1. Introduction
Herpesviruses are large double-stranded DNA viruses that infect cells from humans and other animals and carry out a complicated pattern of expression of hundreds of viral genes to combat the host anti-viral response, replicate the viral genome, and ultimately allow the virus to be packaged and spread to other cells [
1,
2]. All herpesviruses have genes that are transcribed by Pol II very early, early, and late, but beta- and gamma-herpesviruses have a set of six LTFs that are required for the expression of their late genes after viral DNA replication has started [
3,
4]. The very early and early genes are transcribed using the host-encoded GTFs, including TFIID (TBP with TAFs), TFIIA, TFIIB, TFIIE, TFIIF, and TFIIH, to form a preinitiation complex and help with early steps in Pol II initiation. One of the LTFs evidently substitutes for TBP because it interacts with a TATTA or TATAT sequence upstream of the late gene transcription start sites (TSSs) [
5,
6,
7]. Unlike TBP, this LTF also interacts with Pol II through the C-terminal domain (CTD) of the large subunit [
6,
8]. It is not known if some of the other LTFs substitute for some or all of the host GTFs. What is clear from DFF-ChIP [
9] experiments performed on HCMV-infected cells is that the LTF complex containing Pol II is very different from the normal TBP-driven transcription preinitiation complex (PIC) [
10]. The TBP PIC protects 70 to 80 bp of DNA centered just upstream of the TSSs, while the LTFs protect only about 50 bp, all of which is upstream of the late TSSs [
10]. While the LTF complex on late promoters is prevalent, it is relatively less efficient at driving transcription initiation compared to TBP PICs [
10]. A large amount of work has provided incredible details about the structure and function of TBP PICs [
11,
12,
13,
14,
15,
16], but unfortunately, the structure of the LTF complex on a late promoter has not been solved despite efforts to do so [
8].
Major advances in solving the structures of protein nucleic acid complexes have been made over the last two decades, especially through the use of cryo-EM [
17,
18], but many structures remain unsolved due to technical difficulties in producing the proteins and obtaining suitable images. Then, last year, came the Nobel Prize-winning work from Demis Hassabis, John Jumper, and David Baker that resulted in the successful utilization of artificial intelligence to predict protein structures and to design proteins from scratch. The AI algorithms developed by Google DeepMind, AlphaFold, have been demonstrated to accurately predict protein structures, and the most recent incarnation, AlphaFold 3, can predict structures of proteins complexed with nucleic acids and can include post-translational modifications of proteins as well as specific ligands (organic and ionic) [
19]. The goal of the work presented here is to apply AlphaFold 3 to the LTFs to determine if a reasonable structure can be predicted and to use that information to help elucidate how the LTFs might function during initiation on late viral promoters.
3. Results and Discussion
AlphaFold 3 requires specific protein sequences to begin its analysis. Because there are a number of herpesviruses that utilize LTFs, the initial goal was to compile a list of LTFs from six different viruses that included four beta-herpesviruses, HCMV, MCMV, HHV6, and HHV7, and two gamma-herpesviruses, EBV and KSHV. The NCBI protein database has hundreds of sequences for each of the six LTFs in each virus, so the Identical Protein Groups database was used to pick the protein sequence with the largest number of entries which should correspond to the wildtype/prevalent sequence. Unfortunately, the nomenclature for the LTFs is non-uniform and a decision was made to create a common set of names, LTFa, LTFb, LTFc, LTFd, LTFe, and LTFf.
Table 1 indicates how those general names correspond to each virus’s specific names and what the sizes of the proteins are. The sizes of each LTF and the size of the total for the six LTFs varied across the viruses, as shown in
Figure 1. HCMV and MCMV had the most total amino acids (2651 and 2501, respectively) and had the largest LTFc’s. KSHV had the smallest LTFs and the least total amino acids (2036).
COBALT, the constraint-based multiple alignment tool served by NCBI, was used to analyze the similarities between each LTF across the six viruses.
Supplementary Data File S1 has the sequences used in the analyses and the COBALT sequence comparison for each LTF. The numbers of identical amino acids across the six viruses for each LTF were relatively small (LTFa, 35; LTFb, 26; LTFc, 83; LTFd, 2; LTFe, 17; LTFf, 26). Overall, the 189 identities were less than 10% of the total number of amino acids. As expected, the C-terminal domain of LTFc, which is known to interact with the upstream TA-rich sequence found in late promoters, had the highest percent identity. That domain has homology with TBP [
5], but there were only 10 identical amino acids when COBALT analysis was performed on the six LTFc’s and TBP together (see
Supplementary Data File S1).
Hundreds of AlphaFold 3 jobs were created and submitted before settling on the appropriate parameters for generating the highest-confidence structures. Individual LTFs and groups of LTFs were tried. For each virus, the structures of the six LTF complexes with and without a 60 bp DNA sequence from the late promoter driving HCMV UL86 were submitted. The initial results indicated that similar structures were generated from each virus with or without DNA. In addition, a threonine phosphorylation in EBV BGLF3 (LTFf) that has been demonstrated to be required for efficient LTF function [
20] and seven Zn ions for the beta-herpesviruses, five Zn ions for KSHV, and six for EBV were added to the jobs submitted. The structures generated using these parameters for the six LTFs bound to the HCMV UL86 late promoter for the six viruses had a highly conserved arrangement of the LTFs (
Figure 2). Note that HHV6 is not shown because of its very high similarity to HHV7. The ipTM values generated during the structural predictions were between 0.72 and 0.78, which suggests that the predicted structures may have significant accuracy. The HCMV and MCMV complexes had the largest fraction of unstructured regions (loops), and these corresponded with insertions in the protein sequences found in the COBALT analyses. For example, the first 96 residues of HCMV LTFf and the first 28 residues of MCMV LTFf were identified as insertions and were unstructured. Almost no unstructured regions were found in the HHV6, HHV7, EBV, and KSHV LTF/promoter complexes. Of critical importance is the interaction between the LTF complexes and the promoter DNA. The region of LTFc with structural homology to TBP was directly associated with the TATTA sequence in the promoter (colored red), and the DNA was bent at about 120 degrees at that site. This is similar to how TBP interacts with and bends its recognition sequence [
21]. It is likely that the predicted conformation of the DNA was derived from the conservation between LTFc and TBP [
5] coupled with the known structure of TBP bound to its TA-rich recognition sequence, leading to a dramatic bend in the DNA [
22,
23]. Importantly, the AlphaFold 3-predicted structures are completely consistent with the size and positioning of the LTF complex upstream of the transcription start site as determined by DFF-ChIP in HCMV-infected primary human foreskin fibroblasts [
10].
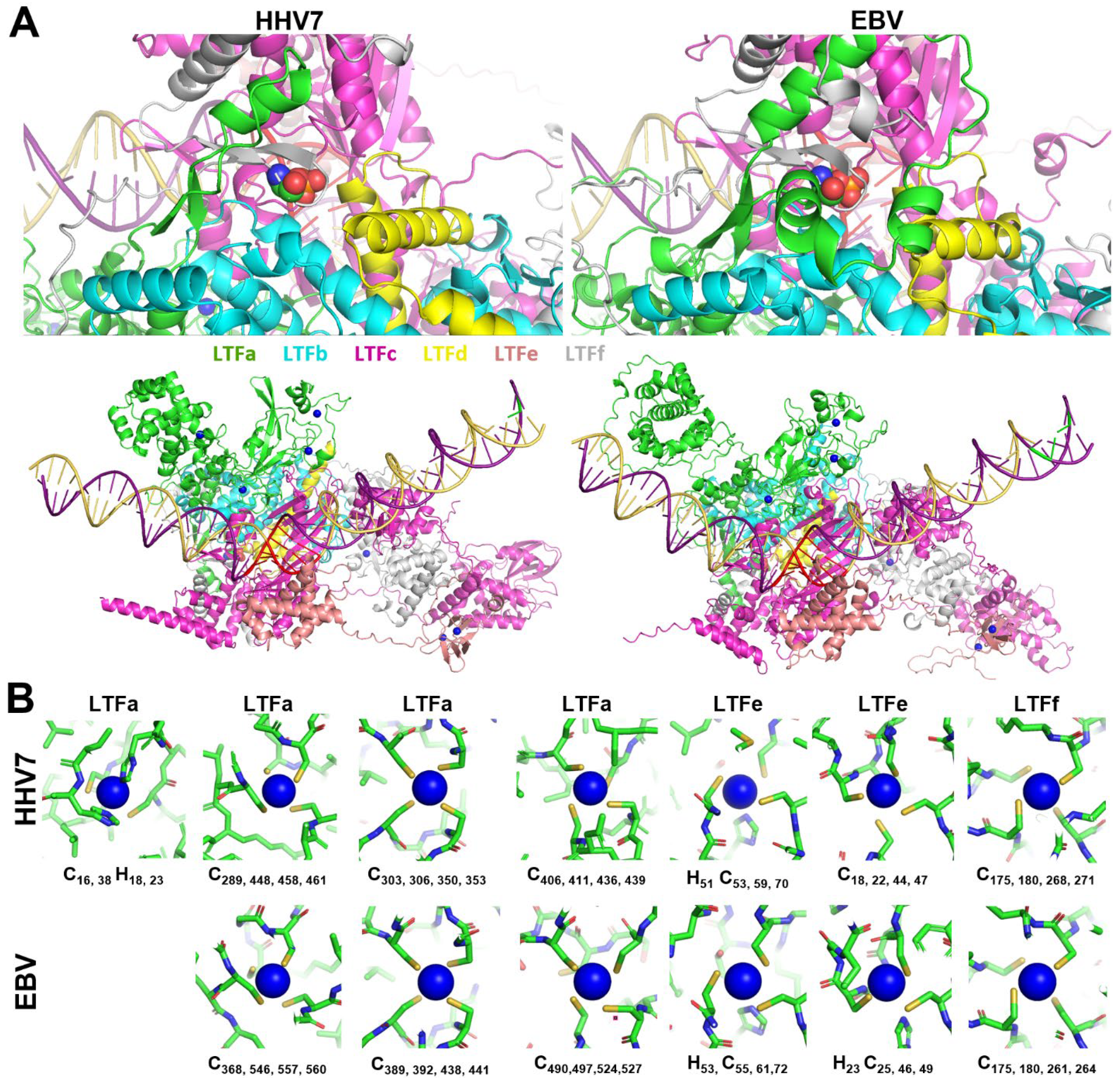
All LTFf’s contain a conserved threonine, and phosphorylation of that residue has been shown to be essential for EBV LTF function [
20]. The phosphorylated threonine was predicted to be in the middle of a conserved N-terminal domain of LTFf, and the position of the phosphate in the LTF/promoter structure is at the junction of five LTFs, as illustrated by HHV7 and EBV (
Figure 3A, top). The phosphate is found in a small, deep pocket at the interface of LTFa, b, c, d, and f in all six of the viral LTF complexes. The pocket is present even in the LTF/promoter structures generated without the phosphorylation. Neutralization of the negative charge of the phosphate is accomplished in part by interactions of several backbone nitrogens and perhaps water molecules that could be accommodated in the deep pocket. The inclusion of threonine phosphorylation in the AlphaFold 3 predictions led to an increase in the confidence values for many residues across the interfaces between the EBV subunits (
Supplementary Figure S1).
Evidence is mounting for metal binding playing a role in the function of the LTFs. Two different groups have demonstrated that conserved Cx
nC motifs found in LTFa [
24] and in LTFf [
25] are important for the function of KSHV LTFs. Because of this, zinc ions were titrated into AlphaFold 3 jobs with all six sets of six LTFs (including LTFf with threonine phosphorylation) with a late promoter. For the two gamma-herpesviruses, there were five (KSHV) or six (EBV) specific sites occupied by zinc. However, for the four beta-herpesviruses, there were seven specific sites of occupancy. The location of the zincs is illustrated by the HHV7 and EBV LTF complexes (
Figure 3A, bottom). At least two zincs were found in LTFa in all viruses, with EBV LTFa containing three and all beta-herpesvirus LTFa’s containing four. Two were found in LTFe and one in LTFf in all viruses (
Figure 3A, bottom). In general, each zinc was chelated by groups of four conserved cysteines and or histidines (
Figure 3B). The lower numbers of zincs in EBV and KSHV were accompanied by the loss of conserved residues in LTFa in those viruses (see sequence comparisons in
Supplemental Data File S1). Another difference between virus types was that one of the zincs in LTFe from KSHV had only two cysteines. Because of this, it is not clear if zinc actually binds to that site. Overall, the phosphorylation of LTFf and the addition of zincs improved the confidence of the prediction of all of the viral LTF complexes, and this correlated with increasing ipTMs for most viruses when the phosphothreonine and zincs were added (
Figure 4). Combined with the earlier experimental data, it seems likely that zinc binding plays a major role in the structure and function of the LTF complex.
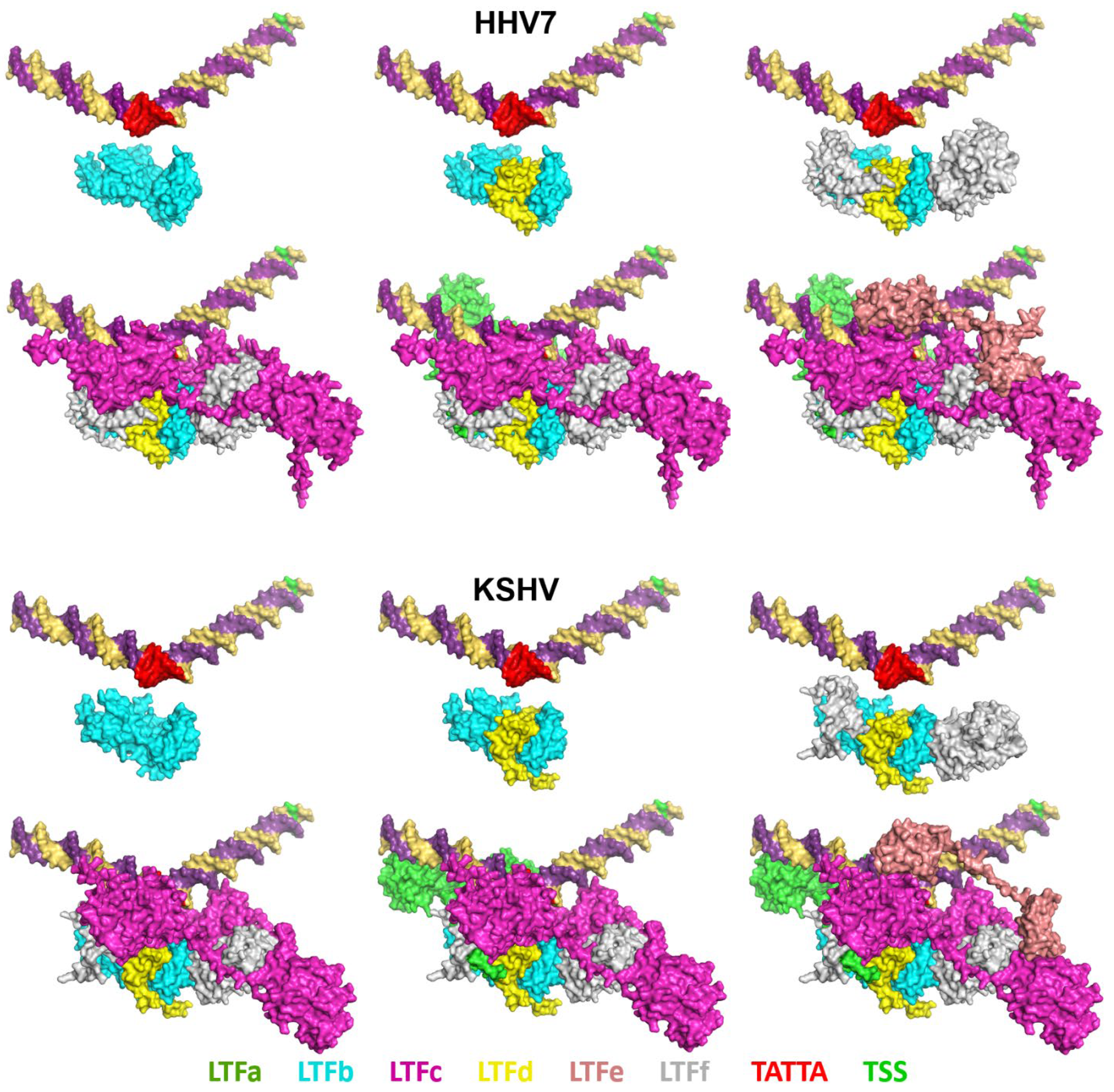
The structures of the LTF complexes have a mix of LTF interactions with each other and with the promoter DNA. The six viral LTF complexes all share the same interactions. For illustrative purposes, the KSHV and HHV7 complexes are compared in
Figure 5. LTFb and LTFd have a major interaction with each other, as has been shown [
26], and LTFf interacts with the LTFbd complex. None of those subunits have any direct interaction with the promoter DNA. LTFc bridges the LTFbdf complex to the promoter through the interaction of LTFc with the upstream TATT element. LTFc extends in a downstream direction with minimal DNA contacts, but with a major interaction with LTFf. LTFa interacts with the DNA mainly upstream of TATTA and interacts with LTFc and LTFb. LTFe binds to the DNA, and each of its two domains binds only to LTFc. The C-terminal domain of LTFe binds near TATTA and interacts with the TBP-like domain of LTFc. The N-terminal domain associates with the downstream lobe of LTFc that has been implicated in binding to the CTD of the large subunit of Pol II [
6,
8]. To quantify the extent of protein/protein interactions, the buried surface area was calculated for the proteins in all viral LTF complexes. The percentage of the protein surface area that was buried was quite high at 20% to 28% (
Supplementary Figure S2). Not surprisingly, HCMV and MCMV had the lowest values due to the inability of the unstructured regions specific for those viral complexes to interact with other regions of the complex.
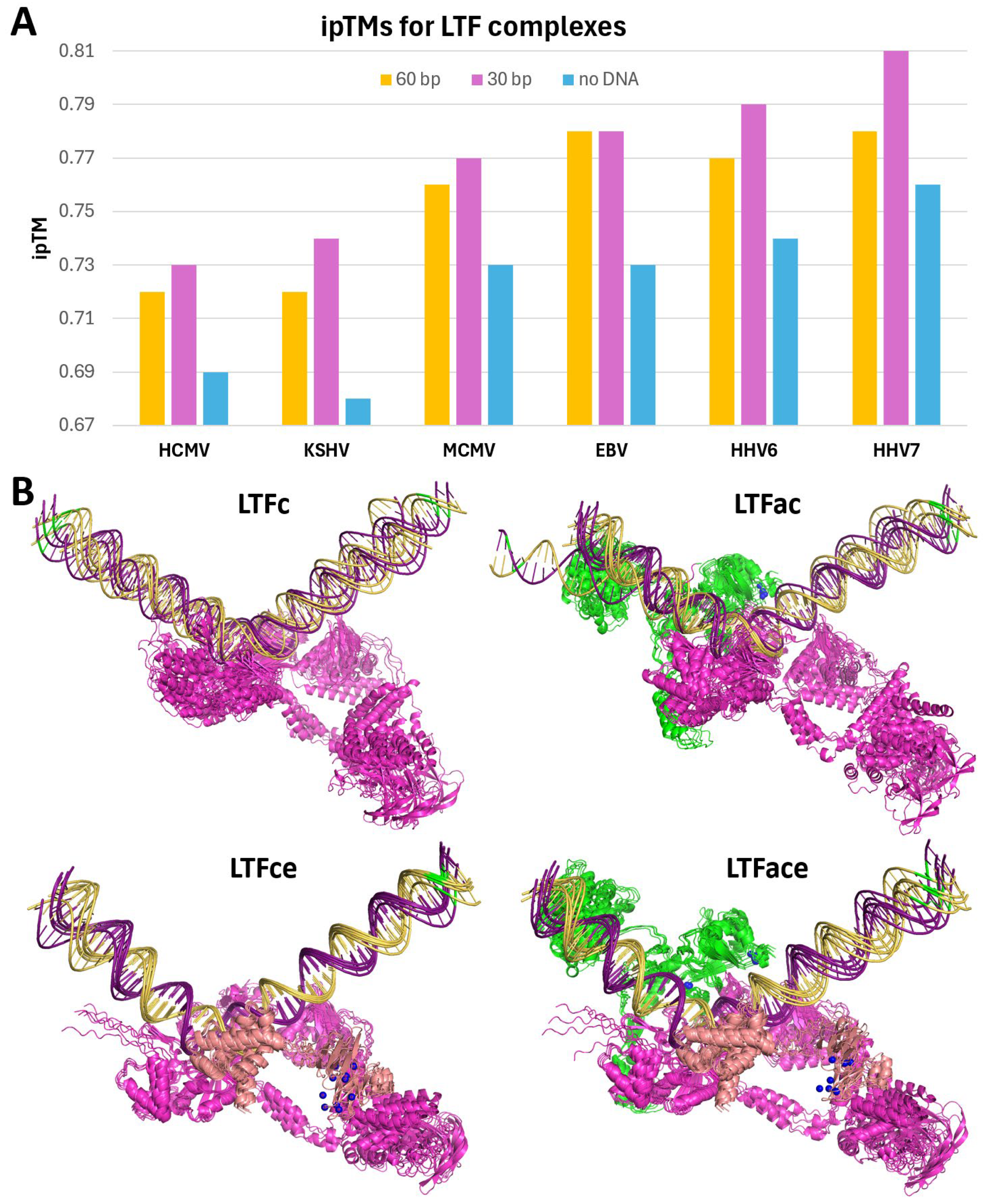
To quantify how DNA might affect the prediction of the LTF complexes, jobs with different lengths of DNA or with no DNA were run while maintaining the LTFf phosphorylation and zinc ions for all six viruses. Compared to the 60 bp promoter, a 30 bp DNA centered on the TA-rich recognition element in the middle improved the ipTM scores of all viral LTF complexes (
Figure 6A). This was somewhat expected since much of the 60 bp DNA was of low confidence and reducing the amount of it would increase the fraction of the atoms with higher confidence. However, when the DNA was removed from the jobs, the ipTMs fell significantly. The percentage of the surface area buried by DNA was between 3% and 5%, again with HCMV and MCMV having the two lowest values due to the presence of unstructured protein segments (
Supplementary Figure S2). Together, these results indicate that DNA plays a significant role in stabilizing the LTF complexes.
To examine the interdependence of the three LTFs that bind to DNA, AlphaFold 3 was used to predict the structures of LTFa, LTFc, and LTFe from KSHV (
Supplementary Figure S3) and HHV7 (
Supplementary Figure S4) with and without promoter DNA and with and without Zn for LTFa and LTFe. LTFa folded into a three-lobe structure, and the addition of Zn improved the confidence of the Zn binding domain. Interestingly, LTFa only bound to late promoter DNA when Zn was included; however, it did not bind in the proper location seen in the full complex. The Zn binding domain of LTFe also folded into a more confident structure in the presence of Zn, but the two domains of LTFe that occupy very different locations in the complete LTF structure were right next to each other. Like LTFa, LTFe was predicted to bind to DNA in the presence of Zn and did not bind in the correct position. AlphaFold 3 predicted that KSHV LTFc alone would bind to the TATTA sequence in the late promoter, but of the five structures generated, only two were bound in the correct orientation seen in the complete LTF structure (
Figure 6B). The addition of LTFa to LTFc improved the directionality of LTFc (four of six structures were in the correct orientation). The addition of LTFe to LTFc led to all six structures being in the proper direction and caused the two domains of LTFe to separate as they are found in the full LTF complex. The LTFace complex maintained the proper positions of the three factors and the correct orientation. Overall, the conclusion drawn is that LTFc is the primary DNA binding factor and that LTFa and especially LTFe are required to provide proper directionality to the interaction with DNA.
There is a vast amount of information about the structures of TBP-containing PICs and the function of the individual GTFs [
11,
12,
13,
14,
15,
16], but how the LTF complex compares to that is mostly unknown. The major difference is that the TBP PIC covers more DNA that includes the transcription start site and downstream sequences [
11,
13,
14,
15,
16,
21], while the LTF complex only covers DNA upstream of the TSS [
10]. This means that Pol II, which must interact with the TSS, is not in position to initiate transcription in the LTF complex. The domain of LTFc that has been shown to interact with Pol II [
8] is the domain that is farthest downstream (
Figure 7A), and its interaction with the CTD of Pol II could help Pol II locate the TSS; however, AlphaFold 3 failed to predict a specific interaction of the CTD with or without phosphorylation with this domain of LTFc. In the TBP PIC, TFIIB is an essential GTF that has two separate domains [
11,
12,
21]. One interacts with upstream DNA and TBP bound to its recognition sequence and the other binds directly to Pol II (
Figure 7B). TFIIA is also involved by binding to upstream DNA and TBP (
Figure 7B). Note that if looking down at the template toward the TSS with TBP on the bottom, TFIIA is on the left side of the DNA and TFIIB is on the right side. By aligning TBP and the TBP-like domain of LTFc, LTFa is in the same position as TFIIA and LTFe is in the same position as TFIIB (
Figure 7C). This suggests that LTFa and LTFe substitute for the two GTFs. Interestingly, both TFIIB and LTFe each have two tethered domains. One interacts with TBP or the TBP-like domain in LTFc and the other interacts with Pol II directly or with the Pol II binding domain of LTFc. Clearly, LTFe would block the entry of TFIIB and LTFa would block the entry of TFIIA, and this was borne out by the failure of AlphaFold 3 to incorporate TFIIB or TFIIA into discrete positions in the complete LTF complex. When TFIIB was used in place of LTFe to generate an LTF complex, TFIIB was positioned exactly as it is in the TBP PIC, with TFIIB interacting with the TBP-like domain of LTFc and the right-hand side of the DNA. It is not clear if TFIIB is required for LTF-driven initiation, but if it is, it would require the removal of LTFe.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}