
The Function of TRIM25 in Antiviral Defense and Viral Immune Evasion


Abstract
1. Introduction
2. General Structure and Gene Regulation of TRIM25
2.1. Gene Regulation of TRIM25
2.2. Structural Insights into and Higher-Order Conformation of the RBCC Domain
2.3. The Structure of the SPRY/PRY Domain and Its Protein Interaction and RNA-Binding Ability
3. Biological Functions of TRIM25
3.1. Basic Functions of TRIM25
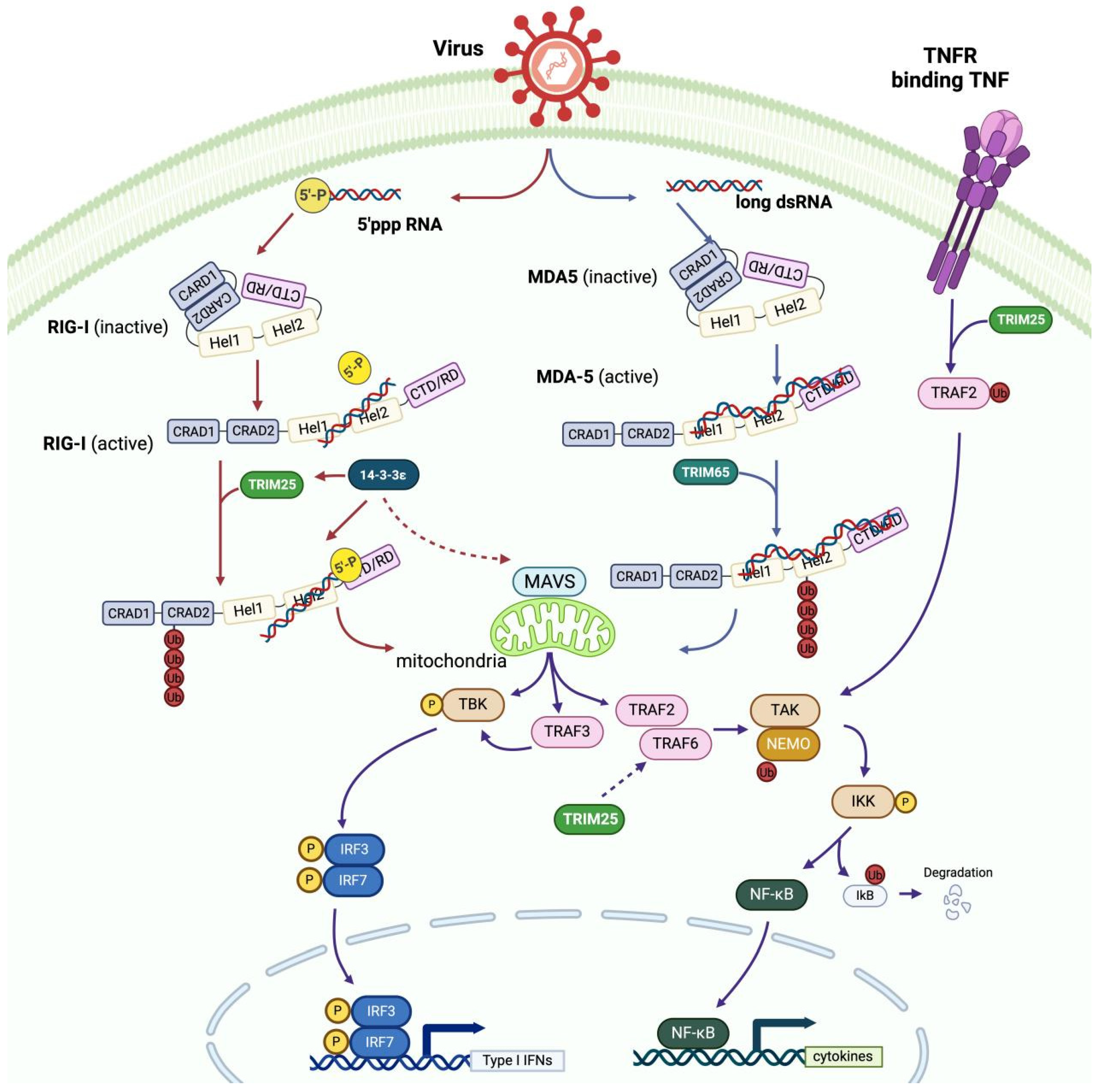
3.2. TRIM25 Is Targeted to Inhibit RIG-I Signaling
3.3. TRIM25 Regulates Viral RNAs for Immune Evasion
4. Interaction Between TRIM25 and Antiviral Host Factors
4.1. TRIM25 Facilitates the Production of IFNs and Cytokines, Mainly Through RIG-I/MDA5 Signaling
4.2. TRIM25 Localizes in Stress Granules During Viral Infection
4.3. TRIM25 Serves as a Co-Factor for the ZAP-Mediated Antiviral Response
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.P.; Haubrich, K.; Perez-Borrajero, C.; Hennig, J. Emerging RNA-binding roles in the TRIM family of ubiquitin ligases. Biol. Chem. 2019, 400, 1443–1464. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.G.; Gao, N.; Yu, H.; et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014, 24, 762–765. [Google Scholar] [CrossRef]
- Sanchez, J.G.; Okreglicka, K.; Chandrasekaran, V.; Welker, J.M.; Sundquist, W.I.; Pornillos, O. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. USA 2014, 111, 2494–2499. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Heikel, G.; Michlewski, G. TRIM25 and its emerging RNA-binding roles in antiviral defense. Wiley Interdiscip. Rev. RNA 2020, 11, e1588. [Google Scholar] [CrossRef]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403.e319. [Google Scholar] [CrossRef]
- Álvarez, L.; Haubrich, K.; Iselin, L.; Gillioz, L.; Ruscica, V.; Lapouge, K.; Augsten, S.; Huppertz, I.; Choudhury, N.R.; Simon, B.; et al. The molecular dissection of TRIM25’s RNA-binding mechanism provides key insights into its antiviral activity. Nat. Commun. 2024, 15, 8485. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Heikel, G.; Trubitsyna, M.; Kubik, P.; Nowak, J.S.; Webb, S.; Granneman, S.; Spanos, C.; Rappsilber, J.; Castello, A.; et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 2017, 15, 105. [Google Scholar] [CrossRef]
- Martín-Vicente, M.; Medrano, L.M.; Resino, S.; García-Sastre, A.; Martínez, I. TRIM25 in the Regulation of the Antiviral Innate Immunity. Front. Immunol. 2017, 8, 1187. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.G.; Sparrer, K.M.J.; Chiang, C.; Reis, R.A.; Chiang, J.J.; Zurenski, M.A.; Wan, Y.; Gack, M.U.; Pornillos, O. TRIM25 Binds RNA to Modulate Cellular Anti-viral Defense. J. Mol. Biol. 2018, 430, 5280–5293. [Google Scholar] [CrossRef]
- Cagliani, R.; Forni, D.; Mozzi, A.; Fuchs, R.; Hagai, T.; Sironi, M. Evolutionary analysis of ZAP and its cofactors identifies intrinsically disordered regions as central elements in host-pathogen interactions. Comput. Struct. Biotechnol. J. 2024, 23, 3143–3154. [Google Scholar] [CrossRef] [PubMed]
- Bohn, J.A.; Meagher, J.L.; Takata, M.A.; Gonçalves-Carneiro, D.; Yeoh, Z.C.; Ohi, M.D.; Smith, J.L.; Bieniasz, P.D. Functional anatomy of zinc finger antiviral protein complexes. Nat. Commun. 2024, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, S.; Tian, X.; Kong, C.; Hong, W.; Xiao, T.; Wang, S.; Wei, Z.; Su, Z.; Ren, H.; et al. The structural basis of TRIM25-mediated regulation of RIG-I. J. Biol. Chem. 2025, 301, 108367. [Google Scholar] [CrossRef]
- Suleman, M.; Sayaf, A.M.; Khan, A.; Khan, S.A.; Albekairi, N.A.; Alshammari, A.; Agouni, A.; Yassine, H.M.; Crovella, S. Molecular screening of phytocompounds targeting the interface between influenza A NS1 and TRIM25 to enhance host immune responses. J. Infect. Public Health 2024, 17, 102448. [Google Scholar] [CrossRef]
- Eberhardt, W.; Nasrullah, U.; Pfeilschifter, J. TRIM25: A Global Player of Cell Death Pathways and Promising Target of Tumor-Sensitizing Therapies. Cells 2025, 14, 65. [Google Scholar] [CrossRef]
- Chiang, C.; Yap, B.K. TRIM25, TRIM28 and TRIM59 and Their Protein Partners in Cancer Signaling Crosstalk: Potential Novel Therapeutic Targets for Cancer. Curr. Issues Mol. Biol. 2024, 46, 10745–10761. [Google Scholar] [CrossRef]
- Rahimi-Tesiye, M.; Zaersabet, M.; Salehiyeh, S.; Jafari, S.Z. The role of TRIM25 in the occurrence and development of cancers and inflammatory diseases. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2023, 1878, 188954. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Abraham-Juarez, M.J.; Solleiro-Villavicencio, H.; Ramirez-Jarquin, J.O. TRIM25: A central factor in breast cancer. World J. Clin. Oncol. 2021, 12, 646–655. [Google Scholar] [CrossRef]
- Nakasato, N.; Ikeda, K.; Urano, T.; Horie-Inoue, K.; Takeda, S.; Inoue, S. A ubiquitin E3 ligase Efp is up-regulated by interferons and conjugated with ISG15. Biochem. Biophys. Res. Commun. 2006, 351, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Heikel, G.; Choudhury, N.R.; Michlewski, G. The role of Trim25 in development, disease and RNA metabolism. Biochem. Soc. Trans. 2016, 44, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, B.S.; Yang, Y.; Li, Y.; Lv, J.L.; Cheng, Y. TRIM25 contributes to the malignancy of acute myeloid leukemia and is negatively regulated by microRNA-137. Open Med. 2021, 16, 95–103. [Google Scholar] [CrossRef]
- Lu, L.F.; Gasteiger, G.; Yu, I.S.; Chaudhry, A.; Hsin, J.P.; Lu, Y.; Bos, P.D.; Lin, L.L.; Zawislak, C.L.; Cho, S.; et al. A Single miRNA-mRNA Interaction Affects the Immune Response in a Context- and Cell-Type-Specific Manner. Immunity 2015, 43, 52–64. [Google Scholar] [CrossRef]
- Wang, J.; Yin, G.; Bian, H.; Yang, J.; Zhou, P.; Yan, K.; Liu, C.; Chen, P.; Zhu, J.; Li, Z.; et al. LncRNA XIST upregulates TRIM25 via negatively regulating miR-192 in hepatitis B virus-related hepatocellular carcinoma. Mol. Med. 2021, 27, 41. [Google Scholar] [CrossRef]
- Vuillier, F.; Li, Z.; Black, I.; Cruciani, M.; Rubino, E.; Michel, F.; Pellegrini, S. IFN-I inducible miR-3614-5p targets ADAR1 isoforms and fine tunes innate immune activation. Front. Immunol. 2022, 13, 939907. [Google Scholar] [CrossRef]
- Li, J.; Xie, Y.; Li, L.; Li, X.; Shen, L.; Gong, J.; Zhang, R. MicroRNA-30a Modulates Type I Interferon Responses to Facilitate Coxsackievirus B3 Replication Via Targeting Tripartite Motif Protein 25. Front. Immunol. 2020, 11, 603437. [Google Scholar] [CrossRef]
- Liu, W.; Jin, Y.; Zhang, W.; Xiang, Y.; Jia, P.; Yi, M.; Jia, K. MiR-202-5p Inhibits RIG-I-Dependent Innate Immune Responses to RGNNV Infection by Targeting TRIM25 to Mediate RIG-I Ubiquitination. Viruses 2020, 12, 261. [Google Scholar] [CrossRef]
- Chen, D.; Ji, Q.; Liu, J.; Cheng, F.; Zheng, J.; Ma, Y.; He, Y.; Zhang, J.; Song, T. MicroRNAs in the Regulation of RIG-I-like Receptor Signaling Pathway: Possible Strategy for Viral Infection and Cancer. Biomolecules 2023, 13, 1344. [Google Scholar] [CrossRef]
- Lee, N.R.; Choi, J.Y.; Yoon, I.H.; Lee, J.K.; Inn, K.S. Positive regulatory role of c-Src-mediated TRIM25 tyrosine phosphorylation on RIG-I ubiquitination and RIG-I-mediated antiviral signaling pathway. Cell Immunol. 2018, 332, 94–100. [Google Scholar] [CrossRef]
- Zou, W.; Wang, J.; Zhang, D.E. Negative regulation of ISG15 E3 ligase EFP through its autoISGylation. Biochem. Biophys. Res. Commun. 2007, 354, 321–327. [Google Scholar] [CrossRef] [PubMed]
- El-Asmi, F.; McManus, F.P.; Thibault, P.; Chelbi-Alix, M.K. Interferon, restriction factors and SUMO pathways. Cytokine Growth Factor. Rev. 2020, 55, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Loo, Y.M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Yla-Anttila, P.; Sandalova, T.; Sun, R.; Achour, A.; Masucci, M.G. 14-3-3 scaffold proteins mediate the inactivation of trim25 and inhibition of the type I interferon response by herpesvirus deconjugases. PLoS Pathog. 2019, 15, e1008146. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, W.; Shi, X.H.; Chang, X.; Han, Y.; Liu, C.; Jiang, Z.; Yang, X. The mechanism of linear ubiquitination in regulating cell death and correlative diseases. Cell Death Dis. 2023, 14, 659. [Google Scholar] [CrossRef]
- Shibata, Y.; Komander, D. LUBAC. Curr. Biol. 2022, 32, R506–R508. [Google Scholar] [CrossRef]
- Oshiumi, H. Recent Advances and Contradictions in the Study of the Individual Roles of Ubiquitin Ligases That Regulate RIG-I-Like Receptor-Mediated Antiviral Innate Immune Responses. Front. Immunol. 2020, 11, 1296. [Google Scholar] [CrossRef]
- Oudshoorn, D.; Versteeg, G.A.; Kikkert, M. Regulation of the innate immune system by ubiquitin and ubiquitin-like modifiers. Cytokine Growth Factor. Rev. 2012, 23, 273–282. [Google Scholar] [CrossRef]
- Horie-Inoue, K.; Inoue, S. Epigenetic and proteolytic inactivation of 14-3-3sigma in breast and prostate cancers. Semin. Cancer Biol. 2006, 16, 235–239. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef]
- Koepke, L.; Gack, M.U.; Sparrer, K.M. The antiviral activities of TRIM proteins. Curr. Opin. Microbiol. 2021, 59, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Koliopoulos, M.G.; Esposito, D.; Christodoulou, E.; Taylor, I.A.; Rittinger, K. Functional role of TRIM E3 ligase oligomerization and regulation of catalytic activity. EMBO J. 2016, 35, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.G.; Chiang, J.J.; Sparrer, K.M.J.; Alam, S.L.; Chi, M.; Roganowicz, M.D.; Sankaran, B.; Gack, M.U.; Pornillos, O. Mechanism of TRIM25 Catalytic Activation in the Antiviral RIG-I Pathway. Cell Rep. 2016, 16, 1315–1325. [Google Scholar] [CrossRef]
- Streich, F.C., Jr.; Ronchi, V.P.; Connick, J.P.; Haas, A.L. Tripartite motif ligases catalyze polyubiquitin chain formation through a cooperative allosteric mechanism. J. Biol. Chem. 2013, 288, 8209–8221. [Google Scholar] [CrossRef]
- Dou, H.; Buetow, L.; Sibbet, G.J.; Cameron, K.; Huang, D.T. Essentiality of a non-RING element in priming donor ubiquitin for catalysis by a monomeric E3. Nat. Struct. Mol. Biol. 2013, 20, 982–986. [Google Scholar] [CrossRef]
- Grütter, C.; Briand, C.; Capitani, G.; Mittl, P.R.; Papin, S.; Tschopp, J.; Grütter, M.G. Structure of the PRYSPRY-domain: Implications for autoinflammatory diseases. FEBS Lett. 2006, 580, 99–106. [Google Scholar] [CrossRef]
- D’Cruz, A.A.; Babon, J.J.; Norton, R.S.; Nicola, N.A.; Nicholson, S.E. Structure and function of the SPRY/B30.2 domain proteins involved in innate immunity. Protein Sci. 2013, 22, 1–10. [Google Scholar] [CrossRef]
- Sanchez-Aparicio, M.T.; Feinman, L.J.; Garcia-Sastre, A.; Shaw, M.L. Paramyxovirus V Proteins Interact with the RIG-I/TRIM25 Regulatory Complex and Inhibit RIG-I Signaling. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef]
- Morita, N.; Tanaka, Y.; Odkhuu, E.; Naiki, Y.; Komatsu, T.; Koide, N. Sendai virus V protein decreases nitric oxide production by inhibiting RIG-I signaling in infected RAW264.7 macrophages. Microbes Infect. 2020, 22, 322–330. [Google Scholar] [CrossRef]
- Tanaka, Y.; Morita, N.; Kitagawa, Y.; Gotoh, B.; Komatsu, T. Human metapneumovirus M2-2 protein inhibits RIG-I signaling by preventing TRIM25-mediated RIG-I ubiquitination. Front. Immunol. 2022, 13, 970750. [Google Scholar] [CrossRef]
- Urano, T.; Saito, T.; Tsukui, T.; Fujita, M.; Hosoi, T.; Muramatsu, M.; Ouchi, Y.; Inoue, S. Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature 2002, 417, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Kang, R.; Tang, D. Cell type-specific induction of ferroptosis to boost antitumor immunity. Oncoimmunology 2023, 12, 2282252. [Google Scholar] [CrossRef]
- Zou, J.; Wang, L.; Tang, H.; Liu, X.; Peng, F.; Peng, C. Ferroptosis in Non-Small Cell Lung Cancer: Progression and Therapeutic Potential on It. Int. J. Mol. Sci. 2021, 22, 13335. [Google Scholar] [CrossRef]
- Liu, Y.; Wan, Y.; Jiang, Y.; Zhang, L.; Cheng, W. GPX4: The hub of lipid oxidation, ferroptosis, disease and treatment. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188890. [Google Scholar] [CrossRef]
- He, Y.M.; Zhou, X.M.; Jiang, S.Y.; Zhang, Z.B.; Cao, B.Y.; Liu, J.B.; Zeng, Y.Y.; Zhao, J.; Mao, X.L. TRIM25 activates AKT/mTOR by inhibiting PTEN via K63-linked polyubiquitination in non-small cell lung cancer. Acta Pharmacol. Sin. 2022, 43, 681–691. [Google Scholar] [CrossRef]
- Yan, H.; Ma, Y.L.; Gui, Y.Z.; Wang, S.M.; Wang, X.B.; Gao, F.; Wang, Y.P. MG132, a proteasome inhibitor, enhances LDL uptake in HepG2 cells in vitro by regulating LDLR and PCSK9 expression. Acta Pharmacol. Sin. 2014, 35, 994–1004. [Google Scholar] [CrossRef]
- Zheng, B.; Qian, F.; Wang, X.; Wang, Y.; Zhou, B.; Fang, L. Neddylation activated TRIM25 desensitizes triple-negative breast cancer to paclitaxel via TFEB-mediated autophagy. J. Exp. Clin. Cancer Res. 2024, 43, 177. [Google Scholar] [CrossRef]
- Nasrullah, U.; Stanke, K.; Recknagel, V.; Bozkurt, S.; Wurzel, P.; Gauer, S.; Imre, G.; Münch, C.; Pfeilschifter, J.; Eberhardt, W. The E3 Ligase TRIM25 Impairs Apoptotic Cell Death in Colon Carcinoma Cells via Destabilization of Caspase-7 mRNA: A Possible Role of hnRNPH1. Cells 2023, 12, 201. [Google Scholar] [CrossRef]
- Nasrullah, U.; Haeussler, K.; Biyanee, A.; Wittig, I.; Pfeilschifter, J.; Eberhardt, W. Identification of TRIM25 as a Negative Regulator of Caspase-2 Expression Reveals a Novel Target for Sensitizing Colon Carcinoma Cells to Intrinsic Apoptosis. Cells 2019, 8, 622. [Google Scholar] [CrossRef]
- Han, Q.; Cheng, P.; Yang, H.; Liang, H.; Lin, F. Altered expression of microRNA-365 is related to the occurrence and development of non-small-cell lung cancer by inhibiting TRIM25 expression. J. Cell Physiol. 2019, 234, 22321–22330. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aparicio, M.T.; Ayllon, J.; Leo-Macias, A.; Wolff, T.; Garcia-Sastre, A. Subcellular Localizations of RIG-I, TRIM25, and MAVS Complexes. J. Virol. 2017, 91, 10-1128. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, K.; Huang, Y.; Sun, M.; Tian, Q.; Zhang, S.; Qin, Y. TRIM25 Promotes TNF-alpha-Induced NF-kappaB Activation through Potentiating the K63-Linked Ubiquitination of TRAF2. J. Immunol. 2020, 204, 1499–1507. [Google Scholar] [CrossRef]
- Khatun, O.; Sharma, M.; Narayan, R.; Tripathi, S. SARS-CoV-2 ORF6 protein targets TRIM25 for proteasomal degradation to diminish K63-linked RIG-I ubiquitination and type-I interferon induction. Cell Mol. Life Sci. 2023, 80, 364. [Google Scholar] [CrossRef]
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef]
- Zheng, X.; Sun, Z.; Yu, L.; Shi, D.; Zhu, M.; Yao, H.; Li, L. Interactome Analysis of the Nucleocapsid Protein of SARS-CoV-2 Virus. Pathogens 2021, 10, 1155. [Google Scholar] [CrossRef]
- Gori Savellini, G.; Anichini, G.; Gandolfo, C.; Cusi, M.G. SARS-CoV-2 N Protein Targets TRIM25-Mediated RIG-I Activation to Suppress Innate Immunity. Viruses 2021, 13, 1439. [Google Scholar] [CrossRef]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Inhibits Type I Interferon Production by Interfering with TRIM25-Mediated RIG-I Ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef]
- Zhao, Y.; Sui, L.; Wu, P.; Wang, W.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q.; Wang, G. A dual-role of SARS-CoV-2 nucleocapsid protein in regulating innate immune response. Signal Transduct. Target. Ther. 2021, 6, 331. [Google Scholar] [CrossRef]
- Oh, S.J.; Shin, O.S. SARS-CoV-2 Nucleocapsid Protein Targets RIG-I-Like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells 2021, 10, 530. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.W.; Xu, Y.; Zhang, J.; Nan, M.L.; Xiao, Y.; Zhan, P.; Liu, X.; et al. SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal Transduct. Target. Ther. 2022, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, J.; Li, J.; Zheng, H.; Li, H.; Lai, Q.; Chen, Y.; Qin, L.; Zuo, Y.; Guo, L.; et al. Engagement of the G3BP2-TRIM25 Interaction by Nucleocapsid Protein Suppresses the Type I Interferon Response in SARS-CoV-2-Infected Cells. Vaccines 2022, 10, 2042. [Google Scholar] [CrossRef]
- Shang, Z.; Zhang, S.; Wang, J.; Zhou, L.; Zhang, X.; Billadeau, D.D.; Yang, P.; Zhang, L.; Zhou, F.; Bai, P.; et al. TRIM25 predominately associates with anti-viral stress granules. Nat. Commun. 2024, 15, 4127. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Trus, I.; Heikel, G.; Wolczyk, M.; Szymanski, J.; Bolembach, A.; Dos Santos Pinto, R.M.; Smith, N.; Trubitsyna, M.; Gaunt, E.; et al. TRIM25 inhibits influenza A virus infection, destabilizes viral mRNA, but is redundant for activating the RIG-I pathway. Nucleic Acids Res. 2022, 50, 7097–7114. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Koliopoulos, M.G.; Lethier, M.; van der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis e Sousa, C.; Cusack, S.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 104502. [Google Scholar] [CrossRef]
- Jiang, J.; Li, J.; Fan, W.; Zheng, W.; Yu, M.; Chen, C.; Sun, L.; Bi, Y.; Ding, C.; Gao, G.F.; et al. Robust Lys63-Linked Ubiquitination of RIG-I Promotes Cytokine Eruption in Early Influenza B Virus Infection. J. Virol. 2016, 90, 6263–6275. [Google Scholar] [CrossRef]
- Galão, R.P.; Wilson, H.; Schierhorn, K.L.; Debeljak, F.; Bodmer, B.S.; Goldhill, D.; Hoenen, T.; Wilson, S.J.; Swanson, C.M.; Neil, S.J.D. TRIM25 and ZAP target the Ebola virus ribonucleoprotein complex to mediate interferon-induced restriction. PLoS Pathog. 2022, 18, e1010530. [Google Scholar] [CrossRef]
- Lin, Y.T.; Chiweshe, S.; McCormick, D.; Raper, A.; Wickenhagen, A.; DeFillipis, V.; Gaunt, E.; Simmonds, P.; Wilson, S.J.; Grey, F. Human cytomegalovirus evades ZAP detection by suppressing CpG dinucleotides in the major immediate early 1 gene. PLoS Pathog. 2020, 16, e1008844. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M.; et al. TRIM25 Enhances the Antiviral Action of Zinc-Finger Antiviral Protein (ZAP). PLoS Pathog. 2017, 13, e1006145. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli, R.; Rahimi, P.; Fateh, A.; Hamidi-Fard, M.; Eaybpoosh, S.; Bahramali, G.; Sadeghi, S.A.; Doroud, D.; Aghasadeghi, M. Exploring the impression of TRIM25 gene expression on COVID-19 severity and SARS-CoV-2 viral replication. J. Infect. Public Health 2024, 17, 102489. [Google Scholar] [CrossRef]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef]
- Goertz, G.P.; Pijlman, G.P. Dengue Non-coding RNA: TRIMmed for Transmission. Cell Host Microbe 2015, 18, 133–134. [Google Scholar] [CrossRef]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Min, J.; Li, Y.; Li, X.; Wang, M.; Li, H.; Bi, Y.; Xu, P.; Liu, W.; Ye, X.; Li, J. The circRNA circVAMP3 restricts influenza A virus replication by interfering with NP and NS1 proteins. PLoS Pathog. 2023, 19, e1011577. [Google Scholar] [CrossRef]
- Diaz-Beneitez, E.; Cubas-Gaona, L.L.; Candelas-Rivera, O.; Benito-Zafra, A.; Sanchez-Aparicio, M.T.; Miorin, L.; Rodriguez, J.F.; Garcia-Sastre, A.; Rodriguez, D. Interaction between chicken TRIM25 and MDA5 and their role in mediated antiviral activity against IBDV infection. Front. Microbiol. 2022, 13, 1068328. [Google Scholar] [CrossRef]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef]
- Okamoto, M.; Kouwaki, T.; Fukushima, Y.; Oshiumi, H. Regulation of RIG-I Activation by K63-Linked Polyubiquitination. Front. Immunol. 2018, 8, 1942. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.-R.; Kim, H.-I.; Choi, M.-S.; Yi, C.-M.; Inn, K.-S. Regulation of MDA5-MAVS Antiviral Signaling Axis by TRIM25 through TRAF6-Mediated NF-κB Activation. Mol. Cells 2015, 38, 759–764. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, R.; Liang, D.; Wang, W.; Min, K.; Luo, T.; Li, X. Uncovering the Interaction between TRAF1 and MAVS in the RIG-I Pathway to Enhance the Upregulation of IRF1/ISG15 during Classical Swine Fever Virus Infection. Cells 2024, 13, 1165. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, Y.; Fu, X.; Bai, G.; Li, X.; Mao, J.; Yan, Y.; Hu, L. Multiple functions of stress granules in viral infection at a glance. Front. Microbiol. 2023, 14, 1138864. [Google Scholar] [CrossRef]
- McVean, G.; Kerns, J.A.; Emerman, M.; Malik, H.S. Positive Selection and Increased Antiviral Activity Associated with the PARP-Containing Isoform of Human Zinc-Finger Antiviral Protein. PLoS Genet. 2008, 4, e21. [Google Scholar] [CrossRef]
- Ficarelli, M.; Neil, S.J.D.; Swanson, C.M. Targeted Restriction of Viral Gene Expression and Replication by the ZAP Antiviral System. Annu. Rev. Virol. 2021, 8, 265–283. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, X.; Tu, F.; Wang, Q.; Fan, Z.; Gao, G.; Diamond, M.S. TRIM25 Is Required for the Antiviral Activity of Zinc Finger Antiviral Protein. J. Virol. 2017, 91, e00088-17. [Google Scholar] [CrossRef]
- Yang, E.; Nguyen, L.P.; Wisherop, C.A.; Kan, R.L.; Li, M.M.H. The Role of ZAP and TRIM25 RNA Binding in Restricting Viral Translation. Front. Cell Infect. Microbiol. 2022, 12, 886929. [Google Scholar] [CrossRef]
- Nchioua, R.; Kmiec, D.; Müller, J.A.; Conzelmann, C.; Groß, R.; Swanson, C.M.; Neil, S.J.D.; Stenger, S.; Sauter, D.; Münch, J.; et al. SARS-CoV-2 Is Restricted by Zinc Finger Antiviral Protein despite Preadaptation to the Low-CpG Environment in Humans. mBio 2020, 11. [Google Scholar] [CrossRef]
- Meyerson, N.R.; Zhou, L.; Guo, Y.R.; Zhao, C.; Tao, Y.J.; Krug, R.M.; Sawyer, S.L. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe 2017, 22, 627–638.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, J.; Pang, X.; Liu, Z.; Li, Q.; Yi, D.; Zhang, Y.; Fang, X.; Zhang, T.; Zhou, R.; et al. An anti-influenza A virus microbial metabolite acts by degrading viral endonuclease PA. Nat. Commun. 2022, 13, 2079. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Viruses | Viral Proteins and/or RNAs | Interactions with TRIM25 | References |
|---|---|---|---|
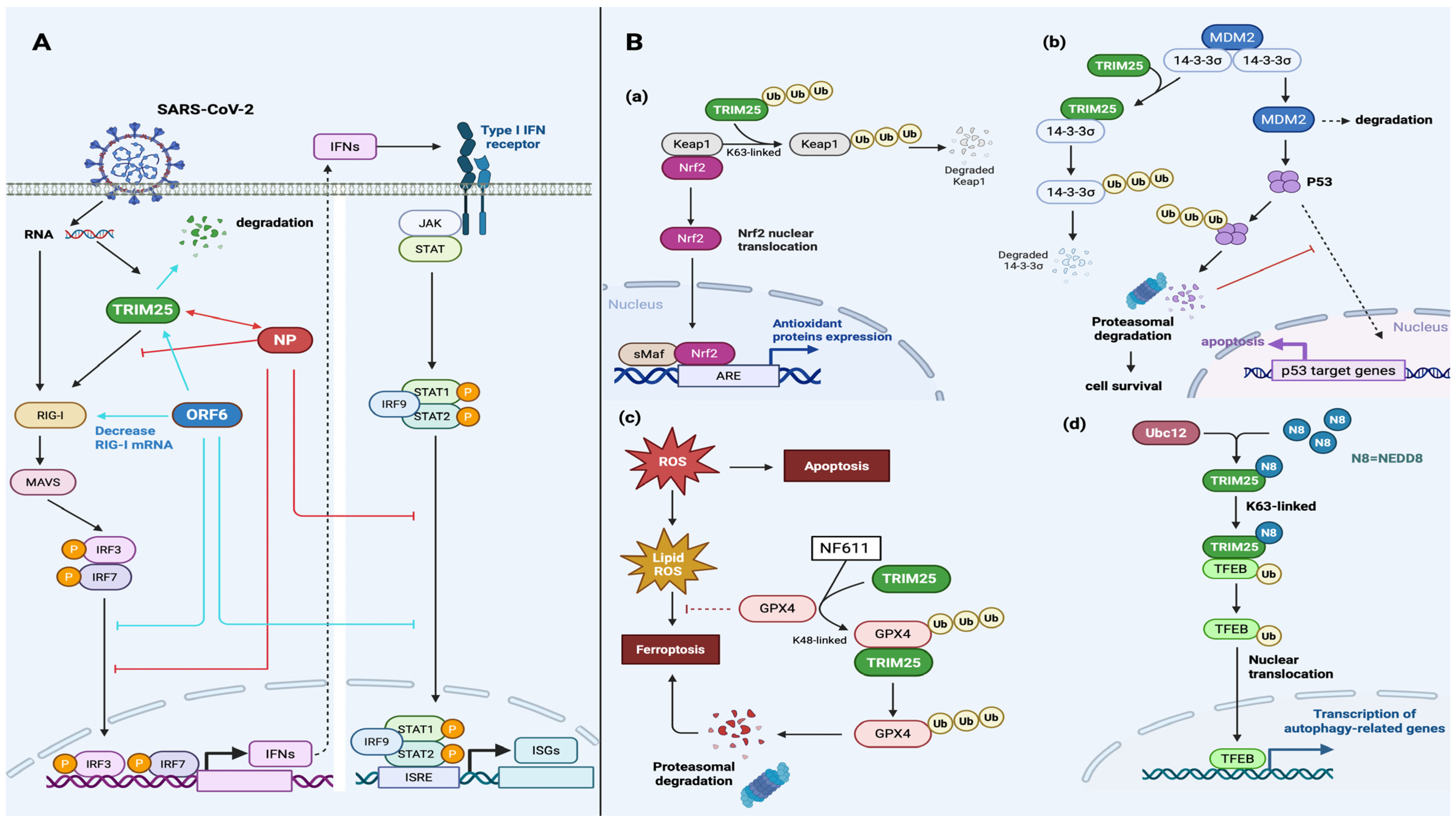
| SARS-CoV-2 | NP | NPs interact with TRIM25 to prevent the binding of TRIM25 to RIG-I in order to inhibit RIG-I polyubiquitination and IFN production. | [70] |
| SARS-CoV-2 NPs recruit TRIM25 and G3BP2 to enhance their interactions and inhibit subsequent RIG-I signaling. | [73] | ||
| SARS-CoV-2 NPs inhibit the formation of SGs by interacting with G3BP1 to prevent the co-condensation of G3BP1 and TRIM25. | [72,74] | ||
| NSP5 | NSP5 inhibits the RLR-mediated IFN response by restricting antiviral SG formation. | [72] | |
| ORF6 | ORF6 inhibits RIG-I activation and IFN production by decreasing the mRNA level of RIG-I and mediating the proteasomal degradation of TRIM25. | [65] | |
| ORF6 directly inhibits the nuclear translocation of IRF3/7 and STAT1 to induce IFN and ISG activity and trigger the IFN-mediated signaling pathway. | [65] | ||
| Influenza A viruses | RNA | TRIM25 inhibits IAV replication and assembly by binding to the positive strand of RNA, promoting RNA instability and degradation. | [75] |
| NS1-A | NS1-A directly interacts with RIG-I to inactivate subsequent IFN transcription. | [76,77] | |
| Influenza B viruses | NS1-B | NS1-B interacts with TRIM25 to relieve the inhibition of the NS1-B CTD region upon RIG-I ubiquitination, enhancing IFN and inflammatory cytokine production. | [78] |
| EBOV | vRNP | TRIM25 ubiquitinates EBOV vRNP NPs and exposes CpG-rich viral genome sequences for ZAP recognition and a subsequent antiviral response. | [79,80] |
| DENV-2 | sfRNA | sfRNA binds to TRIM25 to prevent its deubiquitination, which prevents subsequent activation of RIG-I/IFN signaling. | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Peng, S.; Wei, J.; Xie, Z. The Function of TRIM25 in Antiviral Defense and Viral Immune Evasion. Viruses 2025, 17, 735. https://doi.org/10.3390/v17050735
Liu Q, Peng S, Wei J, Xie Z. The Function of TRIM25 in Antiviral Defense and Viral Immune Evasion. Viruses. 2025; 17(5):735. https://doi.org/10.3390/v17050735
Chicago/Turabian StyleLiu, Qianxun, Shantong Peng, Jiani Wei, and Zhenzhen Xie. 2025. "The Function of TRIM25 in Antiviral Defense and Viral Immune Evasion" Viruses 17, no. 5: 735. https://doi.org/10.3390/v17050735
APA StyleLiu, Q., Peng, S., Wei, J., & Xie, Z. (2025). The Function of TRIM25 in Antiviral Defense and Viral Immune Evasion. Viruses, 17(5), 735. https://doi.org/10.3390/v17050735