
Coronavirus Replication: Genomes, Subgenomic RNAs, and Defective Viral Genomes


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Human Coronaviruses
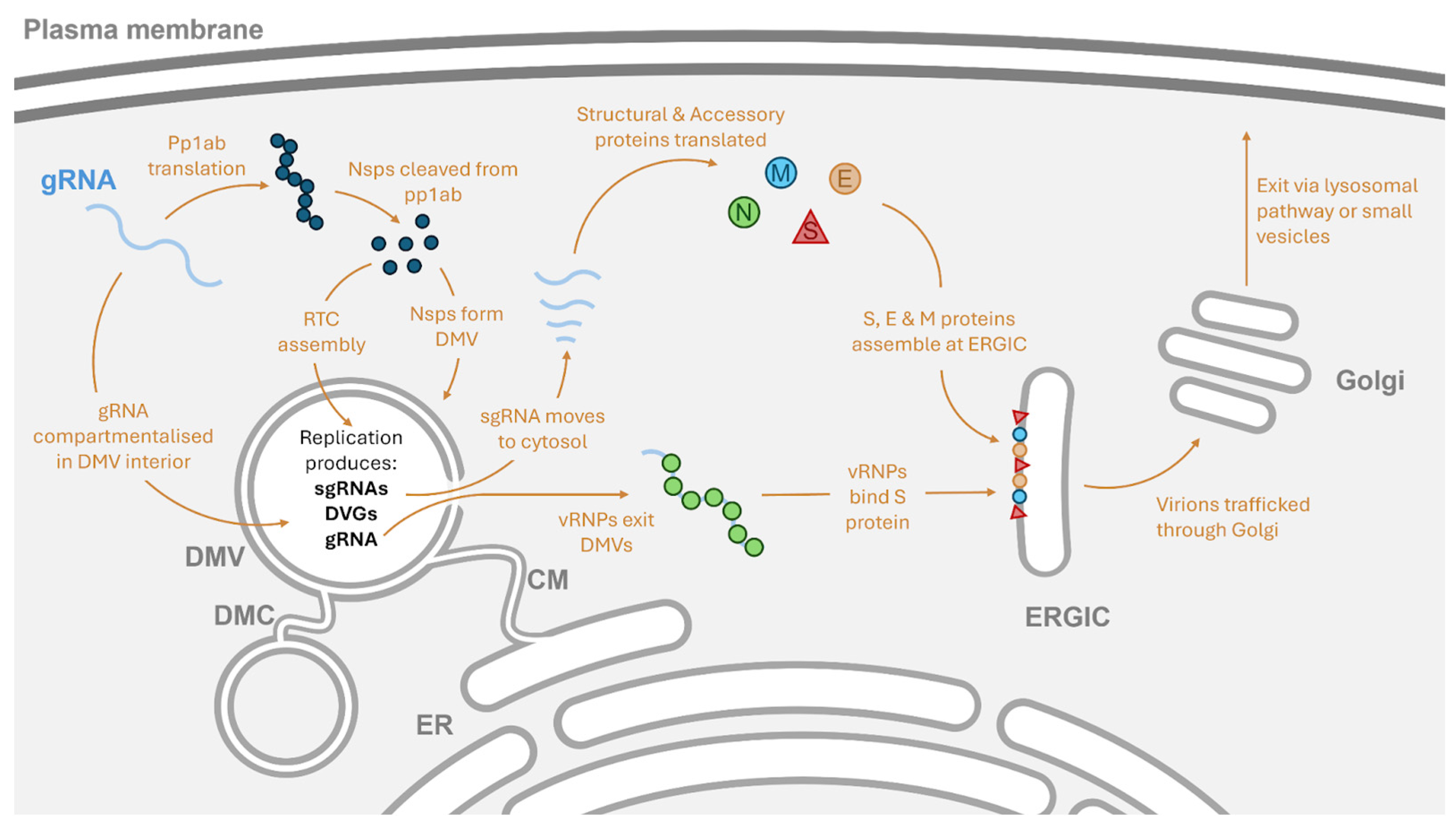
2. Subcellular Location of Coronavirus Replication
3. The Coronavirus Replication Complex
4. The Initiation of Genomic RNA Replication
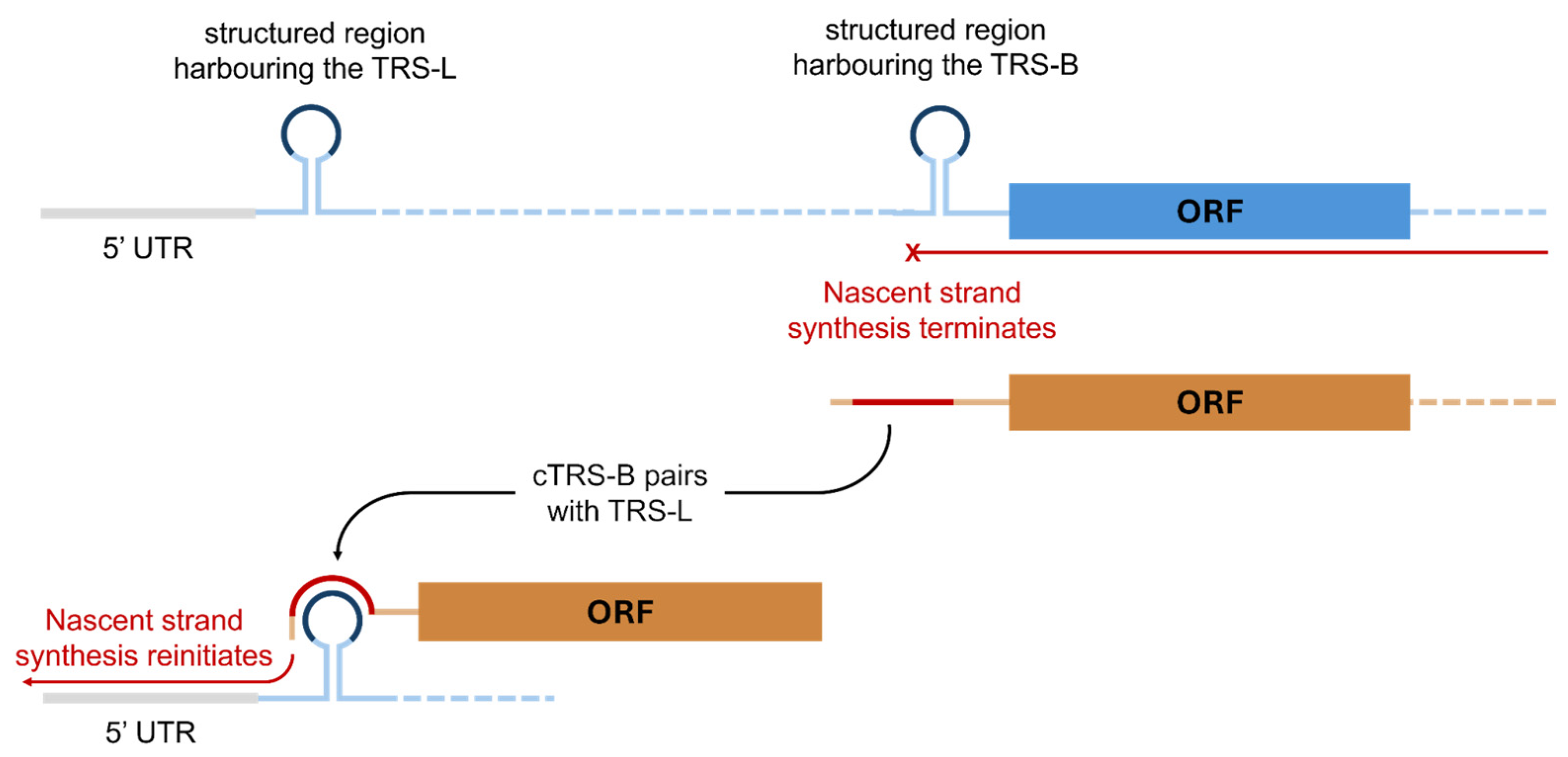
5. The Synthesis of Genomic RNA and Subgenomic RNA
6. Defective Viral Genomes
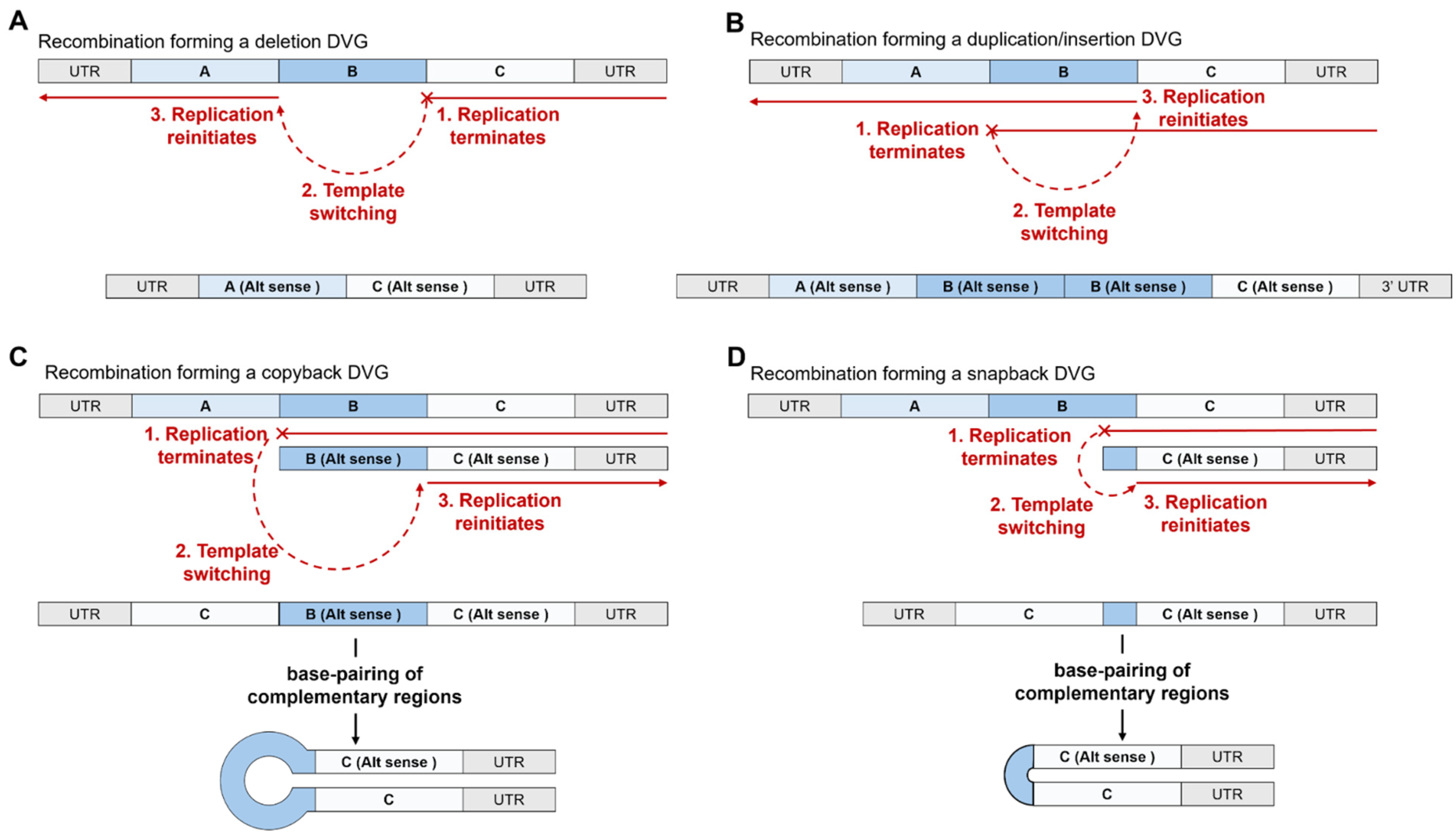
6.1. The Types of Defective Viral Genome Observed in Coronaviruses
6.2. The Impact of Coronaviral DVGs on Coronavirus Replication and Hosts
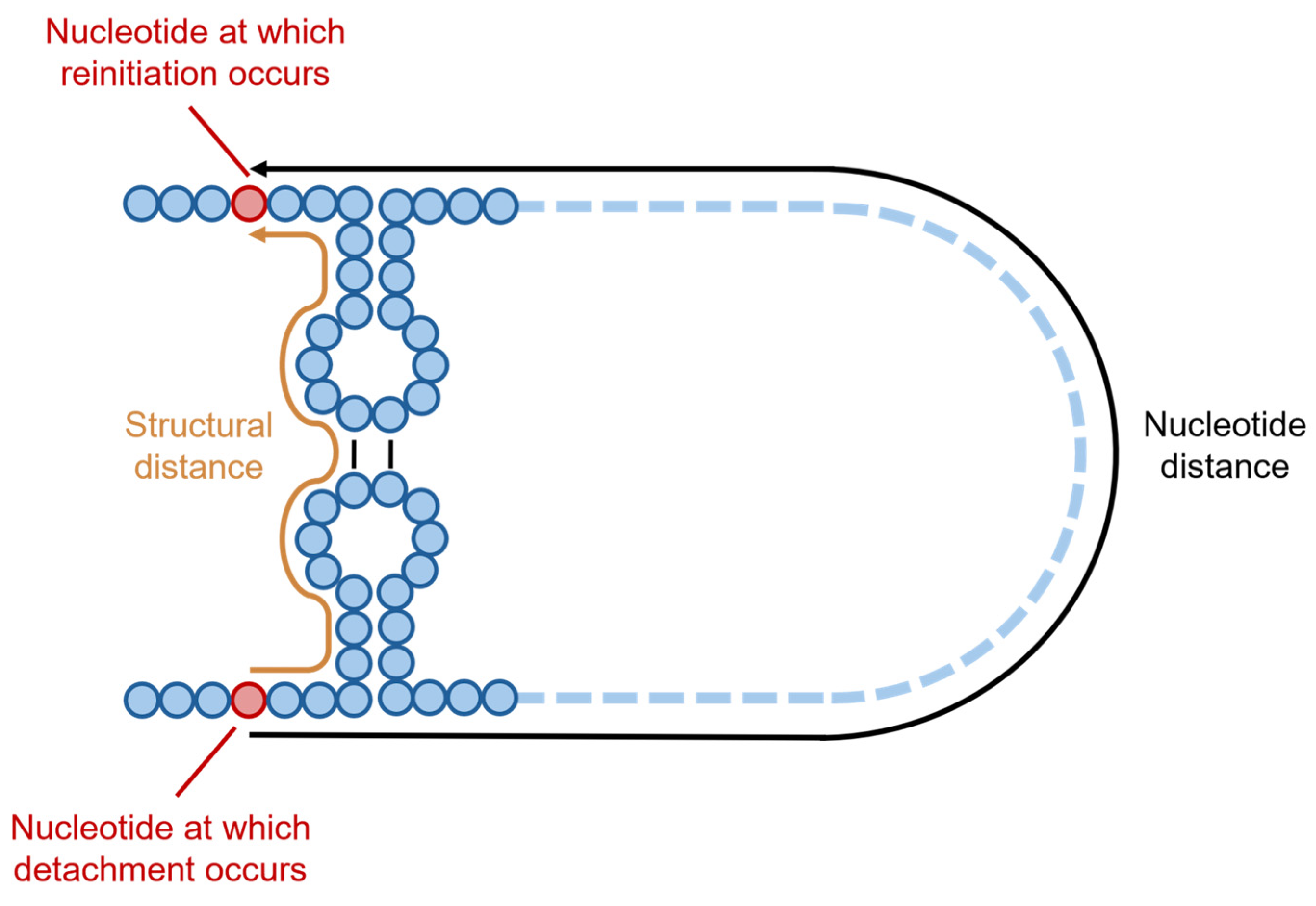
6.3. Mechanisms and Factors Underlying the Formation of DVGs in Coronaviruses
6.4. Propagation and Packaging of Coronavirus RNAs
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HCoV | Human coronavirus |
| MERS-CoV | Middle Eastern respiratory syndrome coronavirus |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | Severe acute respiratory syndrome 2 coronavirus |
| CoV | Coronavirus |
| RTC | Replication/transcription complex |
| gRNA | Genomic RNA |
| ORF | Open reading frame |
| Pp | Polyprotein |
| Nsp | Non-structural protein |
| sgRNA | Subgenomic RNA |
| DMVs | Double-membrane vesicles |
| DMSs | Double-membrane spherules |
| CMs | Convoluted membranes |
| MHV | Mouse hepatitis virus |
| DMCs | Double-membrane connectors |
| PLpro | Papain-like protease |
| Mpro | Main protease |
| RdRp | RNA-dependent RNA polymerase |
| UTR | Untranslated region |
| BCoV | Bovine coronavirus |
| DVG | Defective viral genome |
| TRS | Transcriptional regulatory site |
| TRS-L | Transcriptional regulatory leader site |
| TRS-B | Transcriptional regulatory body site |
| TGEV | Transmissible gastroenteritis virus |
| N | Nucleocapsid |
| E | Envelope |
| M | Membrane |
| S | Spike |
| HE | Haemagglutinin esterase |
| HIV-1 | Human immunodeficiency virus-1 |
| IBV | Infectious bronchitis virus |
| NGS | Next-generation sequencing |
| PHLE | Primary human lung epithelial |
| IFN | Interferon |
| IAV | Influenza A virus |
| MOI | Multiplicity of infection |
| COMRADES | Crosslinking of matched RNAs and deep sequencing |
| vRNP | Viral ribonucleoprotein |
| LLPS | Liquid–liquid phase separation |
| ERGIC | Endoplasmic reticulum–Golgi intermediate compartment |
| ER | Endoplasmic reticulum |
| MHC | Major histocompatibility complex |
References
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Curtis Hendrickson, R.; et al. Recent Changes to Virus Taxonomy Ratified by the International Committee on Taxonomy of Viruses. Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Kesheh, M.M.; Hosseini, P.; Soltani, S.; Zandi, M. An Overview on the Seven Pathogenic Human Coronaviruses. Rev. Med. Virol. 2022, 32, e2282. [Google Scholar] [CrossRef] [PubMed]
- Brucková, M.; McIntosh, K.; Kapikian, A.Z.; Chanock, R.M. The Adaptation of Two Human Coronavirus Strains (OC38 and OC43) to Growth in Cell Monolayers. Proc. Soc. Exp. Biol. Med. 1970, 135, 431–435. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef]
- Chen, B.; Tian, E.-K.; He, B.; Tian, L.; Han, R.; Wang, S.; Xiang, Q.; Zhang, S.; El Arnaout, T.; Cheng, W. Overview of Lethal Human Coronaviruses. Signal Transduct. Target. Ther. 2020, 5, 89. [Google Scholar] [CrossRef]
- Subissi, L.; Imbert, I.; Ferron, F.; Collet, A.; Coutard, B.; Decroly, E.; Canard, B. SARS-CoV ORF1b-Encoded Nonstructural Proteins 12–16: Replicative Enzymes as Antiviral Targets. Antivir. Res. 2014, 101, 122–130. [Google Scholar] [CrossRef]
- Nagy, P.D.; Strating, J.R.P.M.; van Kuppeveld, F.J.M. Building Viral Replication Organelles: Close Encounters of the Membrane Types. PLoS Pathog. 2016, 12, e1005912. [Google Scholar] [CrossRef]
- Snijder, E.J.; Limpens, R.W.A.L.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.F.G.A.; Koster, A.J.; Bárcena, M. A Unifying Structural and Functional Model of the Coronavirus Replication Organelle: Tracking down RNA Synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; van den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.M.; Reggiori, F. Qualitative and Quantitative Ultrastructural Analysis of the Membrane Rearrangements Induced by Coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef]
- de Wilde, A.H.; Raj, V.S.; Oudshoorn, D.; Bestebroer, T.M.; van Nieuwkoop, S.; Limpens, R.W.A.L.; Posthuma, C.C.; van der Meer, Y.; Bárcena, M.; Haagmans, B.L.; et al. MERS-Coronavirus Replication Induces Severe in Vitro Cytopathology and is Strongly Inhibited by Cyclosporin A or Interferon-α Treatment. J. Gen. Virol. 2013, 94, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA Replication of Mouse Hepatitis Virus Takes Place at Double-Membrane Vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A Molecular Pore Spans the Double Membrane of the Coronavirus Replication Organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Serrano, P.; Johnson, M.A.; Almeida, M.S.; Horst, R.; Herrmann, T.; Joseph, J.S.; Neuman, B.W.; Subramanian, V.; Saikatendu, K.S.; Buchmeier, M.J.; et al. Nuclear Magnetic Resonance Structure of the N-Terminal Domain of Nonstructural Protein 3 from the Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2007, 81, 12049–12060. [Google Scholar] [CrossRef]
- Hurst, K.R.; Koetzner, C.A.; Masters, P.S. Characterization of a Critical Interaction between the Coronavirus Nucleocapsid Protein and Nonstructural Protein 3 of the Viral Replicase-Transcriptase Complex. J. Virol. 2013, 87, 9159–9172. [Google Scholar] [CrossRef]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Structure and Replication Characterized by in Situ Cryo-Electron Tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef]
- Zimmermann, L.; Zhao, X.; Makroczyova, J.; Wachsmuth-Melm, M.; Prasad, V.; Hensel, Z.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Nsp3 and Nsp4 Are Minimal Constituents of a Pore Spanning Replication Organelle. Nat. Commun. 2023, 14, 7894. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, T.; Zhong, L.; Zhang, W.; Zhang, Y.; Yu, X.; Yuan, S.; Ni, T. Molecular Architecture of Coronavirus Double-Membrane Vesicle Pore Complex. Nature 2024, 633, 224–231. [Google Scholar] [CrossRef]
- Yang, J.; Tian, B.; Wang, P.; Chen, R.; Xiao, K.; Long, X.; Zheng, X.; Zhu, Y.; Sun, F.; Shi, Y.; et al. SARS-CoV-2 NSP3/4 Control Formation of Replication Organelle and Recruitment of RNA Polymerase NSP12. J. Cell Biol. 2025, 224, e202306101. [Google Scholar] [CrossRef]
- Ricciardi, S.; Guarino, A.M.; Giaquinto, L.; Polishchuk, E.V.; Santoro, M.; Di Tullio, G.; Wilson, C.; Panariello, F.; Soares, V.C.; Dias, S.S.G.; et al. The Role of NSP6 in the Biogenesis of the SARS-CoV-2 Replication Organelle. Nature 2022, 606, 761–768. [Google Scholar] [CrossRef]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The Coding Capacity of SARS-CoV-2. Nature 2021, 589, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Brierley, I.; Digard, P.; Inglis, S.C. Characterization of an Efficient Coronavirus Ribosomal Frameshifting Signal: Requirement for an RNA Pseudoknot. Cell 1989, 57, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-Encoded Proteinases and Proteolytic Processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Thiel, V.; Gorbalenya, A.E. The Autocatalytic Release of a Putative RNA Virus Transcription Factor from Its Polyprotein Precursor Involves Two Paralogous Papain-like Proteases that Cleave the Same Peptide Bond. J. Biol. Chem. 2001, 276, 33220–33232. [Google Scholar] [CrossRef]
- Malone, B.; Chen, J.; Wang, Q.; Llewellyn, E.; Choi, Y.J.; Olinares, P.D.B.; Cao, X.; Hernandez, C.; Eng, E.T.; Chait, B.T.; et al. Structural Basis for Backtracking by the SARS-CoV-2 Replication–Transcription Complex. Proc. Natl. Acad. Sci. USA 2021, 118, e2102516118. [Google Scholar] [CrossRef]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 Proteases for COVID-19 Antiviral Development. Front. Chem. 2022, 9, 819165. [Google Scholar] [CrossRef]
- Posthuma, C.C.; te Velthuis, A.J.W.; Snijder, E.J. Nidovirus RNA Polymerases: Complex Enzymes Handling Exceptional RNA Genomes. Virus Res. 2017, 234, 58–73. [Google Scholar] [CrossRef]
- te Velthuis, A.J.W.; van den Worm, S.H.E.; Snijder, E.J. The SARS-Coronavirus nsp7+nsp8 Complex is a Unique Multimeric RNA Polymerase Capable of Both de Novo Initiation and Primer Extension. Nucleic Acids Res. 2012, 40, 1737–1747. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of Replicating SARS-CoV-2 Polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Pizzato, M.; Baraldi, C.; Boscato Sopetto, G.; Finozzi, D.; Gentile, C.; Gentile, M.D.; Marconi, R.; Paladino, D.; Raoss, A.; Riedmiller, I.; et al. SARS-CoV-2 and the Host Cell: A Tale of Interactions. Front. Virol. 2022, 1, 815388. [Google Scholar] [CrossRef]
- Goebel, S.J.; Hsue, B.; Dombrowski, T.F.; Masters, P.S. Characterization of the RNA Components of a Putative Molecular Switch in the 3′ Untranslated Region of the Murine Coronavirus Genome. J. Virol. 2004, 78, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Hsue, B.; Hartshorne, T.; Masters, P.S. Characterization of an Essential RNA Secondary Structure in the 3′ Untranslated Region of the Murine Coronavirus Genome. J. Virol. 2000, 74, 6911–6921. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Lai, M.M.C. Polypyrimidine Tract-Binding Protein Binds to the Complementary Strand of the Mouse Hepatitis Virus 3′ Untranslated Region, Thereby Altering RNA Conformation. J. Virol. 1999, 73, 9110–9116. [Google Scholar] [CrossRef]
- Williams, G.D.; Chang, R.-Y.; Brian, D.A. A Phylogenetically Conserved Hairpin-Type 3′ Untranslated Region Pseudoknot Functions in Coronavirus RNA Replication. J. Virol. 1999, 73, 8349–8355. [Google Scholar] [CrossRef]
- Züst, R.; Miller, T.B.; Goebel, S.J.; Thiel, V.; Masters, P.S. Genetic Interactions between an Essential 3′ Cis-Acting RNA Pseudoknot, Replicase Gene Products, and the Extreme 3′ End of the Mouse Coronavirus Genome. J. Virol. 2008, 82, 1214–1228. [Google Scholar] [CrossRef]
- Chen, S.-C.; Olsthoorn, R.C.L. Group-Specific Structural Features of the 5′-Proximal Sequences of Coronavirus Genomic RNAs. Virology 2010, 401, 29–41. [Google Scholar] [CrossRef]
- Miao, Z.; Tidu, A.; Eriani, G.; Martin, F. Secondary Structure of the SARS-CoV-2 5′-UTR. RNA Biol. 2021, 18, 447–456. [Google Scholar] [CrossRef]
- Liu, P.; Li, L.; Millership, J.J.; Kang, H.; Leibowitz, J.L.; Giedroc, D.P. A U-Turn Motif-Containing Stem-Loop in the Coronavirus 5′ Untranslated Region Plays a Functional Role in Replication. RNA 2007, 13, 763–780. [Google Scholar] [CrossRef]
- Raman, S.; Bouma, P.; Williams, G.D.; Brian, D.A. Stem-Loop III in the 5′ Untranslated Region Is a Cis-Acting Element in Bovine Coronavirus Defective Interfering RNA Replication. J. Virol. 2003, 77, 6720–6730. [Google Scholar] [CrossRef]
- Raman, S.; Brian, D.A. Stem-Loop IV in the 5′ Untranslated Region Is a Cis-Acting Element in Bovine Coronavirus Defective Interfering RNA Replication. J. Virol. 2005, 79, 12434–12446. [Google Scholar] [CrossRef]
- Escors, D.; Izeta, A.; Capiscol, C.; Enjuanes, L. Transmissible Gastroenteritis Coronavirus Packaging Signal Is Located at the 5′ End of the Virus Genome. J. Virol. 2003, 77, 7890–7902. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the Largest RNA Virus Genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Sola, I.; Moreno, J.L.; Zúñiga, S.; Alonso, S.; Enjuanes, L. Role of Nucleotides Immediately Flanking the Transcription-Regulating Sequence Core in Coronavirus Subgenomic mRNA Synthesis. J. Virol. 2005, 79, 2506–2516. [Google Scholar] [CrossRef]
- Pasternak, A.O.; van den Born, E.; Spaan, W.J.M.; Snijder, E.J. Sequence Requirements for RNA Strand Transfer during Nidovirus Discontinuous Subgenomic RNA Synthesis. EMBO J. 2001, 20, 7220–7228. [Google Scholar] [CrossRef]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef]
- Moreno, J.L.; Zúñiga, S.; Enjuanes, L.; Sola, I. Identification of a Coronavirus Transcription Enhancer. J. Virol. 2008, 82, 3882–3893. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Morales, L.; Zuñiga, S.; Enjuanes, L.; Sola, I. Long-Distance RNA-RNA Interactions in the Coronavirus Genome Form High-Order Structures Promoting Discontinuous RNA Synthesis during Transcription. J. Virol. 2013, 87, 177–186. [Google Scholar] [CrossRef]
- Ziv, O.; Price, J.; Shalamova, L.; Kamenova, T.; Goodfellow, I.; Weber, F.; Miska, E.A. The Short- and Long-Range RNA-RNA Interactome of SARS-CoV-2. Mol. Cell 2020, 80, 1067–1077.e5. [Google Scholar] [CrossRef]
- Wu, C.-H.; Chen, P.-J.; Yeh, S.-H. Nucleocapsid Phosphorylation and RNA Helicase DDX1 Recruitment Enables Coronavirus Transition from Discontinuous to Continuous Transcription. Cell Host Microbe 2014, 16, 462–472. [Google Scholar] [CrossRef]
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.W.A.L.; van der Meer, Y.; Caly, L.; Druce, J.; de Vries, J.J.C.; Kikkert, M.; Bárcena, M.; et al. SARS-Coronavirus-2 Replication in Vero E6 Cells: Replication Kinetics, Rapid Adaptation and Cytopathology. J. Gen. Virol. 2020, 101, 925–940. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, A.; Feng, J.; Li, G.; Guo, D.; Sajid, M.; Wu, K.; Zhang, Q.; Ponty, Y.; Will, S.; et al. The SARS-CoV-2 Subgenome Landscape and its Novel Regulatory Features. Mol. Cell 2021, 81, 2135–2147.e5. [Google Scholar] [CrossRef] [PubMed]
- de Haan, C.A.M.; Volders, H.; Koetzner, C.A.; Masters, P.S.; Rottier, P.J.M. Coronaviruses Maintain Viability despite Dramatic Rearrangements of the Strictly Conserved Genome Organization. J. Virol. 2002, 76, 12491–12502. [Google Scholar] [CrossRef] [PubMed]
- de Groot, R.J. Structure, Function and Evolution of the Hemagglutinin-Esterase Proteins of Corona- and Toroviruses. Glycoconj. J. 2006, 23, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, T.; Arya, S.; Chan, S.-H.; Qi, S.; Dai, N.; Misra, A.; Park, J.-G.; Oladunni, F.; Kovalskyy, D.; Hromas, R.A.; et al. Structural Basis of RNA Cap Modification by SARS-CoV-2. Nat. Commun. 2020, 11, 3718. [Google Scholar] [CrossRef]
- Wu, H.-Y.; Ke, T.-Y.; Liao, W.-Y.; Chang, N.-Y. Regulation of Coronaviral Poly(A) Tail Length during Infection. PLoS ONE 2013, 8, e70548. [Google Scholar] [CrossRef]
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.-M. SARS-CoV-2: From its Discovery to Genome Structure, Transcription, and Replication. Cell Biosci. 2021, 11, 136. [Google Scholar] [CrossRef]
- Tvarogová, J.; Madhugiri, R.; Bylapudi, G.; Ferguson, L.J.; Karl, N.; Ziebuhr, J. Identification and Characterization of a Human Coronavirus 229E Nonstructural Protein 8-Associated RNA 3′-Terminal Adenylyltransferase Activity. J. Virol. 2019, 93, e00291-19. [Google Scholar] [CrossRef]
- Peng, Y.-H.; Lin, C.-H.; Lin, C.-N.; Lo, C.-Y.; Tsai, T.-L.; Wu, H.-Y. Characterization of the Role of Hexamer AGUAAA and Poly(A) Tail in Coronavirus Polyadenylation. PLoS ONE 2016, 11, e0165077. [Google Scholar] [CrossRef]
- Dimmock, N.J.; Easton, A.J. Defective Interfering Influenza Virus RNAs: Time to Reevaluate Their Clinical Potential as Broad-Spectrum Antivirals? J. Virol. 2014, 88, 5217–5227. [Google Scholar] [CrossRef]
- Genoyer, E.; López, C.B. The Impact of Defective Viruses on Infection and Immunity. Annu. Rev. Virol. 2019, 6, 547–566. [Google Scholar] [CrossRef]
- Peccoud, J.; Lequime, S.; Moltini-Conclois, I.; Giraud, I.; Lambrechts, L.; Gilbert, C. A Survey of Virus Recombination Uncovers Canonical Features of Artificial Chimeras Generated During Deep Sequencing Library Preparation. G3 Genes/Genomes/Genet. 2018, 8, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Duhaut, S.D.; Dimmock, N.J. Heterologous Protection of Mice from a Lethal Human H1N1 Influenza A Virus Infection by H3N8 Equine Defective Interfering Virus: Comparison of Defective RNA Sequences Isolated from the DI Inoculum and Mouse Lung. Virology 1998, 248, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kousoulas, K.G.; Storz, J. The Hemagglutinin/Esterase Gene of Human Coronavirus Strain OC43: Phylogenetic Relationships to Bovine and Murine Coronaviruses and Influenza C Virus. Virology 1992, 186, 318–323. [Google Scholar] [CrossRef]
- Luytjes, W.; Bredenbeek, P.J.; Noten, A.F.; Horzinek, M.C.; Spaan, W.J. Sequence of Mouse Hepatitis Virus A59 mRNA 2: Indications for RNA Recombination between Coronaviruses and Influenza C Virus. Virology 1988, 166, 415–422. [Google Scholar] [CrossRef]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural Variant Calling: The Long and the Short of it. Genome Biol. 2019, 20, 246. [Google Scholar] [CrossRef]
- Sapoval, N.; Mahmoud, M.; Jochum, M.D.; Liu, Y.; Elworth, R.A.L.; Wang, Q.; Albin, D.; Ogilvie, H.A.; Lee, M.D.; Villapol, S.; et al. SARS-CoV-2 Genomic Diversity and the Implications for qRT-PCR Diagnostics and Transmission. Genome Res. 2021, 31, 635–644. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Vasen, G.; Pablo, M.; Chen, X.; Beutler, N.; Kumar, A.; Tanner, E.; Illouz, S.; Rahgoshay, D.; Burnett, J.; et al. Identification of a Therapeutic Interfering Particle-A Single-Dose SARS-CoV-2 Antiviral Intervention with a High Barrier to Resistance. Cell 2021, 184, 6022–6036.e18. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Beutler, N.; Vasen, G.; Pablo, M.; Chen, X.; Calia, G.; Buie, L.; Rodick, R.; Smith, D.; Rogers, T.; et al. A Single-Administration Therapeutic Interfering Particle Reduces SARS-CoV-2 Viral Shedding and Pathogenesis in Hamsters. Proc. Natl. Acad. Sci. USA 2022, 119, e2204624119. [Google Scholar] [CrossRef]
- Xiao, Y.; Lidsky, P.V.; Shirogane, Y.; Aviner, R.; Wu, C.-T.; Li, W.; Zheng, W.; Talbot, D.; Catching, A.; Doitsh, G.; et al. A Defective Viral Genome Strategy Elicits Broad Protective Immunity against Respiratory Viruses. Cell 2021, 184, 6037–6051.e14. [Google Scholar] [CrossRef]
- Yao, S.; Narayanan, A.; Majowicz, S.A.; Jose, J.; Archetti, M. A Synthetic Defective Interfering SARS-CoV-2. PeerJ 2021, 9, e11686. [Google Scholar] [CrossRef]
- Routh, A.; Johnson, J.E. Discovery of Functional Genomic Motifs in Viruses with ViReMa–A Virus Recombination Mapper–for Analysis of next-Generation Sequencing Data. Nucleic Acids Res. 2014, 42, e11. [Google Scholar] [CrossRef] [PubMed]
- Sotcheff, S.; Zhou, Y.; Yeung, J.; Sun, Y.; Johnson, J.E.; Torbett, B.E.; Routh, A.L. ViReMa: A Virus Recombination Mapper of next-Generation Sequencing Data Characterizes Diverse Recombinant Viral Nucleic Acids. GigaScience 2023, 12, giad009. [Google Scholar] [CrossRef] [PubMed]
- Beauclair, G.; Mura, M.; Combredet, C.; Tangy, F.; Jouvenet, N.; Komarova, A.V. DI-Tector: Defective Interfering Viral Genomes’ Detector for next-Generation Sequencing Data. RNA 2018, 24, 1285–1296. [Google Scholar] [CrossRef]
- Sun, Y.; Kim, E.J.; Felt, S.A.; Taylor, L.J.; Agarwal, D.; Grant, G.R.; López, C.B. A Specific Sequence in the Genome of Respiratory Syncytial Virus Regulates the Generation of Copy-Back Defective Viral Genomes. PLoS Pathog. 2019, 15, e1007707. [Google Scholar] [CrossRef]
- Achouri, E.; Felt, S.A.; Hackbart, M.; Rivera-Espinal, N.S.; López, C.B. VODKA2: A Fast and Accurate Method to Detect Non-Standard Viral Genomes from Large RNA-Seq Datasets. RNA 2023, 30, 16–25. [Google Scholar] [CrossRef]
- Boussier, J.; Munier, S.; Achouri, E.; Meyer, B.; Crescenzo-Chaigne, B.; Behillil, S.; Enouf, V.; Vignuzzi, M.; van der Werf, S.; Naffakh, N. RNA-Seq Accuracy and Reproducibility for the Mapping and Quantification of Influenza Defective Viral Genomes. RNA 2020, 26, 1905–1918. [Google Scholar] [CrossRef]
- Olmo-Uceda, M.J.; Muñoz-Sánchez, J.C.; Lasso-Giraldo, W.; Arnau, V.; Díaz-Villanueva, W.; Elena, S.F. DVGfinder: A Metasearch Tool for Identifying Defective Viral Genomes in RNA-Seq Data. Viruses 2022, 14, 1114. [Google Scholar] [CrossRef]
- Snijder, E.J.; den Boon, J.A.; Horzinek, M.C.; Spaan, W.J. Characterization of Defective Interfering RNAs of Berne Virus. J. Gen. Virol. 1991, 72, 1635–1643. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Sethna, P.B.; Brian, D.A. Bovine Coronavirus mRNA Replication Continues throughout Persistent Infection in Cell Culture. J. Virol. 1990, 64, 4108–4114. [Google Scholar] [CrossRef]
- Pénzes, Z.; Wroe, C.; Brown, T.D.; Britton, P.; Cavanagh, D. Replication and Packaging of Coronavirus Infectious Bronchitis Virus Defective RNAs Lacking a Long Open Reading Frame. J. Virol. 1996, 70, 8660–8668. [Google Scholar] [CrossRef]
- van der Most, R.G.; Bredenbeek, P.J.; Spaan, W.J. A Domain at the 3′ End of the Polymerase Gene Is Essential for Encapsidation of Coronavirus Defective Interfering RNAs. J. Virol. 1991, 65, 3219–3226. [Google Scholar] [CrossRef] [PubMed]
- Méndez, A.; Smerdou, C.; Izeta, A.; Gebauer, F.; Enjuanes, L. Molecular Characterization of Transmissible Gastroenteritis Coronavirus Defective Interfering Genomes: Packaging and Heterogeneity. Virology 1996, 217, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Gilliam, N.J.; Li, S.; Spandau, S.; Osborn, R.M.; Connor, S.; Anderson, C.S.; Mariani, T.J.; Thakar, J.; Dewhurst, S.; et al. Generation and Functional Analysis of Defective Viral Genomes during SARS-CoV-2 Infection. mBio 2023, 14, e0025023. [Google Scholar] [CrossRef]
- Girgis, S.; Xu, Z.; Oikonomopoulos, S.; Fedorova, A.D.; Tchesnokov, E.P.; Gordon, C.J.; Schmeing, T.M.; Götte, M.; Sonenberg, N.; Baranov, P.V.; et al. Evolution of Naturally Arising SARS-CoV-2 Defective Interfering Particles. Commun. Biol. 2022, 5, 1140. [Google Scholar] [CrossRef]
- Hillung, J.; Olmo-Uceda, M.J.; Muñoz-Sánchez, J.C.; Elena, S.F. Accumulation Dynamics of Defective Genomes during Experimental Evolution of Two Betacoronaviruses. Viruses 2024, 16, 644. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The Coronavirus Proofreading Exoribonuclease Mediates Extensive Viral Recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Molenkamp, R.; Rozier, B.C.D.; Greve, S.; Spaan, W.J.M.; Snijder, E.J. Isolation and Characterization of an Arterivirus Defective Interfering RNA Genome. J. Virol. 2000, 74, 3156–3165. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 Pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired Local Intrinsic Immunity to SARS-CoV-2 Infection in Severe COVID-19. Cell 2021, 184, 4713–4733.e22. [Google Scholar] [CrossRef]
- Cave, D.R.; Hendrickson, F.M.; Huang, A.S. Defective Interfering Virus Particles Modulate Virulence. J. Virol. 1985, 55, 366–373. [Google Scholar] [CrossRef]
- Frensing, T.; Heldt, F.S.; Pflugmacher, A.; Behrendt, I.; Jordan, I.; Flockerzi, D.; Genzel, Y.; Reichl, U. Continuous Influenza Virus Production in Cell Culture Shows a Periodic Accumulation of Defective Interfering Particles. PLoS ONE 2013, 8, e72288. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Zhang, X.; Wu, R.C.; Lai, M.M. The 3′ Untranslated Region of Coronavirus RNA Is Required for Subgenomic mRNA Transcription from a Defective Interfering RNA. J. Virol. 1996, 70, 7236–7240. [Google Scholar] [CrossRef] [PubMed]
- Ranum, J.N.; Ledwith, M.P.; Alnaji, F.G.; Diefenbacher, M.; Orton, R.; Sloan, E.; Güereca, M.; Feltman, E.M.; Smollett, K.; da Silva Filipe, A.; et al. Cryptic Proteins Translated from Deletion-Containing Viral Genomes Dramatically Expand the Influenza Virus Proteome. Nucleic Acids Res. 2024, 52, 3199–3212. [Google Scholar] [CrossRef]
- Ziv, O.; Gabryelska, M.M.; Lun, A.T.L.; Gebert, L.F.R.; Sheu-Gruttadauria, J.; Meredith, L.W.; Liu, Z.-Y.; Kwok, C.K.; Qin, C.-F.; Macrae, I.J.; et al. COMRADES Determines in Vivo RNA Structures and Interactions. Nat. Methods 2018, 15, 785–788. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chen, B.; Chao, D.-Y.; Hsieh, F.-C.; Yang, C.-C.; Hsu, H.-W.; Tam, H.-M.-H.; Wu, H.-Y. Unveiling the Biology of Defective Viral Genomes in Vitro and in Vivo: Implications for Gene Expression and Pathogenesis of Coronavirus. Virol. J. 2023, 20, 225. [Google Scholar] [CrossRef]
- Shapka, N.; Nagy, P.D. The AU-Rich RNA Recombination Hot Spot Sequence of Brome Mosaic Virus is Functional in Tombusviruses: Implications for the Mechanism of RNA Recombination. J. Virol. 2004, 78, 2288–2300. [Google Scholar] [CrossRef]
- Vives, M.C.; Rubio, L.; Sambade, A.; Mirkov, T.E.; Moreno, P.; Guerri, J. Evidence of Multiple Recombination Events between Two RNA Sequence Variants within a Citrus tristeza virus Isolate. Virology 2005, 331, 232–237. [Google Scholar] [CrossRef]
- Ohshima, K.; Tomitaka, Y.; Wood, J.T.; Minematsu, Y.; Kajiyama, H.; Tomimura, K.; Gibbs, A.J. Patterns of Recombination in Turnip Mosaic Virus Genomic Sequences Indicate Hotspots of Recombination. J. Gen. Virol. 2007, 88, 298–315. [Google Scholar] [CrossRef]
- Barr, J.N.; Wertz, G.W. Polymerase Slippage at Vesicular Stomatitis Virus Gene Junctions To Generate Poly(A) Is Regulated by the Upstream 3′-AUAC-5′ Tetranucleotide: Implications for the Mechanism of Transcription Termination. J. Virol. 2001, 75, 6901–6913. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and Functions of Coronavirus Replication–Transcription Complexes and Their Relevance for SARS-CoV-2 Drug Design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- van Marle, G.; van Dinten, L.C.; Spaan, W.J.M.; Luytjes, W.; Snijder, E.J. Characterization of an Equine Arteritis Virus Replicase Mutant Defective in Subgenomic mRNA Synthesis. J. Virol. 1999, 73, 5274–5281. [Google Scholar] [CrossRef] [PubMed]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 Nucleocapsid Protein is Dynamic, Disordered, and Phase Separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef] [PubMed]
- Iserman, C.; Roden, C.A.; Boerneke, M.A.; Sealfon, R.S.G.; McLaughlin, G.A.; Jungreis, I.; Fritch, E.J.; Hou, Y.J.; Ekena, J.; Weidmann, C.A.; et al. Genomic RNA Elements Drive Phase Separation of the SARS-CoV-2 Nucleocapsid. Mol. Cell 2020, 80, 1078–1091.e6. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.S. Coronavirus Genomic RNA Packaging. Virology 2019, 537, 198–207. [Google Scholar] [CrossRef]
- Finke, S.; Conzelmann, K.-K. Virus Promoters Determine Interference by Defective RNAs: Selective Amplification of Mini-RNA Vectors and Rescue from cDNA by a 3′ Copy-Back Ambisense Rabies Virus. J. Virol. 1999, 73, 3818–3825. [Google Scholar] [CrossRef]
- Alnaji, F.G.; Reiser, W.K.; Rivera-Cardona, J.; Te Velthuis, A.J.W.; Brooke, C.B. Influenza A Virus Defective Viral Genomes Are Inefficiently Packaged into Virions Relative to Wild-Type Genomic RNAs. mBio 2021, 12, e0295921. [Google Scholar] [CrossRef]
- Zhang, Z.; Nomura, N.; Muramoto, Y.; Ekimoto, T.; Uemura, T.; Liu, K.; Yui, M.; Kono, N.; Aoki, J.; Ikeguchi, M.; et al. Structure of SARS-CoV-2 Membrane Protein Essential for Virus Assembly. Nat. Commun. 2022, 13, 4399. [Google Scholar] [CrossRef]
- Fan, S.; Sun, W.; Fan, L.; Wu, N.; Sun, W.; Ma, H.; Chen, S.; Li, Z.; Li, Y.; Zhang, J.; et al. The Highly Conserved RNA-Binding Specificity of Nucleocapsid Protein Facilitates the Identification of Drugs with Broad Anti-Coronavirus Activity. Comput. Struct. Biotechnol. J. 2022, 20, 5040–5044. [Google Scholar] [CrossRef]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2N Protein Suggests a Biophysical Basis for Its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e4. [Google Scholar] [CrossRef]
- Mendonça, L.; Howe, A.; Gilchrist, J.B.; Sheng, Y.; Sun, D.; Knight, M.L.; Zanetti-Domingues, L.C.; Bateman, B.; Krebs, A.-S.; Chen, L.; et al. Correlative Multi-Scale Cryo-Imaging Unveils SARS-CoV-2 Assembly and Egress. Nat. Commun. 2021, 12, 4629. [Google Scholar] [CrossRef]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e14. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Post-Translational Modifications of Coronavirus Proteins: Roles and Function. Future Virol. 2018, 13, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Eymieux, S.; Uzbekov, R.; Rouillé, Y.; Blanchard, E.; Hourioux, C.; Dubuisson, J.; Belouzard, S.; Roingeard, P. Secretory Vesicles are the Principal Means of SARS-CoV-2 Egress. Cells 2021, 10, 2047. [Google Scholar] [CrossRef] [PubMed]
- Eymieux, S.; Rouillé, Y.; Terrier, O.; Seron, K.; Blanchard, E.; Rosa-Calatrava, M.; Dubuisson, J.; Belouzard, S.; Roingeard, P. Ultrastructural Modifications Induced by SARS-CoV-2 in Vero Cells: A Kinetic Analysis of Viral Factory Formation, Viral Particle Morphogenesis and Virion Release. Cell. Mol. Life Sci. CMLS 2021, 78, 3565–3576. [Google Scholar] [CrossRef]
- Watts, C. The Endosome–Lysosome Pathway and Information Generation in the Immune System. Biochim. Biophys. Acta 2012, 1824, 14–21. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, R.; Hales, J.; Collier, W.; Gould, P. Coronavirus Replication: Genomes, Subgenomic RNAs, and Defective Viral Genomes. Viruses 2025, 17, 767. https://doi.org/10.3390/v17060767
Williams R, Hales J, Collier W, Gould P. Coronavirus Replication: Genomes, Subgenomic RNAs, and Defective Viral Genomes. Viruses. 2025; 17(6):767. https://doi.org/10.3390/v17060767
Chicago/Turabian StyleWilliams, Rory, Jack Hales, William Collier, and Phillip Gould. 2025. "Coronavirus Replication: Genomes, Subgenomic RNAs, and Defective Viral Genomes" Viruses 17, no. 6: 767. https://doi.org/10.3390/v17060767
APA StyleWilliams, R., Hales, J., Collier, W., & Gould, P. (2025). Coronavirus Replication: Genomes, Subgenomic RNAs, and Defective Viral Genomes. Viruses, 17(6), 767. https://doi.org/10.3390/v17060767