3.1. Evolution of Virulence in an XRV with Maternal Transmission
Using the deterministic model for the XRVs described in the
Section 2, we consider 1100 strains of XRVs, with strain
i having
, which gives values from 0.001 to 1.1. The maternal transmission rates are
. The uninfected birth and death rates are
, and
; hence, the density of individuals in an uninfected population is
. The carrying capacity is nominally
, but the value of
is unimportant because population sizes scale in proportion to
. We begin the simulation with
uninfected individuals and
for all strains. We follow the population sizes forward in time using the standard fourth-order Runge–Kutta method. The mean values of
and
at any point in time are
and
. In the long-time limit, the mean values converge to steady-state values, which we take as the estimate of the optimal virus that will be selected over time.
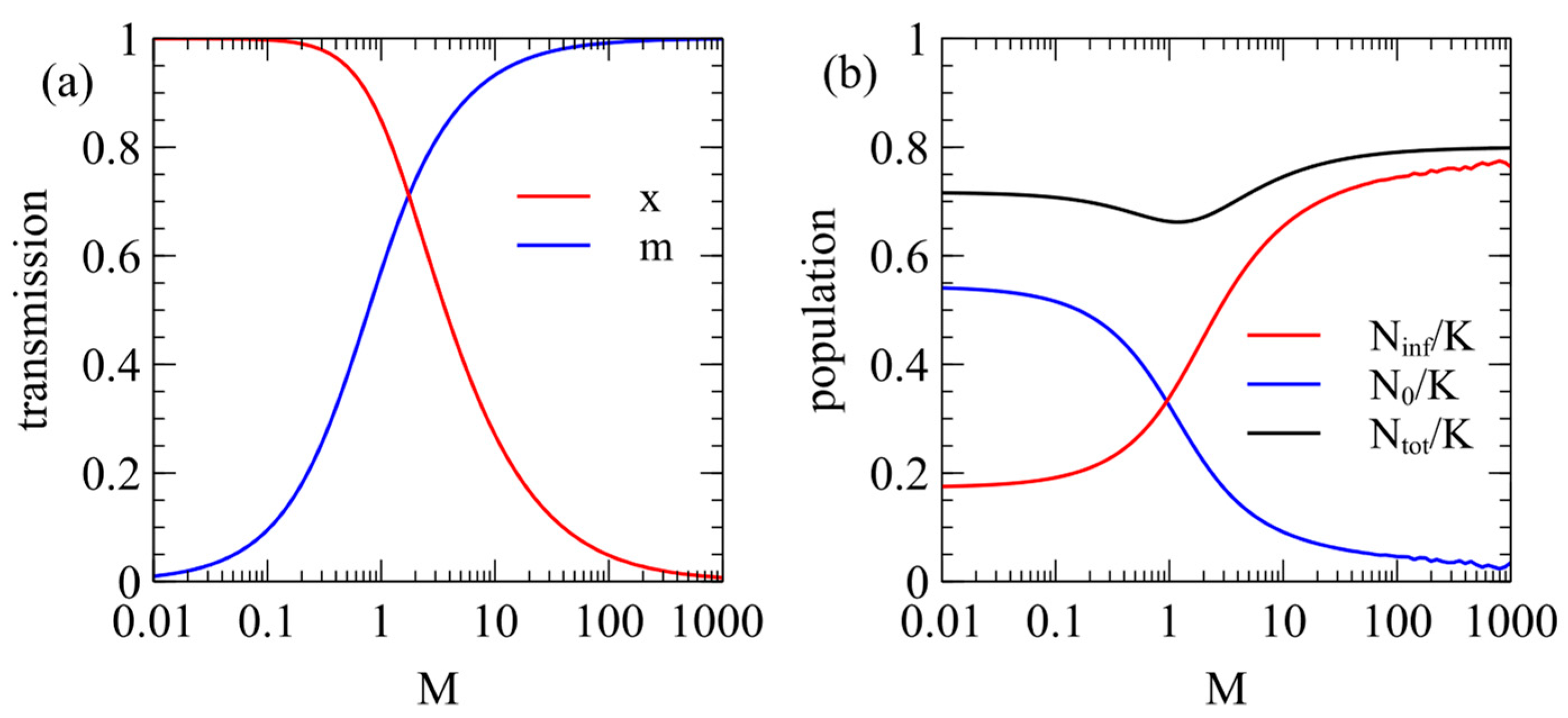
Figure 1a shows the steady-state values of
and
as a function of
with the contact rate fixed at
. When
, there is very little maternal infection,
, and the virulence is high,
. For a larger
,
increases and
decreases. When
,
and
.
Figure 1b shows the steady-state populations of infected and uninfected individuals. The fraction of infected individuals increases with
. For a low
, the virus is virulent, and a fairly small fraction is infected. For a very high
, almost the whole population is infected, but the virus has very little effect on the population.
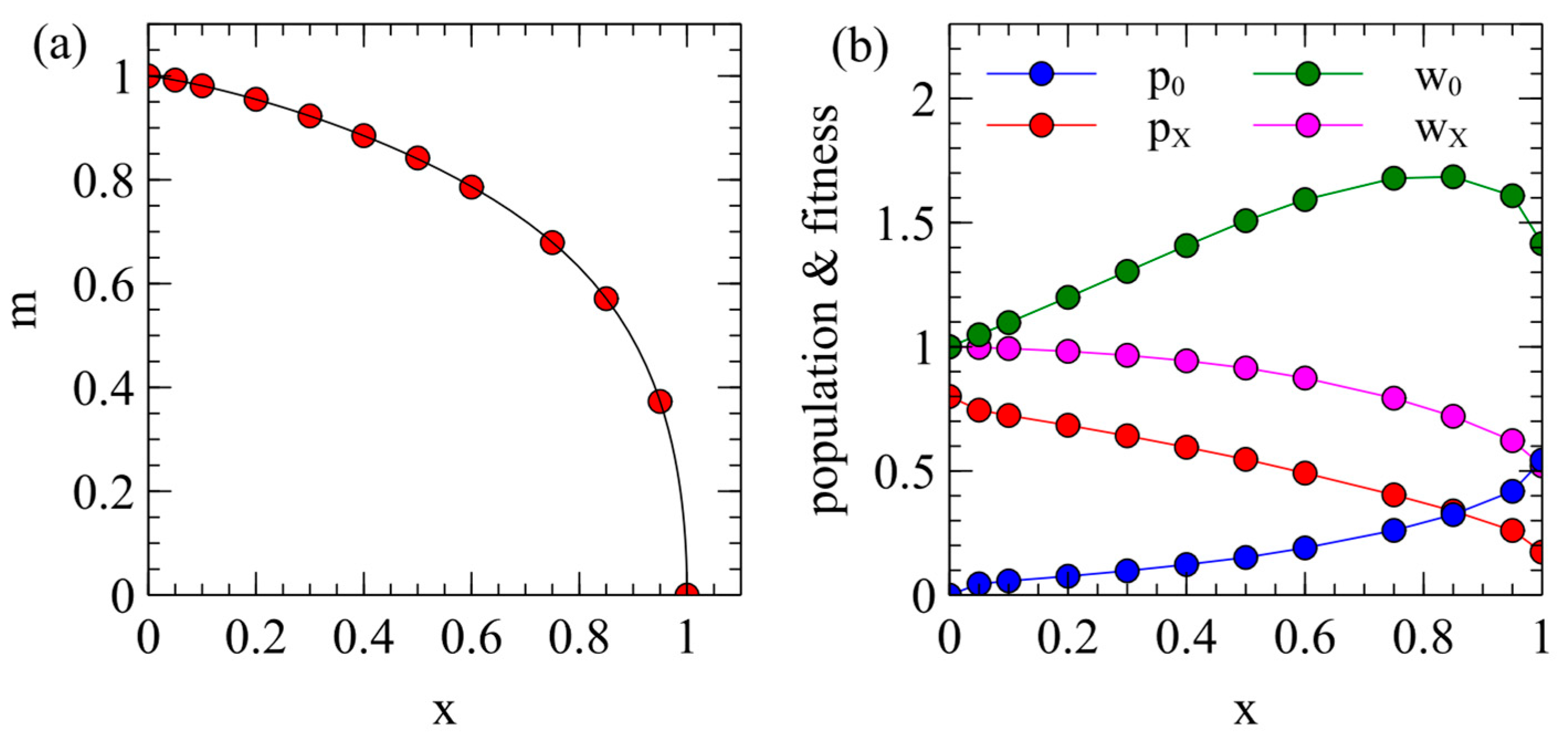
The steady-state
and
values from
Figure 1a can also be plotted as in
Figure 2a, which shows how the maternal transmission rate is expected to coevolve with virulence. Different viruses are expected to evolve to some point on this curve which will depend on the extent that maternal transmission is possible in that virus (i.e., on the
parameter).
Example points
are marked on
Figure 2a. For each of these points, we determined the populations of uninfected and infected individuals
, and
, when there is a single virus strain with the corresponding
and
. This is shown in
Figure 2b. These values are used as starting conditions when we consider the introduction of an ERV later in this paper. The mean death rate of an individual in this population is
. Fitness is inversely proportional to the death rate, so the relative fitness of an uninfected individual is
and the relative fitness of an infected individual is
. These fitness values are shown in
Figure 2b. The value of
decreases steadily with
because
increases with
; however,
has a maximum around
. Although
would be the maximum at
,
is the maximum for smaller
because a larger fraction of the population is infected when
is smaller. This gives a maximum in the
curve which is relevant later when we consider the spread of ERVs.
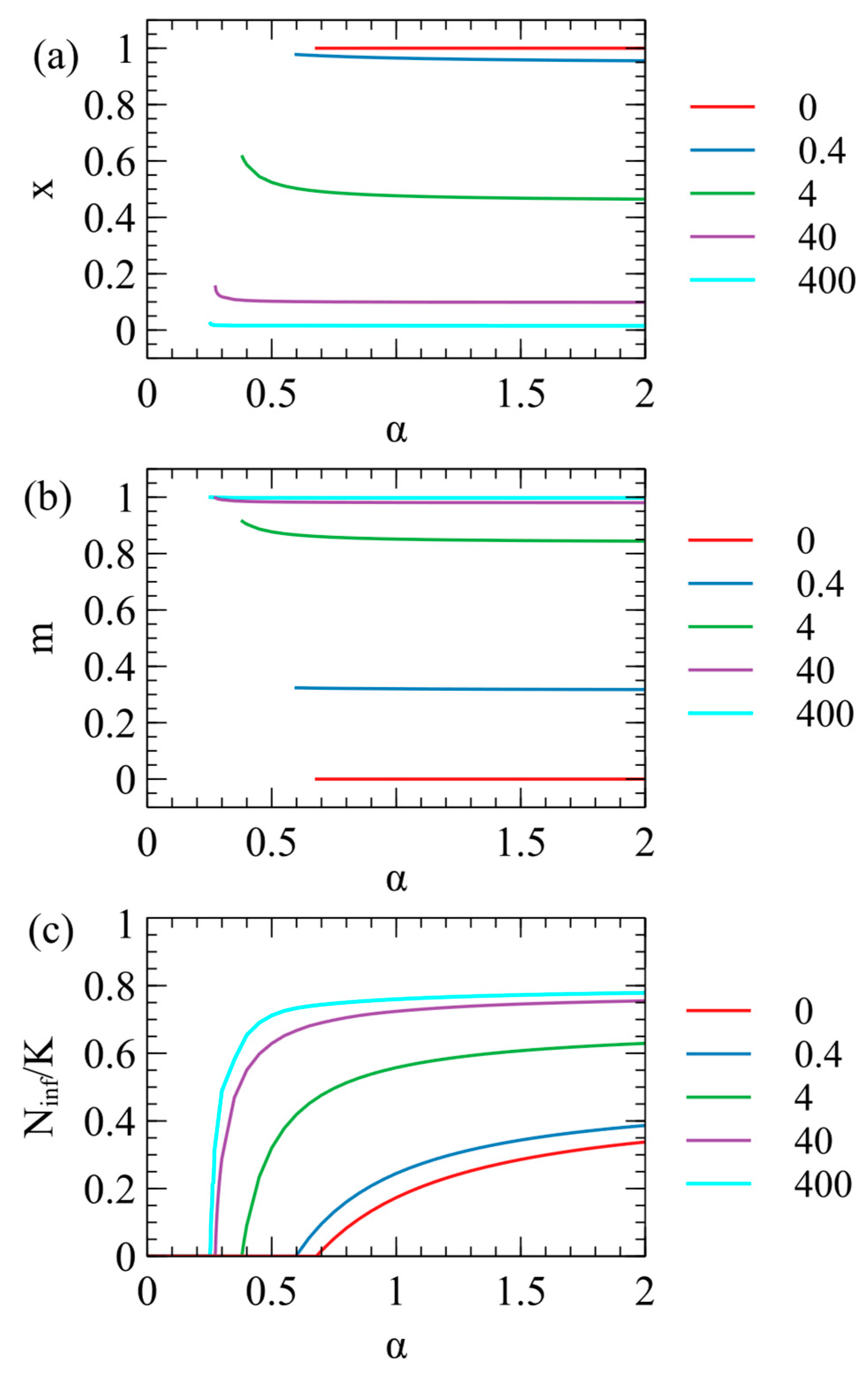
In
Figure 1 we considered a fixed value of the contact rate
. However, the optimal parameters for the virus also depend on
, as shown in
Figure 3. We suppose that
depends on the behaviour of the host population (rate of close contacts between the hosts) and is not controlled by the virus; hence, virus evolution depends on
but
does not change when the virus evolves. We considered the evolution of the optimal virus as a function of
for several fixed values of
. For each value of
, we began with 1100 strategies, as above, and determined the steady-state values of
and
in the long-time limit. The virus survives for
greater than a critical value
, which is different for each value of
. The optimal
and
values are only defined when the virus survives; therefore, they are only plotted for
in
Figure 3a,b. In
Figure 3c, we show the number of infected individuals
, which is zero for
, and increases with
for
. The critical contact rate is
for
and decreases as
increases.
When
, there is no maternal transmission (
) and
for all values of
. Larger
values give larger
and smaller
, i.e., the presence of maternal infection leads to the evolution of lower virulence, as expected. For very large
(e.g.,
in
Figure 3),
is very close to 1, and
is very close to 0, i.e., the virus is almost harmless to the host and relies almost exclusively on maternal transmission. It is not possible to have
strictly equal to 0, however, since a parasite with only vertical transmission can never survive if there is even a tiny deleterious effect on the host.
As
decreases towards
, the optimal
inreases slightly and there is a corresponding increase in
. This is most easily seen for
in
Figure 3a,b, but also occurs in the other curves. As
decreases, there are fewer opportunities for contact between hosts, therefore strains with higher infection rates per contact (higher
) are selected. This means that strains with low virulence and a high maternal transmission rate are most likely to evolve when the opprotunity for horizontal transmission is high (high
), as has been noted previously [
13]. However,
Figure 3 shows that for
there is little change of
and
with
, so we we fix
for all the subsequent examples.
3.2. Spread of an ERV in an Uninfected Population
Using the stochastic model for ERVs described in the
Section 2, we first consider a case where the ERV spreads in a population of uninfected individuals. The carrying capacity is
, therefore the number of ininfected individuals is initially
. At time zero, one of these individuals is turned into a type E individual, and a single copy of the ERV is added to its genome. The number of individuals with at least one ERV copy,
, is followed over time. The simulation is continued until either the ERV is eliminated (
) or the ERV spreads to fixation, so there are no remaining individuals without the ERV (
). We ran
simulations for each parameter set and measured
, the fraction of runs that reaches fixation. In most cases,
. We denote fixation as the point when every individual has at least one ERV copy. This does not mean that the ERV is inserted at every locus, in which case every individual would have
copies. However, once every individual has at least one copy, the ERV continues to multiply slowly by transposition until every individual has
copies. When measuring the fixation probability, we stopped the simulations at the point when every individual has at least one copy, because this saves considerable computer time.
An ERV in an uninfected population is equivalent to a transposable element that spreads by transposition and genetic inheritance. This case is somewhat artificial for an ERV, because we presume the ERV arose by the insertion of an XRV, so the XRV must also be present. However, we begin by considering the spread of the ERV in the absence of the XRV because this provides a baseline with which to compare other cases.
As described in the
Section 2, the death rate of an individual with at least one ERV copy is
, where
is the deleterious effect of the ERV.
Figure 4a shows
as a function of
for several values of the transposition rate
. The fixation probability is highest for the smallest
, because low
ERVs have the least deleterious effect on host reproduction. For a high transposition rate,
, there is a measureable chance of fixation for
as high as 0.7 (i.e., at least 1 out of the 10,000 repeat runs reached fixation). For smaller transposition rates, only lower
values have a measurable chance of fixation.
It is also possible to consider the limit of
, in which case the ERV cannot transpose, but still has some chance of spreading to fixation at the single locus in which it was introduced. If
and
, the ERV is a neutral gene at a single locus. The probability of fixation of a neutral gene beginning with a single copy is
. In this case,
and
. We ran this data point for
, and found 60 cases of fixation, which is consistent with the neutral theory within statistical error. We also ran the case of
for the full range of
with
, and found the fixation probability was too small to measure for non-zero values of
. As shown in
Figure 4a, the fixation probability for ERVs with a non-zero transposition rate is very much higher than expected for a single neutral gene without transposition (
). ERVs with a significant deleterious effect have high fixation probabilities if
is high enough, whereas a single deleterious gene without transposition would have a negligible fixation probability on the scale of
Figure 4a.
The fixation probability also depends on the number of available loci
into which the ERV can insert. We assumed that, if there is currently one ERV copy in a genome, the rate of transposition is proportional to
, the fraction of sites that are not yet occupied by an ERV. This factor gives a significant reduction in the multiplication rate of the ERV if
is small. The examples in
Figure 4a were calculated with
.
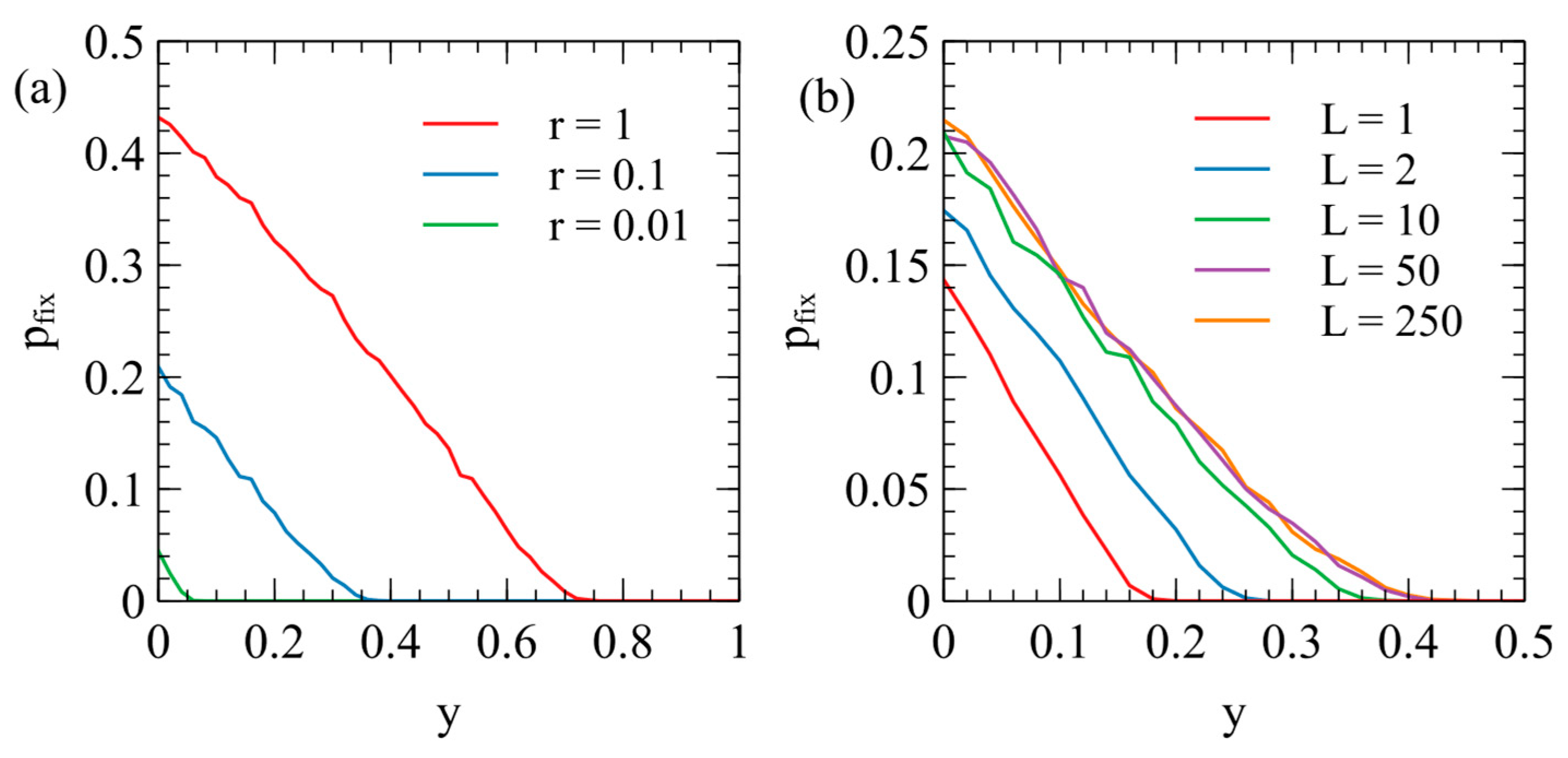
Figure 4b shows cases of
, and
, with
in each case. For
= 1 and 2,
is significantly lower than for
. For
= 50 and 250,
is only slightly higher than for
because the effect is of the order
. For the rest of this paper, we consider only
, because increasing
makes only a small difference to the result and increases the required simulation time by a large amount.
3.3. Spread of an ERV in the Presence of an XRV
We now consider the case where the ERV arises in a population infected by an XRV. The XRV is assumed to have evolved to parameters
and
that are optimal parameters from the model for the evolution of virulence of the XRVs. We fix the contact rate at
. We use the
and
values marked as points in
Figure 2a as parameters for the XRV. The stochastic model begins with numbers of uninfected and infected individuals
and
for the corresponding
and
(shown in
Figure 2b). At time zero, one type X individual is changed to a type EX individual, and one copy of the ERV is added to its genome. We follow
as a function of time until either the ERV is eliminated (
) or reaches fixation (
). We define
as the mean fraction of loci occupied by the ERV, which is the mean number of ERV copies per individual divided by
. The parameters are as previously established, as follows:
.
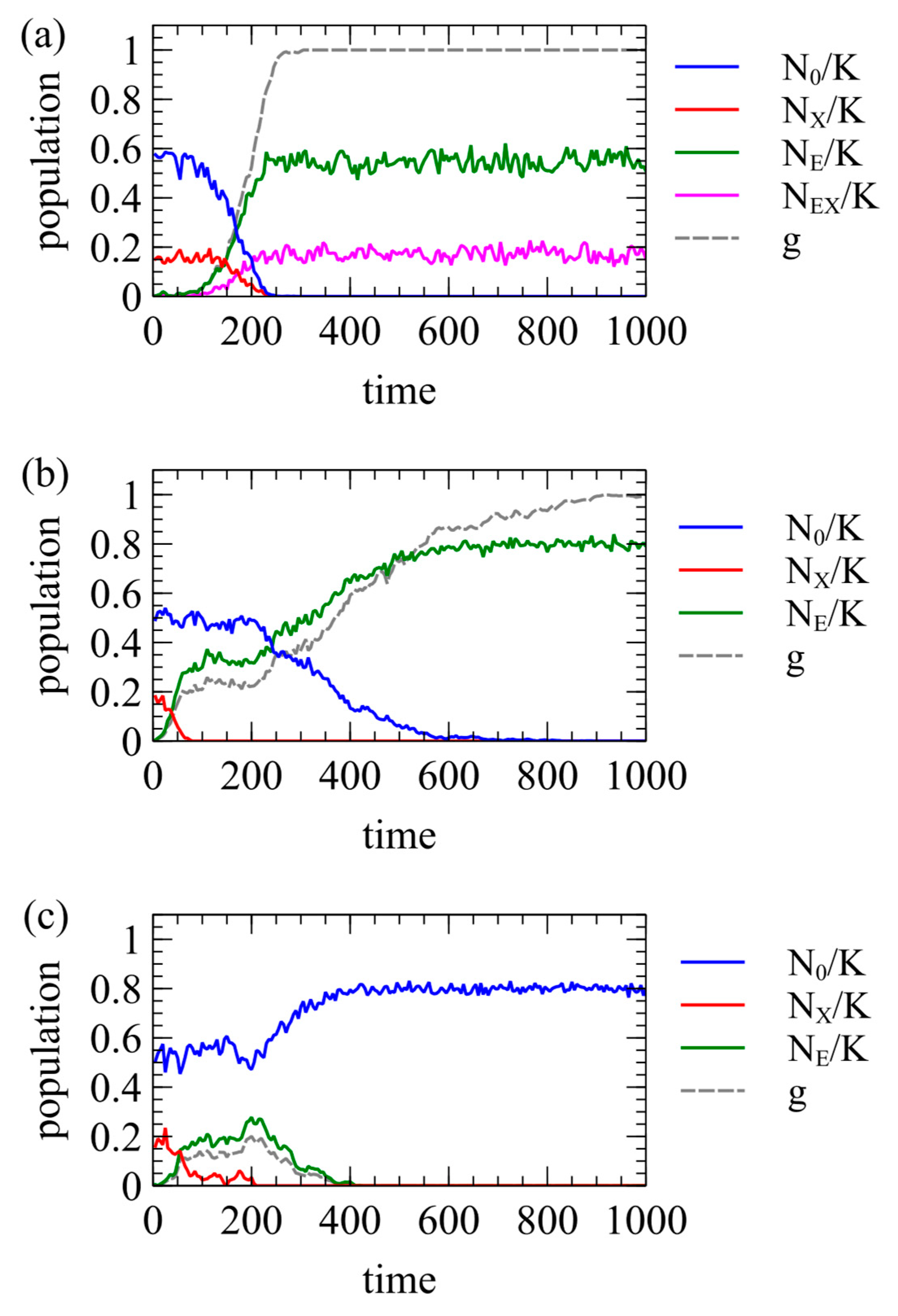
Figure 5 shows three examples of single simulation runs. In all three cases, the XRV has
. In
Figure 5a,
,
, and
, meaning the ERV is completely harmless to the host and offers no resistance to the XRV. In this case, the ERV spreads to fixation quite quickly. All individuals have at least one ERV at time 290, and
shortly afterwards, at time 305. In this case, individuals with the ERV can still be infected by the XRV. Therefore, individuals of type EX and type E remain at the end of the run. In
Figure 5b,
,
, and
, meaning that the ERV offers complete resistance to the XRV. The ERV spreads more slowly because
is lower, but fixation still occurs. All individuals have at least one ERV at time 895, and
at time 1010. In this case, the ERV provides complete resistance to the XRV. The first insertion of the ERV occurs in an infected individual, so one type EX individual is created at time zero, but the offspring inheriting the ERV cannot be infected by the XRV. Therefore, there are no further type EX individuals after the original one dies. At the end of the run, all individuals are type E. For both
Figure 5a,b, we have chosen runs that reach fixation. However, the outcome is stochastic, and there are also many other runs for the same parameters in which the ERV is eliminated almost immediately.
Figure 5c shows what we call “clearance”. In this case,
,
, and
. As
, type E individuals have higher fitness than type X individuals. Therefore, type E individuals increase initially, and type X individuals decrease. After the disappearance of the XRV at time 210, type E individuals have a lower fitness than uninfected individuals because
. Therefore, type E individuals are selected against, and the ERV is eliminated at time 425. Thus, the introduction of the ERV leads to clearance of the XRV from the population, even when it does not itself reach fixation.
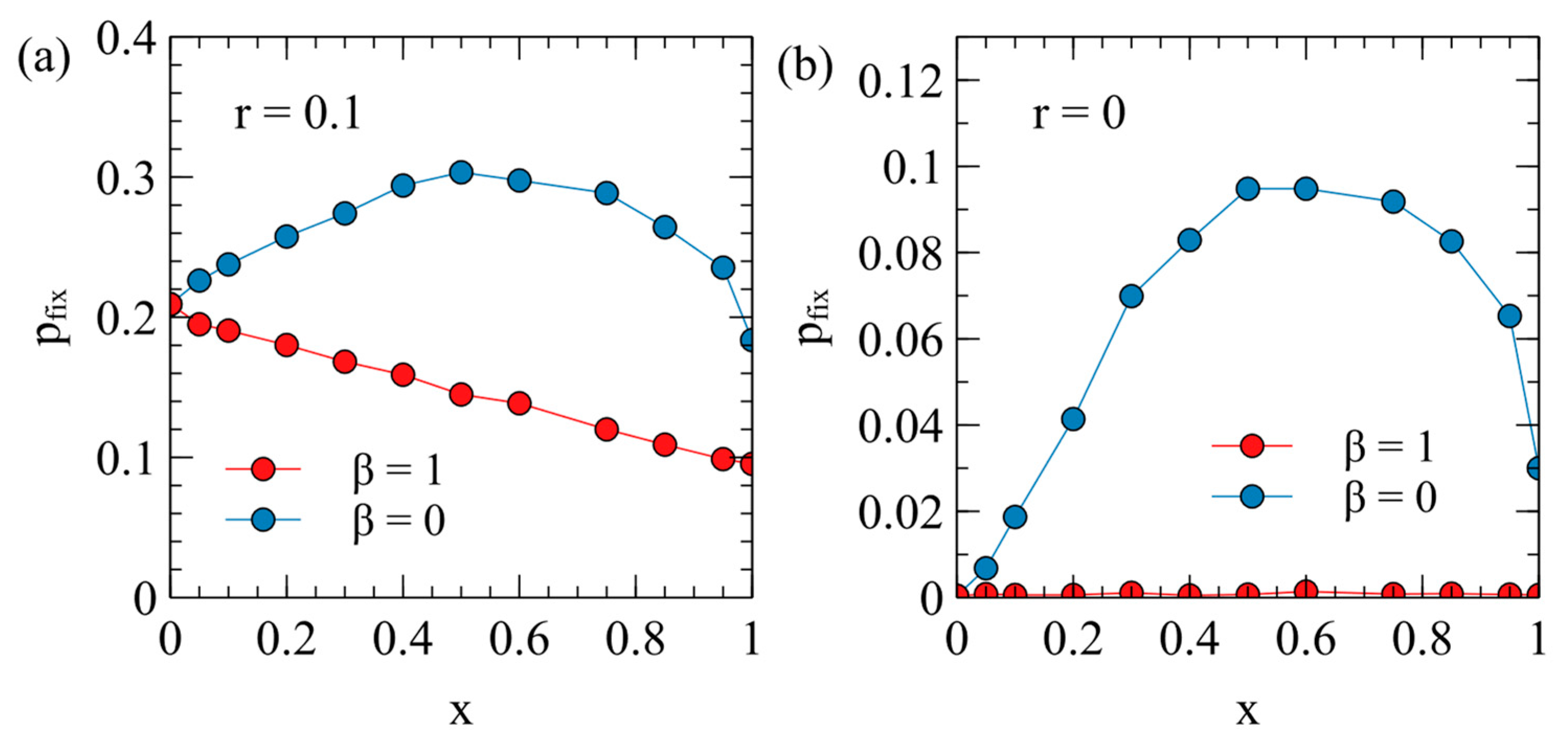
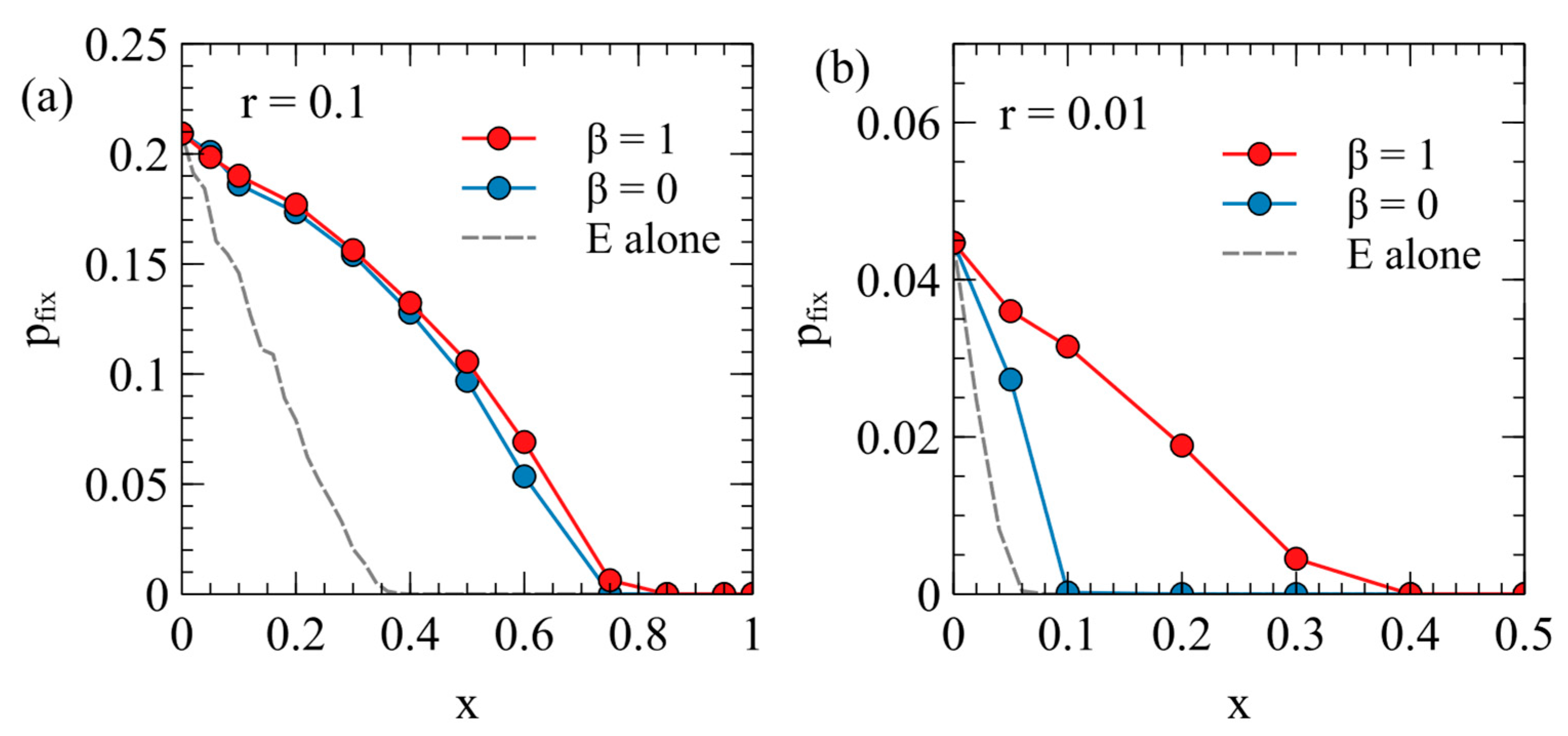
Figure 6 shows the fixation probability of the ERV as a function of its deleterious effect,
, in a population originally infected with a virulent XRV, with
and
. Two different transposition rates are shown in
Figure 6a,b. In each case, the fixation rates are measured separately for
and
, with
for each data point. These are compared with the fixation probability when the ERV spreads alone in an uninfected population (dashed curves are the same as
Figure 4a).
There are two principle factors that cause the fixation probabilities to be different in the presence of the XRV from when the ERV is alone. Firstly, chance events immediately after insertion of the first ERV copy have a significant influence on
. For example, if the first individual with a single ERV copy dies before reproduction, then the ERV disapears immediately. If the first ERV is inserted in an infected individual, this individual is type EX, and has the death rate
(because the death rate is determined by the XRV when
); whereas, if the ERV is alone, the first ERV is in a type E individual with the death rate
. Thus, the ERV is more likely to be eliminated immediately after insertion if it arises in a population with the XRV present. This effect will reduce the fixation probability in the presence of the XRV relative to that in populations without the XRV. Secondly, however, if the first individual successfully passes on the ERV gene, then a new type E individual is created, which has a lower death rate than individuals with the XRV infection. The type E individual is then competing with a mixture of type X and type 0 individuals and may have a higher fitness than the average of the X and 0 types. This effect tends to increase
relative to the case of the uninfected population. In an uninfected population, E types are competing against only uninfected individuals, and they always are less fit than the uninfected individuals. The advantage of ERV in the presence of the XRV is particularly important when
, because in this case, all individuals inheriting the ERV gene are resistant to infection by the XRV. When
, there are both type E and type EX individuals containing ERV genes, so the fitness advantage to the E genes is less. This explains why
is larger for
than it is for
. A comparison of
Figure 6a,b shows that the second effect is larger when the transposition rate
is smaller. For the case
,
with
is very much larger than when
or when the ERV is alone.
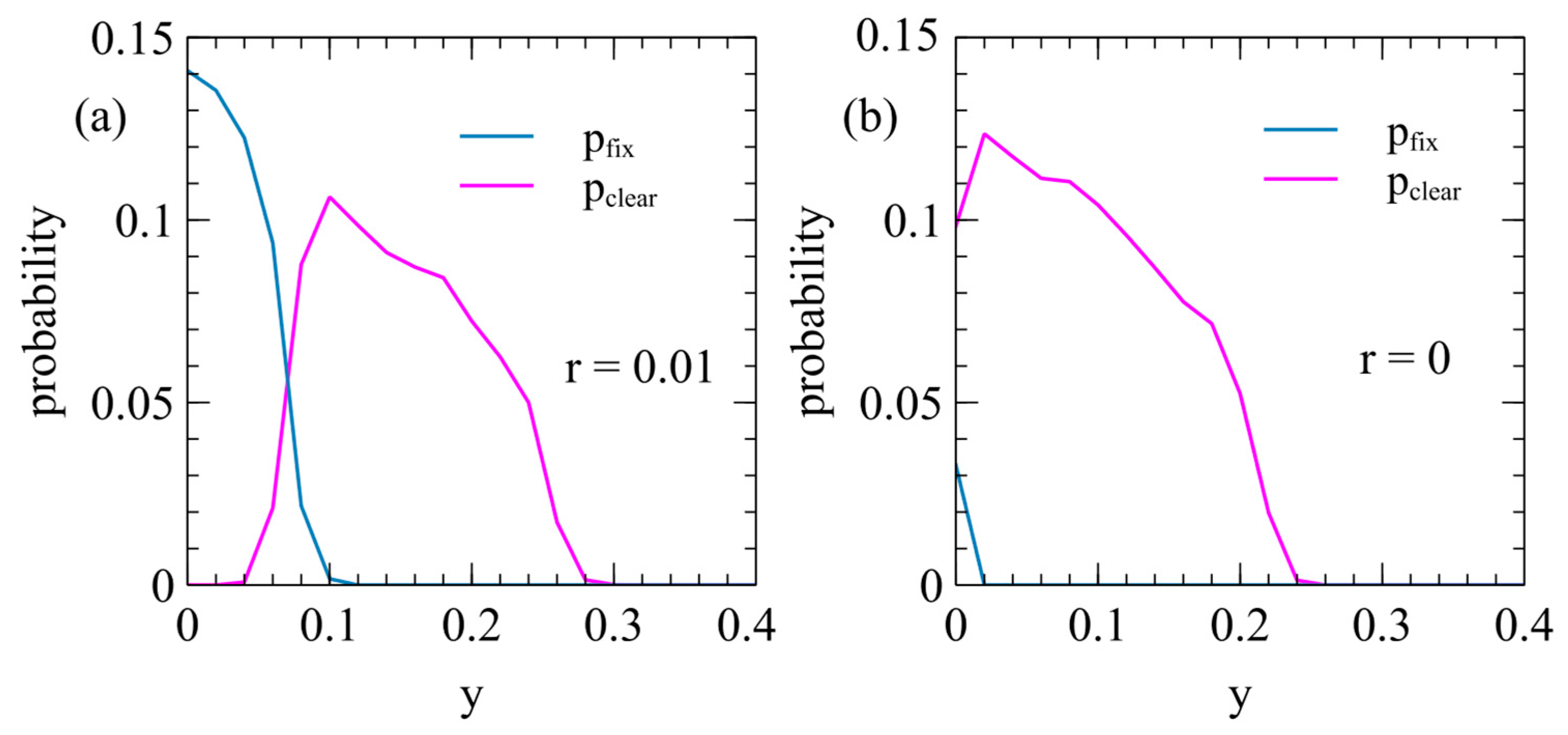
Figure 7 shows the probabilities of clearance and fixation as a function of
in the case where the ERV with the deleterious effect
and
spreads in a population with an XRV with the deleterious effect
. Clearance happens frequently when the transposition rate is small (as in
Figure 7a, with
). Clearance occurs when
is non-zero, but is substantially less than
, so the ERV has a high relative fitness initially while the XRV is still present, but has a low relative fitness compared to uninfected individuals. Clearance also occurs frequently in the limiting case when the ERV cannot transpose (
as in
Figure 7b). In this case, there is a wide range of
where clearance occurs, but fixation occurs only when
.
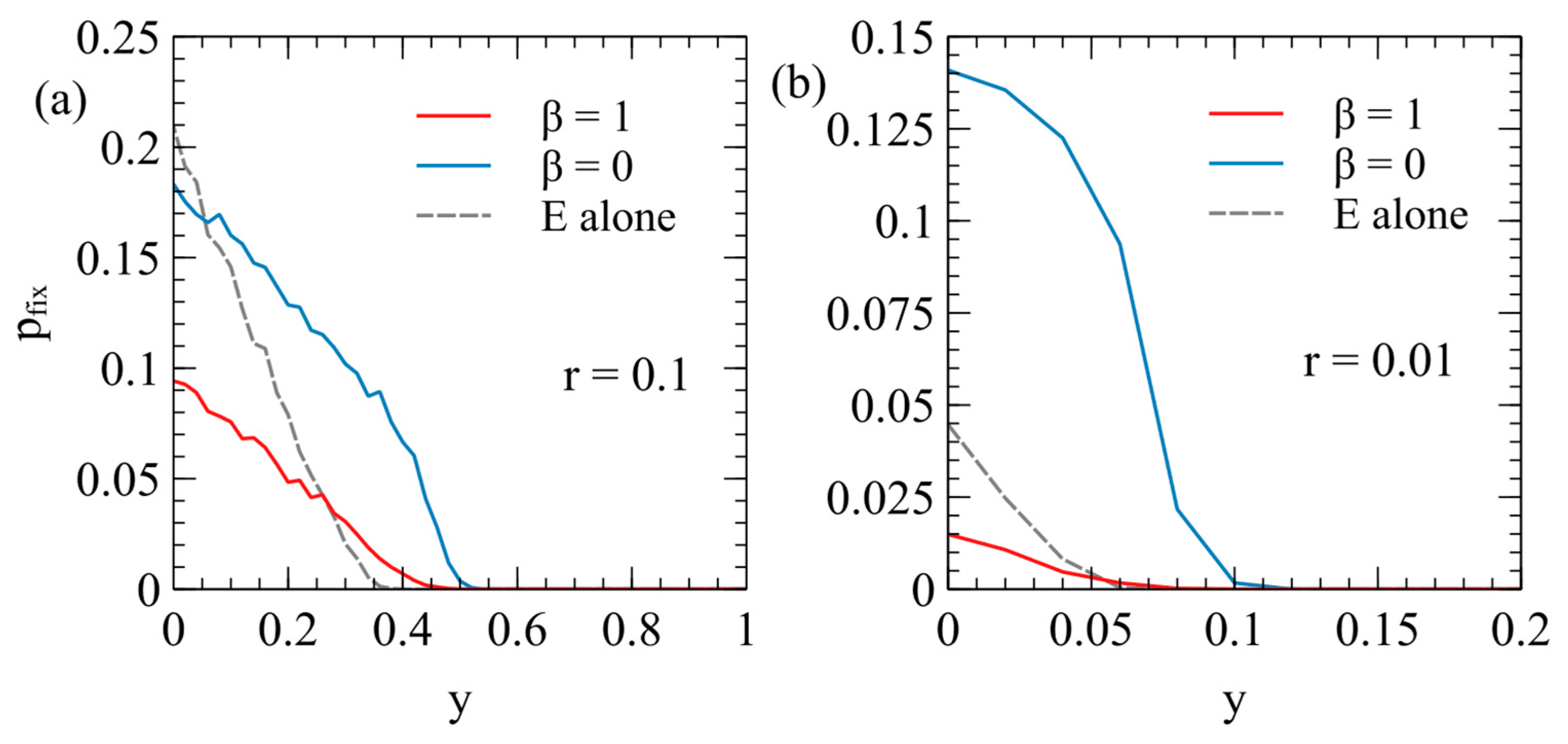
The case that seems most relevant for real endogenous viruses is when the ERV is almost harmless to the host. We therefore consider the fixation probability for an ERV with
as a function of the virulence
of the XRV that is present. For each data point in
Figure 8, a value of
is chosen together with the corresponding maternal transmission rate
from the points on
Figure 2a.
Figure 8a has the transposition rate
. When
, the fixation probability with
is about twice that with
(this case is the same as the point
in
Figure 6a). For
,
decreases steadily with
, while for
,
is largest in the range
= 0.5–0.8. When
, the XRV has no effect on fitness, so the fixation probabilities for
and
are both equal to the fixation probability in absence of an XRV. The shape of the curves in
Figure 8a can be explained by reference to
Figure 2b, where we calculated the relative fitness values
and
for uninfected and infected individuals. When
, type E individuals have the death rate
. The fitness of these individuals relative to the average population shortly after introduction of the ERV is
; therefore, the fixation probability for
in
Figure 8a follows a similar shape to
in
Figure 2b. For
, most of the individuals with ERV genes are type EX and have the death rate
; therefore, the fixation probability for
in
Figure 8a follows a similar shape to
in
Figure 2b.
Figure 8b considers the limit of a zero transposition rate. In this case,
is very small for all values of
when
, but is still quite large when
. A single copy of a non-transposable ERV behaves like a resistance gene whose advantage is proportional to the relative fitness of an uninfected individual with to respect to the mean population fitness. The fixation probability for
therefore has a similar trend to the
curve in
Figure 2b.
If the ERV has
, then it must have changed in some way from the XRV in order to be less harmful to the host. Mutations which limit the harmful effect of an ERV are likely to evolve because this increases the spread rate of the ERV. However, such mutations may not have occurred in the early stages after the introduction of the ERV. Therefore, in
Figure 9 we consider a case where the virulence of the ERV is the same as the virulence of the XRV from which it originates,
. Even so, the ERV and XRV are different in this case, because the XRV has horizontal and maternal transmission, whereas the ERV is transmitted genetically.
Figure 9 shows the fixation probabilities as a function of
in the case
. The fixation probabilities for both
and
are higher than the case when the ERV is alone. This is because type E individuals are competing against the average of the rest of the population, and many of these individuals are type X. By contrast, when the ERV spreads alone, all the competing individuals are type 0.
Figure 9b shows that, for smaller transposition rates,
is lower for
than for
, while the reverse was true in
Figure 6 and
Figure 8. In other words, when
, the ERV has a lower chance of fixation if it confers resistance the the XRV than if it does not. This can be understood by noting that, when
, as type E individuals increase, the fraction of individuals that are susceptible to the XRV decreases, and the XRV infection tends to be eliminated (clearing); so to reach fixation, the type E individuals have to compete against uninfected individuals. Whereas, when
, type E individuals are competing against mostly type X individuals, which have the same fitness as themselves in this case.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}