replication Gives Rise to a Novel Class of Defective Viral Genomes" Viruses 17, no. 6: 856. https://doi.org/10.3390/v17060856
APA StyleAnisi, S., Noble, G., Williams, R., Hales, J., Bridgewater, H. E., Easton, A., Collier, W., & Gould, P. (2025). Intersegment Recombination During Influenza A Virus Replication Gives Rise to a Novel Class of Defective Viral Genomes. Viruses, 17(6), 856. https://doi.org/10.3390/v17060856